
Physico-chemical profiles of the wobble ↔ Watson–Crick G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) tautomerisations: a QM/QTAIM comprehensive survey†
Ol'ha O.
Brovarets'
 *ab,
Ivan S.
Voiteshenko
b and
Dmytro M.
Hovorun
ab
*ab,
Ivan S.
Voiteshenko
b and
Dmytro M.
Hovorun
ab
aDepartment of Molecular and Quantum Biophysics, Institute of Molecular Biology and Genetics, National Academy of Sciences of Ukraine, 150 Akademika Zabolotnoho Str., 03680 Kyiv, Ukraine. E-mail: o.o.brovarets@imbg.org.ua
bDepartment of Molecular Biotechnology and Bioinformatics, Institute of High Technologies, Taras Shevchenko National University of Kyiv, 2-h Akademika Hlushkova Ave., 03022 Kyiv, Ukraine
First published on 23rd November 2017
Abstract
This study is intended to clarify in detail the tautomeric transformations of the wobble (w) G*·2AP(w) and A·2AP(w) nucleobase mispairs involving 2-aminopurine (2AP) into the Watson–Crick (WC) G·2AP(WC) and A*·2AP(WC) base mispairs (asterisks denote mutagenic tautomers of the DNA bases), respectively, by quantum-mechanical methods and Bader's Quantum Theory of Atoms in Molecules. Our previously reported methodology has been used, which allows the evolution of the physico-chemical parameters to be tracked along the entire internal reaction coordinate (IRC), not exclusively in the stationary states of these reactions. These biologically important G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisations, which are involved in mutagenic tautomerically-conformational pathways, determine the origin of the transitions and transversions induced by 2AP. In addition, it is established that they proceed through planar, highly stable, zwitterionic transition states and they exhibit similar physico-chemical profiles and stages of sequential intrapair proton transfer, followed by spatial rearrangement of the nucleobases relative to each other within the base pairs. These w ↔ WC tautomerisations occur non-dissociatively and are accompanied by a significant alteration in geometry (from wobble to Watson–Crick and vice versa) and redistribution of the specific intermolecular interactions, which can be divided into 10 patterns including AH⋯B H-bonds and loosened A–H–B covalent bridges along the IRC of tautomerisation. Based on the redistribution of the geometrical and electron-topological parameters of the intrapair hydrogen bonds, exactly 9 key points have been allocated to characterize the evolution of these reactions.
Introduction
Establishment of the fundamental mechanisms of the origin of point mutations induced by analogues of DNA bases is an intriguing topic in biophysics of DNA due to their importance in human health and numerous technical applications.1–8To date, several experimental9–22 and theoretical23–26 studies have reported the mechanisms of action of the classic mutagen 2-aminopurine (2AP), which is also used as a fluorescent probe,18–21 inducing A·T → G·C and G·C → A·T transitions.11,12 However, these results are essentially independent and do not represent a complete picture of the mechanisms of the origin of the point mutations in DNA induced by 2AP.
In recent papers,27–35 a solution of this extremely important and very difficult biological problem was comprehensively and thoroughly addressed by analyzing the structural mechanisms of the 2AP mutagenic pressure on DNA due to the latest breakthrough in the precise understanding of the origin of spontaneous point mutagenesis in DNA36–42 within the framework of the classical Watson–Crick tautomeric hypothesis.43
Moreover, our theoretical data on spontaneous point mutations and that induced by analogues of the DNA bases, in particular transitions and transversions, are in good agreement with the experimental observations.9–22,44–48
In particular, our results prove that 2AP produces transversions by forming a wobble (w) mispair with A DNA base, A·2AP(w), followed by further A·2AP(w) → A*·2AP(WC) (WC – Watson–Crick) tautomerisation and subsequent A*·2AP(WC) → A*·2APsyn conformational transition, thus acquiring the A*·2APsyn enzymatically-competent conformation33 (herein, the asterisk denotes the mutagenic tautomers of the DNA bases49–57).
It was also shown for the first time that 2AP* may also produce another transversion when the 2AP* mutagenic tautomer pairs with the G base to form the G·2AP*(w) mispair, which converts to the G·2APsyn enzymatically-competent structure via consecutive tautomeric and conformational transformations: G·2AP*(w) → G*·2AP(w) → G·2AP(WC) → G·2APsyn.32 In this case, the long G·2AP(WC) WC-like mispair acts as a precursor to the enzymatically-competent conformation, the G·2APsyn mispair.
The estimated ratio of probabilities PA·2AP/PA·A = 40.533 and PG·2AP*/PG·A* = 1.9 × 107![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 indicates that these structural transformation routes are mutagenic. Notably, the formed A*·2APsyn and G·2APsyn base pairs quite easily acquire an enzymatically-competent WC conformation during the process of thermal fluctuations, which enables their successful incorporation into the DNA double helix by the high-fidelity replicative DNA-polymerase.
32 indicates that these structural transformation routes are mutagenic. Notably, the formed A*·2APsyn and G·2APsyn base pairs quite easily acquire an enzymatically-competent WC conformation during the process of thermal fluctuations, which enables their successful incorporation into the DNA double helix by the high-fidelity replicative DNA-polymerase.
The present work presents a detailed quantum-mechanical/Quantum Theory of Atoms in Molecules (QM/QTAIM) investigation of the physico-chemical parameters at each point of the intrinsic reaction coordinate (IRC) of the acquisition by the G*·2AP(w) and A·2AP(w) nucleobase mispairs involving the 2AP mutagen27–30 with Watson–Crick geometry due to the G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisations. This investigation enables the monitoring of the energetic, polar, geometric, charge, electron-topological and natural bond orbital (NBO) characteristics of the base mispairs and intrapair interactions along the IRC. It is worthy to note that the profiles of the geometric parameters, including the distance R(H9–H9) between the H9 and H9 glycosidic hydrogens, glycosidic angles α1/α2, dA⋯B distances between the electronegative A and B atoms, ∠AH⋯B angles of the AH⋯B hydrogen (H) bonds and patterns of the energy of the intermolecular H-bonds EHB indicate significant rebuilding of the geometry of the nucleobase pairs and the non-dissociative character of these biologically important w ↔ WC tautomerisations, which occur without breakage of the pairs. Using the obtained evolutions of the physico-chemical parameters along the IRC, 9 key points (KPs) have been established, which allow the phases of the reactions to be distinguish and demonstrate that in the course of these w ↔ WC tautomerisations, protons move sequentially without a stable intermediate, followed by spatial rearrangement of the nucleobases relative to each other within the base pairs.
Computational methods
All calculations were performed using the Gaussian'09 program package.58 The geometries and harmonic vibrational frequencies of the considered base mispairs and transition states (TSs) of their mutual tautomeric conversions were obtained in our previous work at the B3LYP/6-311++G(d,p) level of theory, which suggested that it is suitable for the investigation of similar systems,59,60 followed by single point energy calculations at the MP2/aug-cc-pVDZ level of theory.27–35In order to consider the surrounding effect on the investigated tautomerisation processes, we used the conductor-like polarizable continuum model (CPCM),61,62 choosing the continuum with a dielectric constant ε = 4, which is typical for the active center of DNA-polymerase.63,64
The reaction pathway for the proton transfer (PT) tautomerisation of the wobble base mispairs was obtained by following the IRC in the forward and reverse directions from the transition state (TS) using the Hessian-based predictor–corrector integration algorithm65 with tight convergence criteria. We obtained the profiles of the energetic, geometric, polar, charge, electron-topological and NBO characteristics of the H-bonds and complexes along the pathway of the tautomerisation reactions by calculating them at each point of the IRC using our previously reported methodology.66–70 The profiles of the NBO charges along the IRC were obtained using the NBO program,71 which was implemented in the Gaussian'0958 program package.
Bader's quantum theory of atoms in molecules (QTAIM) was applied to analyse the electron density distribution72 using the program package AIMAll73 with all default options. The presence of a bond critical point (BCP), namely the so-called (3,−1) BCP, and a bond path between hydrogen donor and acceptor, as well as the positive value of the Laplacian at this BCP (Δρ > 0), were considered as criteria for H-bond formation.74 Wave functions were obtained at the level of theory used for geometry optimisation.
The H-bond energy, EHB, was calculated using the empirical Espinosa–Molins–Lecomte (EML) formula75,76 based on the electron density distribution at the (3,−1) BCPs of the H-bonds:
| EHB = 0.5·V(r) |
The atomic numbering scheme for the nucleotide bases is conventional.77
Results and discussion
Exhaustive analysis of the potential energy surface
Comprehensive analysis of the potential energy surface and IRC calculations revealed that the G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisations occur via the initial transfer of the single proton localized at the O6/N6 atoms of the G*/A DNA bases in the wobble G*·2AP(w)/A·2AP(w) mispairs along the upper O6/N6H⋯N1 H-bonds to the 2AP base followed by the shifting of the 2AP base according the G*/A DNA bases into the minor groove of DNA. This results in the localized transition states TSG−·2AP+G*·2AP(w)↔G·2AP(WC)/TSA−·2AP+A·2AP(w)↔A*·2AP(WC) since the G−/A−·2AP+ tight ion pairs are stabilized by the participation of the network of the C6+H⋯O6−/N6−, N1+H⋯O6−/N6−, N1+H⋯N1−, N2+H⋯N1−, and N2+H⋯N2− H-bonds (Fig. 1 and 5) with the ΔΔGTS/ΔΔETS barriers of 18.04/16.58 and 29.85/31.00 kcal mol−1 (Tables 2–5), respectively. Further, the reagent complexes finally acquire the Watson–Crick geometry and the mobile proton at the N1 atom of the 2AP+ base moves back to the N1 atom of the G*/A DNA bases along the middle N1H⋯N1 H-bond. This compensates the excessive charge of the 2AP+ nucleobase and leads to the formation of the G·2AP(WC)/A*·2AP(WC) Watson–Crick-like nucleobase mispairs joined by the C6H⋯O6/N6, N1H⋯N1, and N2/C2H⋯N2/HN2 H-bonds with relative ΔΔG/ΔΔE energies of 1.33/1.07 and 13.71/13.57 kcal mol−1 (Table 1), respectively. It was established that for the minima, which are the initial G*·2AP(w)/A·2AP(w) and terminal G·2AP(WC)/A*·2AP(WC) complexes, all frequencies of the normal vibrations are real, whereas the TSs possess one imaginary frequency, corresponding to the shifting motion of the bases relative to each other into the minor and major DNA grooves.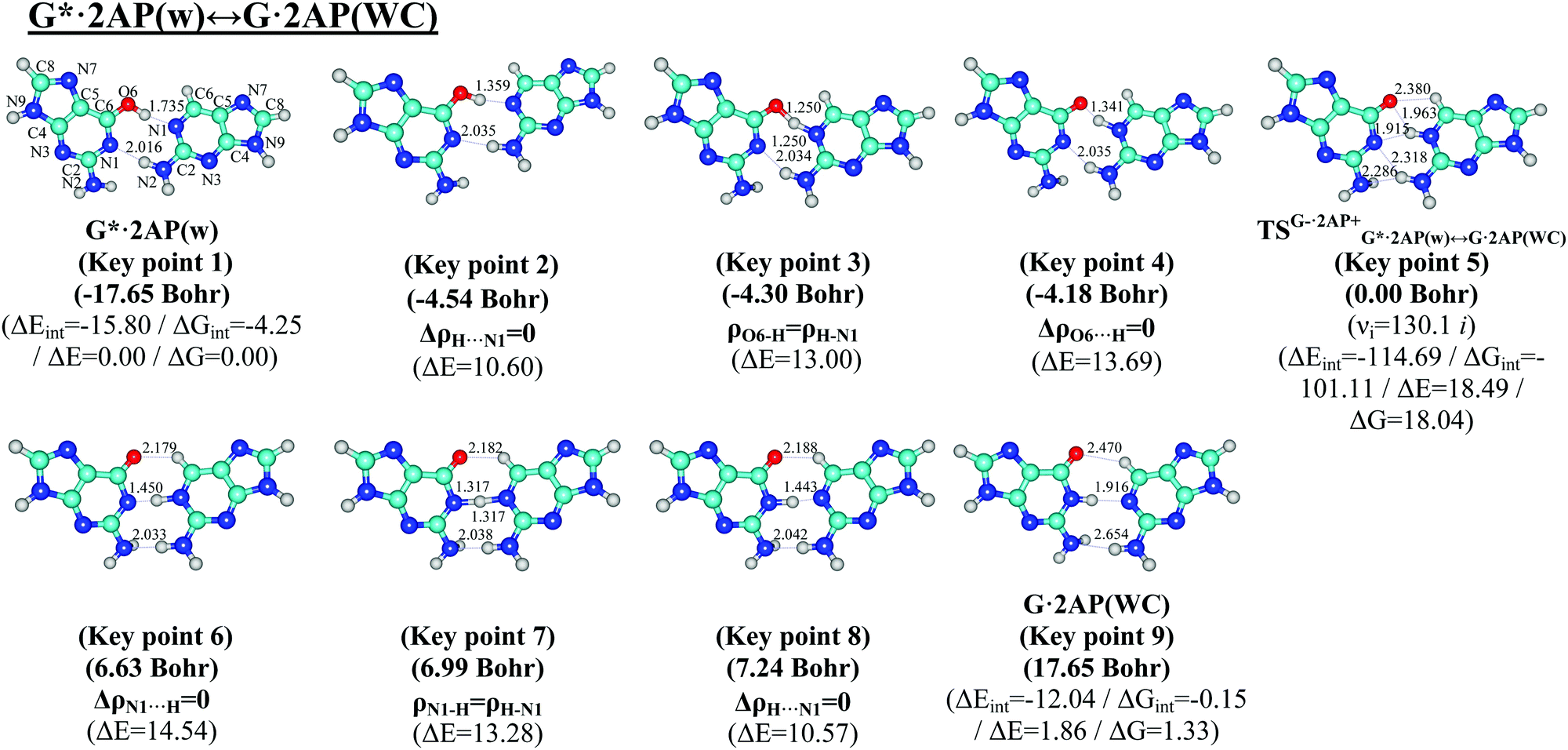 | ||
| Fig. 1 Geometrical structures of the 9 key points describing the evolution of the G*·2AP(w) ↔ G·2AP(WC) w ↔ WC tautomerisation via the sequential PT along the IRC obtained at the B3LYP/6-311++G(d,p) level of theory in vacuo. The coordinates of the 9 key points, their relative Gibbs free energies ΔG or relative electronic energies ΔE (in kcal mol−1 obtained at the MP2/aug-cc-pVDZ//B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1 at T = 298.15 K) and imaginary frequencies νi (cm−1) at the TSs of their interconversions are presented below them in brackets. The dotted lines indicate AH⋯B H-bonds, while the continuous lines show covalent bonds (their lengths are presented in angstroms). Carbon atoms are in light-blue, nitrogen in dark-blue, hydrogen in grey and oxygen in red. | ||
| Tautomeric transformation | ν i | ΔGb | ΔEc | ΔΔGTSd | ΔΔETSe | ΔΔGf | ΔΔEg | k f | k r | τ 99.9% | τ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Note:a Imaginary frequency at the TSs of the tautomerisation reaction, cm−1.b The Gibbs free energy of the product relative to the reactant of the tautomerisation reaction (T = 298.15 K), kcal mol−1.c The electronic energy of the product relative to the reactant of the tautomerisation reaction, kcal mol−1.d The Gibbs free energy barrier for the forward reaction of tautomerisation, kcal mol−1.e The electronic energy barrier for the forward reaction of the tautomerisation, kcal mol−1.f The Gibbs free energy barrier for the reverse reaction of the tautomerisation, kcal mol−1.g The electronic energy barrier for the reverse reaction of the tautomerisation, kcal mol−1.h The forward rate constant for the tautomerisation reaction, s−1.i The reverse rate constant for the tautomerisation reaction, s−1.j The time necessary to reach 99.9% of the equilibrium concentration between the reactant and the product of the tautomerization reaction, s.k The lifetime of the product of the tautomerisation reaction, s. | |||||||||||
| ε = 1 | |||||||||||
| G*·2AP(w) ↔ G·2AP(WC) | 130.1 | 1.33 | 1.07 | 18.04 | 16.58 | 16.70 | 15.51 | 0.37 | 3.53 | 1.77 | 0.28 |
| G*·A(w) ↔ G·A(WC) | 107.2 | −4.30 | −6.73 | 12.33 | 9.97 | 16.64 | 16.70 | 5.64 × 103 | 3.94 | 1.22 × 10−3 | 0.25 |
| A·2AP(w) ↔ A*·2AP(WC) | 117.0 | 13.71 | 13.57 | 29.85 | 31.00 | 16.14 | 17.43 | 8.01 × 10−10 | 9.10 | 0.76 | 0.11 |
| A·A(w) ↔ A*·A(WC) | 152.4 | 3.63 | 1.09 | 25.86 | 22.56 | 22.24 | 21.47 | 6.79 × 10−7 | 3.10 × 10−4 | 2.22 × 104 | 3.22 × 103 |
| ε = 4 | |||||||||||
| G*·2AP(w) ↔ G·2AP(WC) | 104.6 | −1.20 | −1.09 | 12.30 | 11.61 | 13.50 | 12.71 | 5.93 × 103 | 7.80 × 102 | 1.03 × 10−3 | 1.28 × 10−3 |
| G*·A(w) ↔ G·A(WC) | 84.5 | −4.87 | −6.90 | 8.42 | 6.08 | 13.29 | 12.98 | 4.20 × 106 | 1.11 × 103 | 8.97 × 10−4 | 1.65 × 10−6 |
| A·2AP(w) ↔ A*·2AP(WC) | 95.9 | 12.62 | 11.88 | 26.53 | 26.33 | 13.91 | 14.45 | 2.18 × 10−7 | 3.95 × 102 | 1.75 × 10−2 | 2.53 × 10−3 |
| A·A(w) ↔ A*·A(WC) | 108.3 | 6.82 | 2.37 | 21.82 | 19.19 | 15.01 | 16.82 | 6.17 × 10−4 | 61.71 | 0.11 | 1.62 × 10−2 |
| Complex | AH⋯B H-bond/A–H/H–B covalent bond | ρ | Δρb | 100·εc | d A⋯B | d H⋯B | ∠AH···Bf | μ |
|---|---|---|---|---|---|---|---|---|
| Notes:a The electron density at the (3,−1) BCP, a.u.b The Laplacian of the electron density at the (3,−1) BCP, a.u.c The ellipticity at the (3,−1) BCP.d The distance between the A (H-bond donor) and B (H-bond acceptor) atoms of the AH⋯B H-bonds, Å.e The distance between the H and B atoms of the AH⋯B H-bonds, Å.f The H-bond angle, degree.g The dipole moment of the complex, D. | ||||||||
| Key point 1 (−17.65 Bohr): G*·2AP(w) | O6H⋯N1 | 0.049 | 0.100 | 5.14 | 2.728 | 1.735 | 168.4 | 4.58 |
| N1⋯HN2 | 0.026 | 0.077 | 7.43 | 3.029 | 2.016 | 171.3 | ||
| Key point 2 (−4.54 Bohr): ΔρH⋯N1 = 0 | O6H⋯N1 | 0.126 | −0.037 | 3.27 | 2.492 | 1.359 | 164.3 | 5.64 |
| N1⋯HN2 | 0.025 | 0.076 | 6.60 | 2.989 | 2.035 | 155.0 | ||
| Key point 3 (−4.30 Bohr): ρO6–H = ρH–N1 | O6–H/H–N1 | 0.169 | −0.317 | 2.79 | 2.500 | 1.250 | 161.9 | 6.82 |
| N1⋯HN2 | 0.025 | 0.075 | 6.65 | 2.989 | 2.034 | 154.8 | ||
| Key point 4 (−4.18 Bohr): ΔρO6⋯H = 0 | O6⋯HN1 | 0.124 | 0.008 | 2.25 | 2.507 | 1.341 | 160.5 | 7.41 |
| N1⋯HN2 | 0.025 | 0.074 | 6.64 | 2.990 | 2.035 | 154.6 | ||
| Key point 5 (0.00 Bohr): TSG−·2AP+G*·2AP(w)↔G·2AP(WC) | O6−⋯HC6+ | 0.013 | 0.049 | 67.40 | 2.961 | 2.380 | 112.0 | 10.62 |
| O6−⋯HN1+ | 0.028 | 0.092 | 26.10 | 2.754 | 1.963 | 129.7 | ||
| N1−⋯HN1+ | 0.031 | 0.088 | 6.86 | 2.910 | 1.915 | 156.9 | ||
| N1−⋯HN2+ | 0.014 | 0.045 | 8.67 | 3.166 | 2.318 | 139.9 | ||
| N2−⋯HN2+ | 0.013 | 0.044 | 9.18 | 3.240 | 2.286 | 155.5 | ||
| Key point 6 (6.63 Bohr): ΔρN1⋯H = 0 | O6⋯HC6 | 0.018 | 0.064 | 7.54 | 2.963 | 2.179 | 127.1 | 9.50 |
| N1⋯HN1 | 0.104 | 0.006 | 4.74 | 2.628 | 1.450 | 173.5 | ||
| N2⋯HN2 | 0.025 | 0.072 | 5.73 | 3.039 | 2.033 | 167.3 | ||
| Key point 7 (6.99 Bohr): ρN1–H = ρH–N1 | O6⋯HC6 | 0.018 | 0.064 | 7.52 | 2.965 | 2.182 | 127.0 | 8.47 |
| N1–H/H–N1 | 0.145 | −0.168 | 3.56 | 2.612 | 1.317 | 172.4 | ||
| N2⋯HN2 | 0.025 | 0.073 | 6.23 | 3.038 | 2.038 | 166.5 | ||
| Key point 8 (7.24 Bohr): ΔρH⋯N1 = 0 | O6⋯HC6 | 0.017 | 0.063 | 7.64 | 2.967 | 2.188 | 126.7 | 7.79 |
| N1H⋯N1 | 0.105 | 0.024 | 4.32 | 2.610 | 1.443 | 170.9 | ||
| N2⋯HN2 | 0.025 | 0.074 | 6.51 | 3.039 | 2.042 | 166.3 | ||
| Key point 9 (17.65 Bohr): G·2AP(WC) | O6⋯HC6 | 0.010 | 0.029 | 2.90 | 3.366 | 2.470 | 138.9 | 7.72 |
| N1H⋯N1 | 0.033 | 0.088 | 6.76 | 2.948 | 1.916 | 176.2 | ||
| N2⋯HN2 | 0.008 | 0.026 | 110.77 | 3.400 | 2.654 | 130.7 | ||
| Patterns | IRC range, Bohr | Specific intermolecular interactions, forming patterns |
|---|---|---|
| I | [−17.65 to −4.54) | (G)O6H⋯N1(2AP), (G)N1⋯HN2(2AP) |
| II | [−4.54 to −4.18) | (G)O6-H-N1(2AP), (G)N1⋯HN2(2AP) |
| III | [−4.18 to −2.46) | (G)O6⋯HN1(2AP), (G)N1⋯HN2(2AP) |
| IV | [−2.46 to −1.97) | (G)O6⋯HN1(2AP), (G)N1⋯HN2(2AP), (G)N2⋯HN2(2AP) |
| V | [−1.97 to −0.86) | (G)O6⋯HN1(2AP), (G)N1⋯HN1(2AP), (G)N1⋯HN2(2AP), (G)N2⋯HN2(2AP) |
| VI | [−0.86 to 2.21) | (G)O6⋯HC6(2AP), (G)O6⋯HN1(2AP), (G)N1⋯HN1(2AP), (G)N1⋯HN2(2AP), (G)N2⋯HN2(2AP) |
| VII | [2.21 to 3.56) | (G)O6⋯HC6(2AP), (G)N1⋯HN1(2AP), (G)N1⋯HN2(2AP), (G)N2⋯HN2(2AP) |
| VIII | [3.56 to 6.75) | (G)O6⋯HC6(2AP), (G)N1⋯HN1(2AP), (G)N2⋯HN2(2AP) |
| IX | [6.75 to 7.24) | (G)O6⋯HC6(2AP), (G)N1-H-N1(2AP), (G)N2⋯HN2(2AP) |
| X | [7.24 to 17.65] | (G)O6⋯HC6(2AP), (G)N1H⋯N1(2AP), (G)N2⋯HN2(2AP) |
| Complex | AH⋯B H-bond/A–H/H–B covalent bond | ρ | Δρ | 100·ε | d A···B | d H···B | ∠AH···B | μ |
|---|---|---|---|---|---|---|---|---|
| Notes: for footnote definitions see Table 2. | ||||||||
| Key point 1 (−18.38 Bohr): A·2AP(w) | N6H⋯N1 | 0.029 | 0.081 | 7.56 | 3.002 | 1.976 | 179.4 | 1.09 |
| N1⋯HN2 | 0.026 | 0.076 | 7.89 | 3.043 | 2.021 | 179.7 | ||
| Key point 2 (−5.21 Bohr): ΔρH⋯N1 = 0 | N6H⋯N1 | 0.105 | 0.033 | 3.41 | 2.598 | 1.426 | 171.9 | 3.37 |
| N1⋯HN2 | 0.028 | 0.083 | 7.18 | 2.957 | 1.977 | 159.5 | ||
| Key point 3 (−4.96 Bohr): ρN6–H = ρH–N1 | N6–H/H–N1 | 0.142 | −0.146 | 2.79 | 2.602 | 1.313 | 168.7 | 4.91 |
| N1⋯HN2 | 0.029 | 0.082 | 7.20 | 2.957 | 1.974 | 159.5 | ||
| Key point 4 (−4.60 Bohr): ΔρN6⋯H = 0 | N6⋯HN1 | 0.098 | 0.011 | 4.80 | 2.621 | 1.475 | 163.0 | 7.06 |
| N1⋯HN2 | 0.029 | 0.080 | 7.18 | 2.959 | 1.974 | 159.2 | ||
| Key point 5 (0.00 Bohr): TSA−·2AP+A·2AP(w)↔A*·2AP(WC) | N6−⋯HC6+ | 0.016 | 0.052 | 9.19 | 3.023 | 2.344 | 119.0 | 9.98 |
| N6−⋯HN1+ | 0.020 | 0.071 | 47.64 | 2.887 | 2.131 | 126.4 | ||
| N1−⋯HN1+ | 0.039 | 0.089 | 1.05 | 2.860 | 1.834 | 162.3 | ||
| N1−⋯HN2+ | 0.015 | 0.048 | 4.15 | 3.154 | 2.277 | 144.0 | ||
| Key point 6 (4.00 Bohr): ΔρN1⋯H = 0 | N6⋯HC6 | 0.020 | 0.063 | 1.63 | 3.012 | 2.205 | 129.1 | 8.54 |
| N1⋯HN1 | 0.097 | 0.016 | 4.34 | 2.661 | 1.482 | 179.2 | ||
| C2H⋯HN2 | 0.008 | 0.029 | 25.88 | 2.689 | 2.049 | 114.3 | ||
| Key point 7 (4.35 Bohr): ρN1–H = ρH–N1 | N6⋯HC6 | 0.020 | 0.063 | 1.55 | 3.014 | 2.209 | 129.0 | 6.98 |
| N1–H/H–N1 | 0.145 | −0.181 | 3.36 | 2.645 | 1.317 | 177.8 | ||
| Key point 7 (4.60 Bohr): ΔρH⋯N1 = 0 | C2H⋯HN2 | 0.008 | 0.028 | 18.64 | 2.701 | 2.051 | 115.2 | |
| Key point 8 (4.60 Bohr): ΔρH⋯N1 = 0 | N6⋯HC6 | 0.019 | 0.063 | 1.45 | 3.015 | 2.215 | 128.6 | 5.79 |
| N1H⋯N1 | 0.106 | 0.012 | 4.02 | 2.642 | 1.440 | 176.2 | ||
| C2H⋯HN2 | 0.008 | 0.028 | 15.70 | 2.704 | 2.052 | 115.4 | ||
| Key point 9 (16.31 Bohr): A*·2AP(WC) | N6⋯HC6 | 0.013 | 0.039 | 5.58 | 3.332 | 2.388 | 144.2 | 4.55 |
| N1H⋯N1 | 0.029 | 0.082 | 7.01 | 2.991 | 1.961 | 175.2 | ||
| C2H⋯HN2 | 0.001 | 0.005 | 75.42 | 3.668 | 2.987 | 125.7 | ||
| Patterns | IRC range, Bohr | Specific intermolecular interactions, forming patterns |
|---|---|---|
| I | [−18.34 to −5.09) | (A)N6H⋯N1(2AP), (A)N1⋯HN2(2AP) |
| II | [−5.09 to −4.60) | (A)N6–H–N1(2AP), (A)N1⋯HN2(2AP) |
| III | [−4.60 to −3.51) | (A)N6⋯HN1(2AP), (A)N1⋯HN2(2AP) |
| IV | [−3.51 to −2.91) | (A)N6⋯HN1(2AP), (A)N1⋯HN1(2AP), (A)N1⋯⋯HN2(2AP) |
| V | [–2.91 to 1.21) | (A)N6⋯HC6(2AP), (A)N6⋯HN1(2AP), (A)N1⋯HN1(2AP), (A)N1⋯HN2(2AP) |
| VI | [1.21 to 2.30) | (A)N6⋯HC6(2AP), (A)N1⋯HN1(2AP), (A)N1⋯HN2(2AP) |
| VII | [2.30 to 3.26) | (A)N6⋯HC6(2AP), (A)N1⋯HN1(2AP), (A)N1⋯HN2(2AP), (A)C2H⋯HN2(2AP) |
| VIII | [3.26 to 4.23) | (A)N6⋯HC6(2AP), (A)N1⋯HN1(2AP), (A)C2H⋯HN2(2AP) |
| IX | [4.23 to 4.60) | (A)N6⋯HC6(2AP), (A)N1–H–N1(2AP), (A)C2H⋯HN2(2AP) |
| X | [4.60 to 16.31] | (A)N6⋯HC6(2AP), (A)N1H⋯N1(2AP), (A)C2H⋯HN2(2AP) |
Analysis of the potential energy surface shows that these reactions proceed through the sequential intrapair PT.
The profiles of the electronic energy of the G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) tautomerisation reactions are quite smooth in the vicinity of the TSs. In contrast, peaks and fractures are present on both sides of the TSs, which correspond to the range of existence of the G−/A−·2AP+ ion pairs during their transformations, namely to KP 4 and KP 6 (Fig. 2a and 6a, respectively).
The transition from vacuum to weakly polar continuum with ε = 4, which is typical for the interfaces of specific protein–nucleic acid interactions, does not qualitatively affect the course and structure of the transition state of the tautomerisation reactions (Fig. S1, ESI†). This could be explained by the fact that this transition does not disturb the structure of the TSs of these tautomerisations, since it is a highly stable, tight ion pair. Simultaneously, this transition accelerates the w ↔ WC tautomerisation, while the shape of the curves dependent on the energy of the studied pairs from the IRC does not significantly change (Fig. S1, ESI†).
The calculated first derivatives of the electronic energy E by the IRC, in particular, the peaks on its curve, enable the reaction pathways of these reactions to be divided into regions of reagent (13.11/13.17 Bohr), transition state (11.78/9.81 Bohr) and product (10.41/11.71 Bohr) based on the data collected by us31–37 and our colleagues78–80 (Fig. 2b and 6b). The widest is the preparation reagent stage corresponding to the intrapair proton transfer, whereas the narrowest is the TS region, where mutual reorientation and shifting of the bases relative to each other occur. Consequently, the direct initiation of the G*·2AP(w) → G·2AP(WC) and A·2AP(w) → A*·2AP(WC) reactions requires the electronic energy ΔEKP2 − ΔEKP1[G*·2AP(w)/A·2AP(w)] = 10.60/18.75 kcal mol−1, which represents 57.3%/56.4% of the electronic energies of the TSs. In contrast, much less energy is required for the final relaxation of the reaction complexes to the terminal mispairs: ΔEKP8 − ΔEKP9[G·2AP(WC)/A*·2AP(WC)] = 8.71/13.85 kcal mol−1, which represents 47.1%/41.4% of the energies of TSG−·2AP+G*·2AP(w)↔G·2AP(WC)/TSA−·2AP+A·2AP(w)↔A*·2AP(WC). These observations indicate that more energy is required to initiate the reaction due to the preparatory PT before the structurally-electronic rebuilding of the nucleobases within the mispairs in the TS region, especially in the case of the A·2AP(w) → A*·2AP(WC) w → WC tautomerisation (∼2 times), than for the final relaxation to the terminal mispairs (Fig. 2b).
The established data show the resemblance between the courses of these processes and their electronic energy profiles regardless of their structural difference (Fig. 1, 2, 5 and 6). Notably, these conversions with the participation of the 2AP analogue are established to be the same as that with the participation of the canonical A DNA bases G*·A(w) ↔ G·A(WC)41 and A·A(w) ↔ A*·A(WC)42 (Table 1).
Polar and charge characteristics
The profiles of the dipole moment reflecting the changes in the polarities along the IRC are bell-shaped near the TS (4.40–10.67/1.04–10.00 D) and almost plateau on each side of it with average values of 4.56/1.11 D on the left and 7.43/4.75 D on the right (Fig. 2c and 6c), respectively.The calculated profiles of the NBO charges (0.402–0.508/0.375–0.493 e) of the hydrogen atoms involved in the intermolecular H-bonds are dome-shaped in the TS region with fractures, whereas they are almost constant lines at the beginning and ending of these tautomerisation reactions (Fig. 2d and 6d, respectively). The notable feature of the A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisation is that the NBO charges for the H atoms located at the N2 and N6 atoms coincide at the beginning of the reaction (0.435 and 0.434 e), while their difference increases at the ending of the reaction (0.384 and 0.451 e) (Fig. 2d and 6d), respectively.
Geometric and electron-topological characteristics of the mispairs and intermolecular H-bonds
Using our previously reported methodology81–85 we calculated the changes in the main physico-chemical characteristics at each point of the IRC, which are presented in Fig. 2–4 and 6–8, respectively.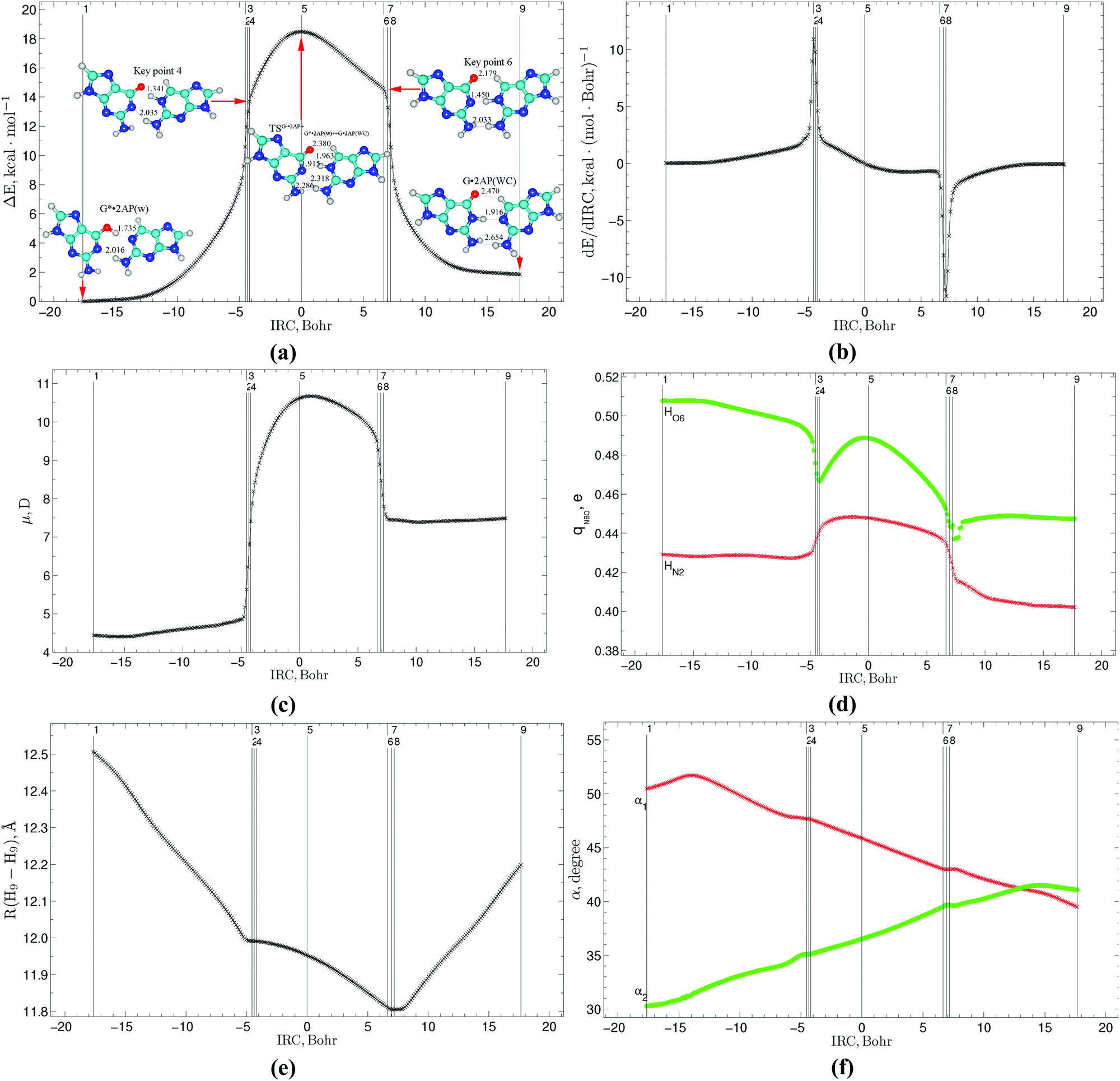 | ||
| Fig. 2 Profiles of (a) relative electronic energy ΔE together with the stationary states (G*·2AP(w), TSG−·2AP+G*·2AP(w)↔G·2AP(WC), G·2AP(WC)) and KPs 4 and 6, (b) first derivative of the electronic energy with respect to the IRC (dE/dIRC), (c) dipole moment μ, (d) NBO charges qNBO, (e) distance R(H9–H9) between the H9 and H9 glycosidic hydrogens and (f) α1 (∠N9H9(2AP)H9(G)) and α2 (∠N9H9(G)H9(2AP)) glycosidic angles along the IRC of the G*·2AP(w) ↔ G·2AP(WC) w ↔ WC tautomerisation via the sequential PT obtained at the B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1. | ||
 | ||
| Fig. 3 Profiles of (a) electron density ρ; (b) Laplacian of the electron density Δρ, (c) ellipticity ε at the (3,−1) BCPs, (d) distance dA⋯B between the electronegative A and B atoms; (e) distance dAH/HB between the hydrogen and electronegative A or B atoms and (f) angle ∠AH⋯B of the H-bonds along the IRC of the G*·2AP(w) ↔ G·2AP(WC) w ↔ WC tautomerisation via the sequential PT obtained at the B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1. | ||
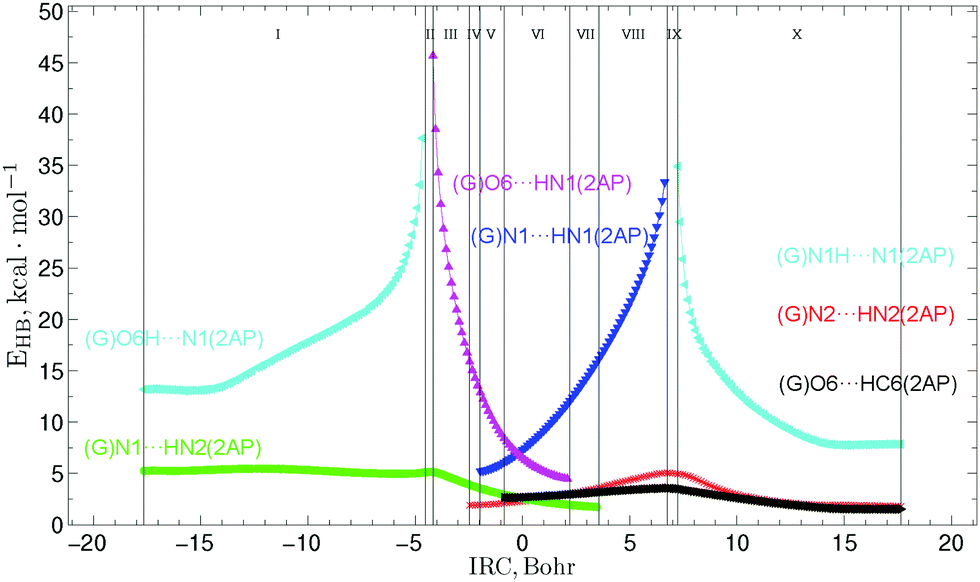 | ||
| Fig. 4 Profiles of the energy of the intermolecular H-bonds EHB estimated by the EML formula at the (3,−1) BCPs along the IRC of the G*·2AP(w) ↔ G·2AP(WC) w ↔ WC tautomerisation via the sequential PT obtained at the B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1 (see Table 3). | ||
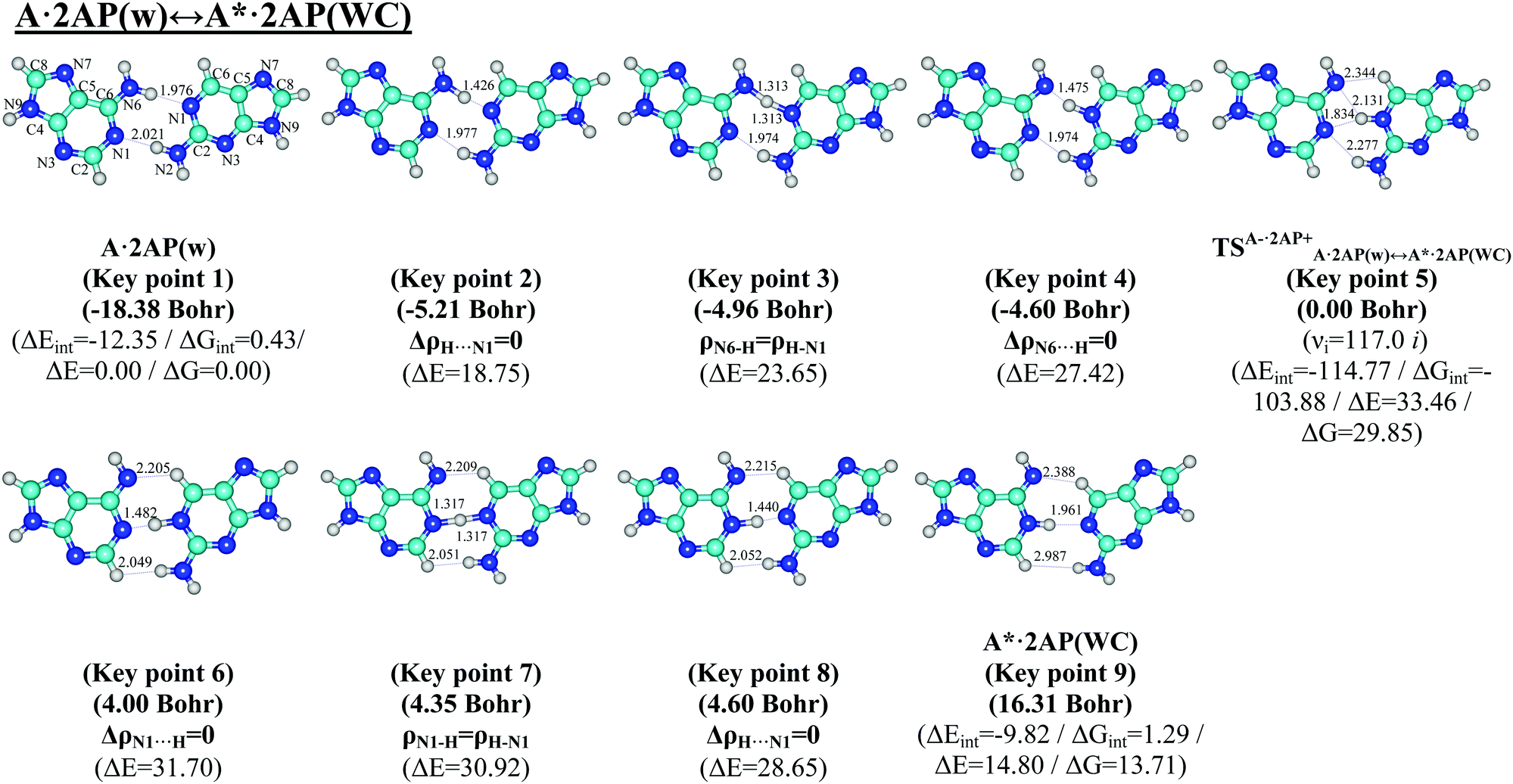 | ||
| Fig. 5 Geometric structures of the 9 key points describing the evolution of the A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisation via the sequential PT along the IRC obtained at the B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1. For designations see Fig. 1. | ||
 | ||
| Fig. 6 Profiles of (a) relative electronic energy ΔE together with stationary states (A·2AP(w), TSA−·2AP+A·2AP(w)↔A*·2AP(WC), A*·2AP(WC)) and KPs 4 and 6, (b) first derivative of the electronic energy with respect to the IRC (dE/dIRC), (c) dipole moment μ, (d) NBO charges qNBO, (e) distance R(H9–H9) between the H9 and H9 glycosidic hydrogens and (f) α1 (∠N9H9(2AP)H9(A)) and α2 (∠N9H9(A)H9(2AP)) glycosidic angles along the IRC of the A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisation via the sequential PT obtained at the B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1. | ||
 | ||
| Fig. 7 Profiles of (a) electron density ρ; (b) Laplacian of the electron density Δρ, (c) ellipticity ε at the (3,−1) BCPs, (d) distance dA⋯B between the electronegative A and B atoms; (e) distance dAH/HB between the hydrogen and electronegative A or B atoms and (f) angle ∠AH⋯B of the AH⋯B H-bonds along the IRC of the A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisation via the sequential PT obtained at the B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1. | ||
 | ||
| Fig. 8 Profiles of the energy of the intermolecular H-bonds EHB estimated by the EML formula at the (3,−1) BCPs along the IRC of the A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisation via the sequential PT obtained at the B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1 (see Table 5). | ||
Based on the evolution of the geometrical parameters (R(H9–H9), dAH/HB and dA⋯B distances, α1/α2 and ∠AH⋯B angles) along the IRC, it was found that the investigated reactions are followed by significant geometry changes in the base pairs, thus reflecting the large-scale movement of the bases relative to each other within the mispairs (Fig. 2e, f, 3d–f, 6e, f and 7d–f). The distinguished feature of the profiles of the glycosidic distances R(H9–H9) (11.805–12.506/12.142–12.766 Å), distances between the hydrogen and electronegative atoms dAH/HB (1.004–3.353/1.003–3.696 Å), distances between the electronegative atoms dA⋯B (2.490–4.023/2.597–4.209 Å) and angles of the H-bonding ∠AH⋯B (89.4–173.9/89.0–179.7°) is the presence of kinks in the vicinity of the 2nd and 8th key points, which could be associated with the structural changes in the base mispairs from the wobble to the Watson–Crick architecture due to the shifting of the 2AP base downwards into the minor DNA groove according the G*/A bases. The same tendencies have also been observed for the 2AP·T(WC) ↔ 2AP·T*(w) and 2AP·C*(WC) ↔ 2AP·C(w) WC ↔ w tautomeric transformations.34
The curves of the α1 glycosidic angle of 2AP gradually decrease, whereas the curves of the α2 glycosidic angle of the G*/A bases gradually increase along the IRC, which vary in a broad range of values (30.3–51.7/31.7–53.3°) and equalize (41.2/39.7°) at IRC = 13.11/8.57 Bohr (Fig. 2f and 6f, respectively).
The calculations of the profiles of the electron-topological parameters at each point of the IRC, namely the electron density ρ (0.009–0.316/0.001–0.322 a.u.) and Laplacian of the electron density Δρ (−2.141–0.124/−1.703–0.127 a.u.), as well as the dAH/HB distances reveal their crossings at the transformations of the H-bonds along the IRC: (G/A)O6/N6H⋯N1(2AP) → (G/A)O6/N6⋯HN1(2AP) and (G/A)N1⋯HN1(2AP) → (G/A)N1H⋯N1(2AP) (Fig. 3a, b, e, 7a, b and e). These data correspond to the maximal and minimal values of ρ (0.169/0.142 and 0.145/0.145 a.u.), Δρ (−0.317/−0.146 and −0.168/−0.181 a.u.) and dAH/HB (1.250/1.313 and 1.317/1.317 Å) and enable the 2 key points containing loosened O6/N6–H–N1 and N1–H–N1 covalent bridges to be distinguished (Fig. 1 and 5), respectively, which are the 3rd (−4.30/−4.96 Bohr) and 7th (6.99/4.35 Bohr) key points. Another 4 key points have been allocated as that where the Laplacian of the electron density Δρ equals zero, that is the H-bond converts to a covalent bond and vice versa, which are the 2nd (−4.54/−5.21 Bohr), 4th (−4.18/−4.60 Bohr), 6th (6.63/4.00 Bohr) and 8th (7.24/4.60 Bohr) key points. The other 3 key points, the 1st, 5th and 9th, represent stationary structures of the initial wobble G*·2AP(w)/A·2AP(w) base mispairs, the TSG−·2AP+G*·2AP(w)↔G·2AP(WC)/TSA−·2AP+A·2AP(w)↔A*·2AP(WC) transition states and terminal Watson–Crick-like G·2AP(WC)/A*·2AP(WC) base mispairs (Fig. 1 and 5) respectively.
The profiles of the ellipticity ε at the (3,−1) BCPs of the (G/A)N1⋯HN2(2AP), (G/A)O6/N6⋯HC6(2AP), (G/A)N2/C2H⋯N2/HN2(2AP) intermolecular H-bonds gradually increase or decrease along the IRC, rapidly reaching their maxima on the borders, whereas the (G/A)O6/N6H⋯N1(2AP) and (G/A)N1H⋯N1(2AP) H-bonds remain almost stable along the IRC (Fig. 3c and 7c, respectively).
Similarly to the previously investigated 2AP·T(WC) ↔ 2AP·T*(w) and 2AP·C*(WC) ↔ 2AP·C(w) WC ↔ w tautomeric transformations,34 the calculated evolutions of the energies EHB of the intermolecular H-bonds could be divided into 10 patterns which sequentially change each other along the IRC (Tables 3, 5 and Fig. 4, 8).
The G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisations are accompanied by the formation and breakage of the intermolecular H-bonds, including simultaneously maximum 5 H-bonds in the first case, (G)O6⋯HC6(2AP), (G)O6⋯HN1(2AP), (G)N1⋯HN1(2AP), (G)N1⋯HN2(2AP) and (G)N2⋯HN2(2AP), within the ranges of IRC −0.86 to 2.21 Bohr and 4 H-bonds in the second case, (A)N6⋯HC6(2AP), (A)N6⋯HN1(2AP), (A)N1⋯HN1(2AP) and (A)N1⋯HN2(2AP) and (A)N6⋯HC6(2AP), (A)N1⋯HN1(2AP), (A)N1⋯HN2(2AP) and (A)C2H⋯HN2(2AP), within the ranges of IRC from −2.91 to 1.21 and from 2.30 to 3.26 Bohr, respectively. The curves of the (G/A)O6/N6H⋯N1(2AP) and (G/A)N1⋯HN1(2AP) H-bonds gradually increase from 13.19/6.10 to 37.64/36.11 and from 5.22/4.29 to 33.36/33.74 kcal mol−1, curves of the (G/A)O6/N6⋯HN1(2AP) and (G/A)N1H⋯N1(2AP) H-bonds gradually decrease from 45.66/30.37 to 4.47/3.57 and from 34.90/36.64 to 7.86/6.11 kcal mol−1, while the profiles of the (G/A)N1⋯HN2(2AP), (G/A)O6/N6H⋯N1(2AP) and (G/A)N2/C2H⋯HN2(2AP) H-bonds monotonically change along the IRC reaching their maximal values at the borders of the II and III (5.47/6.25 kcal mol−1), VIII and IX (3.56/3.58 kcal mol−1), and IX and X regions (5.04/1.36 kcal mol−1) (Fig. 4 and 8, Tables 3 and 5).
It is worth mentioning that at the beginning of the A·2AP(w) ↔ A*·2AP(WC) reaction, the geometrical, energetic and electron-topological parameters almost coincide, that demonstrates their close values (Fig. 3, 4, 7 and 8, and Tables 2 and 4).
Close comparison of the obtained profiles for the w ↔ WC tautomeric conversions with the participation of the 2AP nucleobase analogue (Fig. 2–4, 6–8 and Fig. S1, ESI†) and canonical DNA bases40,41 reflects high similarity between the courses of these processes and their scanning along the IRC. This represents an especially valuable observation since it could be used for the extension of the data for the canonical conversions to that with the participation of 2AP.
Notably, in all considered tautomerisation reactions, we did not fix the non-planar deformations of the DNA bases, despite their susceptibility to such processes.86–88
Conclusions
The current study is intended to give insight into the physico-chemical mechanism of the G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisations, which are involved into the mutagenic G·2AP*(w) → G*·2AP(w) → G·2AP(WC) → G·2APsyn32,35 and A·2AP(w) → A*·2AP(WC) → A*·2APsyn33 tautomerically-conformational pathways originating from the induced transitions and transversions.The profiles of the electronic energy E, the first derivative of the electronic energy with respect to the IRC, dE/dIRC, the dipole moment of the base pair μ, the NBO charges qNBO of the hydrogen atoms involved in the tautomerisations, the distance R(H9–H9) between the H9 glycosidic hydrogens, the glycosidic angles α1/α2, the electron density ρ, the Laplacian of the electron density Δρ and the ellipticity ε at the (3,−1) BCPs of the intrapair covalent and hydrogen bonds, the distances dA⋯B, dAH/HB, the angles ∠AH⋯B and the energy of the intermolecular H-bonds EHB along the IRC were established.
This detailed analysis enabled us to follow all the changes in the physico-chemical characteristics, including energetic, structural, polar, charge, and electron-topological, along the IRC at each point of these reactions. Moreover, it was found that the G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisations proceed via the followed by the subsequent shifting of the G/A and 2AP bases relative to each other.
Nine key points along the IRC were established, three of them (1st, 5th and 9th) correspond to the stationary structures and the six others (2nd, 3rd, 4th, 6th, 7th and 8th) reflect the rearrangement of the intermolecular H-bonds, O6/N6H⋯N1, N2H⋯N1 (G*·2AP(w)/A·2AP(w)) → C6+H⋯O6−/N6−, N1+H⋯O6−/N6−, N1+H⋯N1−, N2+H⋯N1−, N2+H⋯N2− (TSG−·2AP+G*·2AP(w)↔G·2AP(WC)/TSA−·2AP+A·2AP(w)↔A*·2AP(WC)) → C6H⋯O6/N6, N1H⋯N1 and N2/C2H⋯N2/HN2 (G·2AP(WC)/A*·2AP(WC)), which are grouped into 10 patterns of interactions including AH⋯B H-bonds and loosened A–H–B covalent bridges.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully appreciate technical support and computational facilities of joint computer cluster of SSI “Institute for Single Crystals” of the National Academy of Sciences of Ukraine (NASU) and Institute for Scintillation Materials of the NASU incorporated into the Ukrainian National Grid. This work was partially supported by the Grant of the NASU for young scientists for 2017 year, Grant of the President of Ukraine to support the research of young scientists [project number F70] from the State Fund for Fundamental Research of Ukraine of the Ministry of the Education and Science of Ukraine and by the Personal Scholarship of Verkhovna Rada (Parliament) of Ukraine for the talented young scientists in 2017 year given to DrSci Ol'ha O. Brovarets'. O. O. B. expresses sincere gratitude to organizing committee for financial support of the participation in the “EMBO/FEBS Lecture Course Spetsai Summer School 2017 for Proteins and Organized Complexity” (September 24–October 1, 2017, Spetses, Greece), to Lawyers Association “AVER Lex” (Kyiv, Ukraine) for the sponsorship of the presenting the plenary lecture as invited speaker at the “EMN Meeting on Computation and Theory” (November 6–10, 2017, Dubai, United Arab Emirates) and to Max Planck Institute of Plant Physiology (hosted by Dr. Yariv Brotman) for the invitation and financial support of the invited talk (November 29, 2017, Potsdam, Germany).References
- C. Janion, The efficiency and extent of mutagenic activity of some new mutagens of base-analogue type, Mutat. Res., 1978, 56, 225–234 CrossRef CAS PubMed.
- M. F. Goodman, S. Creighton, L. B. Bloom, J. Petruska and T. A. Kunkel, Biochemical basis of DNA replication fidelity, Crit. Rev. Biochem. Mol. Biol., 1993, 28, 83–126 CrossRef CAS PubMed.
- A. N. Nedderman, M. J. Stone, P. K. T. Lin, D. M. Brown and D. H. Williams, Base pairing of cytosine analogues with adenine and guanine in oligonucleotide duplexes: evidence for exchange between Watson–Crick and wobble base pairs using 1H NMR spectroscopy, J. Chem. Soc., Chem. Commun., 1991, 1357–1359 RSC.
- A. N. Nedderman, M. J. Stone, D. H. Williams, P. K. Y. Lin and D. M. Brown, Molecular basis for methoxyamine-initiated mutagenesis: 1H nuclear magnetic resonance studies of oligonucleotide duplexes containing base-modified cytosine residues, J. Mol. Biol., 1993, 230, 1068–1076 CrossRef CAS PubMed.
- V. I. Danilov, T. van Mourik, N. Kurita, H. Wakabayashi, T. Tsukamoto and D. M. Hovorun, On the mechanism of the mutagenic action of 5-bromouracil: a DFT study of uracil and 5-bromouracil in a water cluster, J. Phys. Chem. A, 2009, 113, 2233–2235 CrossRef CAS PubMed.
- C. S. Peng, B. I. Fedeles, V. Singh, D. Li, T. Amariuta, J. M. Essigmann and A. Tokmakoff, Two-dimensional IR spectroscopy of the anti-HIV agent KP1212 reveals protonated and neutral tautomers that influence pH-dependent mutagenicity, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3229–3234 CrossRef CAS PubMed.
- Ch.W. Lawrence, Induced mutagenesis: molecular mechanisms and their implications for environmental protection, Basic Life Sci., 1983, 433 Search PubMed.
- A. Hollaender, Chemical mutagens: principles and methods for their detection, V.1, Plenum Press, New York, 1971, p. 310 Search PubMed.
- M. J. Bessman, N. Muzyczka, M. F. Goodman and R. L. Schnaar, Studies on the biochemical basis of spontaneous mutation. II. The incorporation of a base and its analogue into DNA by wild-type, mutator and antimutator DNA polymerases, J. Mol. Biol., 1974, 88, 409–421 CrossRef CAS PubMed.
- R. Rein and R. Garduno, in Energetics and mechanism of 2-aminopurine induced mutations chapter quantum science, ed. J.-L. Calais, O. Goscinski, J. Linderberg and Y. Öhrn, Springer, 1976, pp. 549–560 Search PubMed.
- M. F. Goodman, R. Hopkins and W. C. Gore, 2-Aminopurine-induced mutagenesis in T4 bacteriophage: a model relating mutation frequency to 2-aminopurine incorporation in DNA, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4806–4810 CrossRef CAS.
- A. Ronen, 2-Aminopurine, Mutat. Res., 1979, 75, 1–47 Search PubMed.
- S. M. Watanabe and M. F. Goodman, On the molecular basis of transition mutations: frequencies of forming 2-aminopurine·cytosine and adenine·cytosine base mispairs in vitro, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2864–2868 CrossRef CAS.
- S. M. Watanabe and M. F. Goodman, Kinetic measurement of 2-aminopurine cytosine and 2-aminopurine thymine base pairs as a test of DNA polymerase fidelity mechanisms, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 6429–6433 CrossRef CAS.
- B. W. Glickman, 2-aminopurine mutagenesis in Escherichia coli, in Genetic consequences of nucleotide pool imbalance. V.31 of the series Basic Life Sciences, ed. F. J. de Serres, Springer, 1985, pp. 353–379 Search PubMed.
- L. C. Sowers, Y. Boulard and G. V. Fazakerley, Multiple structures for the 2-aminopurine-cytosine mispairs, Biochemistry, 2000, 39, 7613–7620 CrossRef CAS PubMed.
- P. Pitsikas, J. M. Patapas and C. G. Cupples, Mechanism of 2-aminopurine-stimulated mutagenesis in Escherichia coli, Mutat. Res., 2004, 550, 25–32 CrossRef CAS PubMed.
- E. A. Alemán, C. de Silva, E. M. Patrick, K. Musier-Forsyth and D. Rueda, Single-molecule fluorescence using nucleotide analogs: a proof-of-principle, J. Phys. Chem. Lett., 2014, 5, 777–781 CrossRef PubMed.
- A. Elvin, E. A. Alemán and D. Rueda, 2-Aminopurine single-molecule fluorescence, Biophys. J., 2011, 100(Suppl. 1), 474a Search PubMed.
- C. Wan, T. Fiebig, O. Schiemann, J. K. Barton and A. H. Zewail, Femtosecond direct observation of charge transfer between bases in DNA, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14052–14055 CrossRef CAS PubMed.
- E. L. Rachofsky, R. Osman and J. B. Ross, Probing structure and dynamics of DNA with 2-aminopurine: effects of local environment on fluorescence, Biochemistry, 2001, 40, 946–949 CrossRef CAS PubMed.
- J. M. Jean and K. B. Hall, 2-Aminopurine fluorescence quenching and lifetimes: role of base stacking, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 37–41 CrossRef CAS.
- V. I. Danilov, Iu. A. Krugliak, V. A. Kuprievich and O. V. Shramko, O mekhanizme mutagennogo deistviia 2-aminopurina [According the mechanism of the mutagenic action of 2-aminopurine], Biofizika, 1967, 12, 726–729 CAS.
- A. Broo and A. Holmén, Ab initio MP2 and DFT calculations of geometry and solution tautomerism of purine and some purine derivatives, Chem. Phys., 1996, 211, 147–161 CrossRef CAS.
- E. C. Sherer and C. J. Cramer, Quantum chemical characterization of the cytosine: 2-aminopurine base pair, J. Comput. Chem., 2001, 22, 1167–1179 CrossRef CAS.
- R. Ramaekers, L. Adamowicz and G. Maes, Tautomery and H-bonding characteristics of 2-aminopurine: a combined experimental and theoretical study, Eur. Phys. J. D, 2002, 20, 375–388 CrossRef CAS.
- O. O. Brovarets' and D. M. Hovorun, Molecular mechanisms of the mutagenic action of 2-aminopurine on DNA, Ukr. Bioorg. Acta, 2010, 9, 11–17 Search PubMed.
- O. O. Brovarets' and D. M. Hovorun, IR vibrational spectra of H-bonded complexes of adenine, 2-aminopurine and 2-aminopurine+ with cytosine and thymine: quantum-chemical study, Opt. Spectrosc., 2010, 111, 750–757 CrossRef.
- O. O. Brovarets', PhD thesis, “Physico-chemical nature of the spontaneous and induced by the mutagens transitions and transversions”, Taras Shevchenko National University of Kyiv, Kyiv, Ukraine, 2010.
- O. O. Brovarets', DrSci thesis, “Microstructural mechanisms of the origin of the spontaneous point mutations”, Taras Shevchenko National University of Kyiv, Kyiv, Ukraine, 2015.
- O. O. Brovarets', H. E. Pérez-Sánchez and D. M. Hovorun, Structural grounds for the 2-aminopurine mutagenicity: a novel insight into the old problem of the replication errors, RSC Adv., 2016, 6, 99546–99557 RSC.
- O. O. Brovarets' and H. E. Pérez-Sánchez, Whether the amino-imino tautomerism of 2-aminopurine is involved into its mutagenicity? Results of a thorough QM investigation, RSC Adv., 2016, 110, 108255–108264 RSC.
- O. O. Brovarets' and H. E. Pérez-Sánchez, Whether 2-aminopurine induces incorporation errors at the DNA replication? A quantum-mechanical answer on the actual biological issue, J. Biomol. Struct. Dyn., 2017, 35, 3398–3411 Search PubMed.
- O. O. Brovarets', I. S. Voiteshenko, H. E. Pérez-Sánchez and D. M. Hovorun, A QM/QTAIM detailed look at the Watson-Crick ↔ wobble tautomeric transformations of the 2-aminopurine·pyrimidine mispairs, J. Biomol. Struct. Dyn., 2017 DOI:10.1080/07391102.2017.1331864.
- O. O. Brovarets', I. S. Voiteshenko, H. E. Pérez-Sánchez and D. M. Hovorun, A QM/QTAIM research under the magnifying glass of the DPT tautomerisation of the wobble mispairs involving 2-aminopurine, New J. Chem., 2017, 41, 7232–7243 RSC.
- O. O. Brovarets' and D. M. Hovorun, New structural hypostases of the A·T and G·C Watson-Crick DNA base pairs caused by their mutagenic tautomerisation in a wobble manner: a QM/QTAIM prediction, RSC Adv., 2015, 5, 99594–99605 RSC.
- O. O. Brovarets' and D. M. Hovorun, Tautomeric transition between wobble A·C DNA base mispair and Watson-Crick-like A·C* mismatch: microstructural mechanism and biological significance, Phys. Chem. Chem. Phys., 2015, 17, 15103–15110 RSC.
- O. O. Brovarets' and D. M. Hovorun, By how many tautomerisation routes the Watson-Crick-like A·C* DNA base mispair is linked with the wobble mismatches? A QM/QTAIM vision from a biological point of view, Struct. Chem., 2016, 27, 119–131 CrossRef.
- O. O. Brovarets' and D. M. Hovorun, How many tautomerisation pathways connect Watson-Crick-like G*·T DNA base mispair and wobble mismatches?, J. Biomol. Struct. Dyn., 2015, 33, 2297–2315 Search PubMed.
- O. H. O. Brovarets’ and D. M. Hovorun, Wobble ↔ Watson-Crick tautomeric transitions in the homo-purine DNA mismatches: a key to the intimate mechanisms of the spontaneous transversions, J. Biomol. Struct. Dyn., 2015, 33, 2710–2715 Search PubMed.
- O. O. Brovarets' and D. M. Hovorun, Novel physico-chemical mechanism of the mutagenic tautomerisation of the Watson–Crick-like A·G and C·T DNA base mispairs: a quantum-chemical picture, RSC Adv., 2015, 5, 66318–66333 RSC.
- O. O. Brovarets' and D. M. Hovorun, A novel conception for spontaneous transversions caused by homo-pyrimidine DNA mismatches: a QM/QTAIM highlight, Phys. Chem. Chem. Phys., 2015, 17, 21381–21388 RSC.
- J. D. Watson and F. H. C. Crick, The structure of DNA, Cold Spring Harbor Symp. Quant. Biol., 1953, 18, 123–131 CrossRef CAS PubMed.
- M. M. Huang, N. Arnheim and M. F. Goodman, Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR, Nucleic Acids Res., 1992, 20, 4567–4573 CrossRef CAS PubMed.
- M. Lynch, Rate, molecular spectrum, and consequences of human mutation, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 961–968 CrossRef CAS PubMed.
- H. Lee, E. Popodi, H. Tang and P. L. Foster, Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR, Nucleic Acids Res., 2012, 20, 4567–4573 Search PubMed.
- I. J. Kimsey, K. Petzold, B. Sathyamoorthy, Z. W. Stein and H. M. Al-Hashimi, Visualizing transient Watson-Crick-like mispairs in DNA and RNA duplexes, Nature, 2015, 519, 315–320 CrossRef CAS PubMed.
- E. S. Szymanski, I. J. Kimsey and H. M. Al-Hashimi, Direct NMR evidence that transient tautomeric and anionic states in dG·dT form Watson–Crick-like base pairs, J. Am. Chem. Soc., 2017, 139, 4326–4329 CrossRef CAS PubMed.
- O. O. Brovarets' and D. M. Hovorun, Quantum-chemical investigation of tautomerization ways of Watson-Crick DNA base pair guanine-cytosine, Ukr. Biochem. J., 2010, 82, 55–60 Search PubMed.
- O. O. Brovarets' and D. M. Hovorun, Quantum-chemical investigation of the elementary molecular mechanisms of pyrimidine·purine transversions, Ukr. Biochem. J., 2010, 82, 57–67 Search PubMed.
- O. O. Brovarets, R. O. Zhurakivsky and D. M. Hovorun, Is there adequate ionization mechanism of the spontaneous transitions? Quantum-chemical investigation, Biopolym. Cell, 2010, 26, 398–405 CrossRef CAS.
- O. O. Brovarets' and D. M. Hovorun, Intramolecular tautomerization and the conformational variability of some classical mutagens – cytosine derivatives: Quantum chemical study, Biopolym. Cell, 2011, 27, 221–230 CrossRef.
- O. O. Brovarets' and D. M. Hovorun, Does the G·G*syn DNA mismatch containing canonical and rare tautomers of the guanine tautomerise through the DPT? A QM/QTAIM microstructural study, Mol. Phys., 2014, 112, 3033–3046 CrossRef.
- Ya.R Mishchuk, A. L. Potyagaylo and D. M. Hovorun, Structure and dynamics of 6-azacytidine by MNDO/H quantum-chemical method, J. Mol. Struct., 2000, 552, 283–289 CrossRef CAS.
- I. V. Kondratyuk, S. P. Samijlenko, I. M. Kolomiets' and D. M. Hovorun, Prototropic molecular-zwitterionic tautomerism of xanthine and hypoxanthine, J. Mol. Struct., 2000, 523, 109–118 CrossRef CAS.
- S. P. Samijlenko, O. M. Krechkivska, D. A. Kosach and D. M. Hovorun, Transitions to high tautomeric states can be induced in adenine by interactions with carboxylate and sodium ions: DFT calculation data, J. Mol. Struct., 2004, 708, 97–104 CrossRef CAS.
- M. O. Platonov, S. P. Samijlenko, O. O. Sudakov, I. V. Kondratyuk and D. M. Hovorun, To what extent can methyl derivatives be regarded as stabilized tautomers of xanthine?, Spectrochim. Acta, Part A, 2005, 62, 112–114 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman and J. A. Pople, GAUSSIAN 09 (Revision B.01), Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- M. A. Palafox, Molecular structure differences between the antiviral nucleoside analogue 5-iodo-2′-deoxyuridine and the natural nucleoside 2′-deoxythymidine using MP2 and DFT methods: conformational analysis, crystal simulations, DNA pairs and possible behavior, J. Biomol. Struct. Dyn., 2014, 32, 831–851 Search PubMed.
- A. A. El-Sayed, A. Tamara Molina, M. C. Alvarez-Ros and M. Alcolea Palafox, Conformational analysis of the anti-HIV Nikavir prodrug: comparisons with AZT and thymidine, and establishment of structure-activity relationships/tendencies in other 6′-derivatives, J. Biomol. Struct. Dyn., 2015, 33, 723–748 CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- E. L. Mertz and L. I. Krishtalik, Low dielectric response in enzyme active site, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2081–2086 CrossRef CAS PubMed.
- J. Petrushka, L. C. Sowers and M. Goodman, Comparison of nucleotide interactions in water, proteins, and vacuum: Model for DNA polymerase fidelity, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 1559–1562 CrossRef.
- H. P. Hratchian and H. B. Schlegel, Finding minima, transition states, and following reaction pathways on ab initio potential energy surfaces, in Theory and applications of computational chemistry: The first 40 years, ed. Dykstra C. E., Frenking G., Kim K. S. and Scuseria G., Amsterdam, Elsevier, 2005, pp. 195–249 Search PubMed.
- O. O. Brovarets' and D. M. Hovorun, Can tautomerisation of the A·T Watson–Crick base pair via double proton transfer provoke point mutations during DNA replication? A comprehensive QM and QTAIM analysis, J. Biomol. Struct. Dyn., 2014, 32, 127–154 Search PubMed.
- O. O. Brovarets' and D. M. Hovorun, Why the tautomerization of the G·C Watson–Crick base pair via the DPT does not cause point mutations during DNA replication? QM and QTAIM comprehensive analysis, J. Biomol. Struct. Dyn., 2014, 32, 1474–1499 Search PubMed.
- O. O. Brovarets', R. O. Zhurakivsky and D. M. Hovorun, DPT tautomerisation of the wobble guanine·thymine DNA base mispair is not mutagenic: QM and QTAIM arguments, J. Biomol. Struct. Dyn., 2015, 33, 674–689 Search PubMed.
- O. O. Brovarets' and D. M. Hovorun, The physicochemical essence of the purine·pyrimidine transition mismatches with Watson-Crick geometry in DNA: A·C* versa A*·C. A QM and QTAIM atomistic understanding, J. Biomol. Struct. Dyn., 2015, 33, 28–55 Search PubMed.
- O. O. Brovarets' and D. M. Hovorun, The nature of the transition mismatches with Watson-Crick architecture: the G*·T or G·T* DNA base mispair or both? A QM/QTAIM perspective for the biological problem, J. Biomol. Struct. Dyn., 2015, 33, 925–945 Search PubMed.
- NBO, 3.1, ed. Glendening E. D., Reed A. E., Carpenter J. E. and Weinhold F., Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 1994 Search PubMed.
- R. F. W. Bader, Atoms in molecules: a quantum theory, Oxford University Press, Oxford, 1990 Search PubMed.
- T. A. Keith, AIMAll (Version 10.07.01), 2010, Retrieved from http://aim.tkgristmill.com.
- C. F. Matta and J. Hernández-Trujillo, Bonding in polycyclic aromatic hydrocarbons in terms of the electron density and of electron delocalization, J. Phys. Chem. A, 2005, 109, 10798 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- I. Mata, I. Alkorta, E. Espinosa and E. Molins, Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields, Chem. Phys. Lett., 2011, 507, 185–189 CrossRef CAS.
- W. Saenger, Principles of nucleic acid structure, Springer, New York, 1984 Search PubMed.
- J. C. Hargis, E. Vöhringer-Martinez, H. L. Woodcock, A. Toro-Labbé and H. F. Schaefer, 3rd, Characterizing the mechanism of the double proton transfer in the formamide dimer, J. Phys. Chem. A, 2011, 115, 2650–2657 CrossRef CAS PubMed.
- F. Duarte, E. Vöhringer-Martinez and A. Toro-Labbé, Insights on the mechanism of proton transfer reactions in amino acids, Phys. Chem. Chem. Phys., 2011, 13, 7773–7782 RSC.
- D. Guzmán-Angel, R. Inostroza-Rivera, S. Gutiérrez-Oliva, B. Herrera and A. Toro-Labbé, Role of water in intramolecular proton transfer reactions of formamide and thioformamide, Theor. Chem. Acc., 2016, 135, 37 CrossRef.
- O. O. Brovarets' and D. M. Hovorun, DPT tautomerization of the long A·A* Watson-Crick base pair formed by the amino and imino tautomers of adenine: combined QM and QTAIM investigation, J. Mol. Model., 2013, 19, 4223–4237 CrossRef PubMed.
- O. O. Brovarets', R. O. Zhurakivsky and D. M. Hovorun, Structural, energetic and tautomeric properties of the T·T*/T*·T DNA mismatch involving mutagenic tautomer of thymine: a QM and QTAIM insight, Chem. Phys. Lett., 2014, 592, 247–255 CrossRef.
- O. O. Brovarets' and D. M. Hovorun, How does the long G·G* Watson-Crick DNA base mispair comprising keto and enol tautomers of the guanine tautomerise? The results of a QM/QTAIM investigation, Phys. Chem. Chem. Phys., 2014, 16, 15886–15899 RSC.
- O. O. Brovarets', R. O. Zhurakivsky and D. M. Hovorun, Is the DPT tautomerisation of the long A·G Watson-Crick DNA base mispair a source of the adenine and guanine mutagenic tautomers? A QM and QTAIM response to the biologically important question, J. Comput. Chem., 2014, 35, 451–466 CrossRef PubMed.
- O. O. Brovarets' and D. M. Hovorun, DPT tautomerisation of the G·Asyn and A*·G*syn DNA mismatches: a QM/QTAIM combined atomistic investigation, Phys. Chem. Chem. Phys., 2014, 16, 9074–9085 RSC.
- T. Y. Nikolaienko, L. A. Bulavin and D. M. Hovorun, How flexible are DNA constituents? The quantum-mechanical study, J. Biomol. Struct. Dyn., 2011, 29, 563–575 CAS.
- D. M. Hovorun, L. Gorb and J. Leszczynski, From the nonplanarity of the amino group to the structural nonrigidity of the molecule: a post-Hartree-Fock ab initio study of 2-aminoimidazole, Int. J. Quantum Chem., 1999, 75, 245–253 CrossRef CAS.
- D. N. Govorun, V. D. Danchuk, Ya.R. Mishchuk, I. V. Kondratyuk, N. F. Radomsky and N. V. Zheltovsky, AM1 calculation of the nucleic acid bases structure and vibrational spectra, J. Mol. Struct., 1992, 267, 99–103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Profiles of the relative electronic energy along the IRC of the G*·2AP(w) ↔ G·2AP(WC) and A·2AP(w) ↔ A*·2AP(WC) w ↔ WC tautomerisations in the continuum with ε = 4, characteristic of the active center of the DNA-polymerase. See DOI: 10.1039/c7cp05139e |
| This journal is © the Owner Societies 2018 |