
Comment on “Thermal compaction of the intrinsically disordered protein tau: entropic, structural, and hydrophobic factors” by A. Battisti, G. Ciasca, A. Grottesi and A. Tenenbaum, Phys. Chem. Chem. Phys., 2017, 19, 8435
Giuseppe
Graziano

Dipartimento di Scienze e Tecnologie, Università del Sannio, Via Port’Arsa 11, 82100 Benevento, Italy. E-mail: graziano@unisannio.it; Fax: +39 0824 23013; Tel: +39 0824 305133
First published on 30th November 2017
Abstract
In a recent article, A. Battisti et al., Phys. Chem. Chem. Phys., 2017, 19, 8435, results from SAXS measurements, metadynamic trajectories and classic MD trajectories at different temperatures have been used to study the temperature-induced compaction of the intrinsically disordered protein tau. The analysis, though technically sound, does not provide a clear explanation of hydrophobic interaction strengthening on increasing the temperature and its relationship with the population increase of secondary structural elements. Actually, hydrophobic interaction is driven by the gain in translational entropy of water molecules associated with the decrease in solvent-excluded volume due to chain compaction. The magnitude of this solvent-excluded volume effect increases with temperature in water because the density of water is almost temperature-independent due to the strength of H-bonds. Since α-helix formation leads to a significant decrease in the solvent-excluded volume, the connection with hydrophobic interaction and chain compaction emerges directly.
It is well established that both intrinsically disordered proteins, IDPs, and unfolded proteins undergo a temperature-induced compaction in aqueous solutions.1–3 A robust explanation of this phenomenon is still lacking. In an interesting work,4 Battisti and colleagues have investigated the temperature-induced compaction of protein tau, a large IDP involved in an important biological function, by performing both SAXS measurements over the 20–60 °C temperature range, and metadynamics simulations using an implicit water model and classic MD simulations using the SPC/E water model5 at 20 °C and 60 °C. Analysis of SAXS data indicated that the radius of gyration, Rg, of protein tau decreases from 70 Å at 20 °C to 57 Å at 60 °C. Analysis of both metadynamics and classic MD trajectories led to a quantitatively similar decrease in Rg.4 To provide a molecular-level rationalization of their findings, the authors wrote: “There are three relevant factors that account for the thermal compaction process in unfolded globular proteins or IDPs: an entropic contribution, the hydrophobic interaction, and the secondary and tertiary structures’ landscape.”
However, the role of one of these factors does not emerge in a clear-cut way. For instance, Battisti and colleagues found that, on increasing the temperature, protein tau populates conformations characterized by a smaller solvent-accessible surface area, SASA, and attributed this SASA decrease to hydrophobic interaction without assigning a clear meaning to the latter expression. The sentence is the following:4 “the enhancement of hydrophobic interaction associated with an increase in temperature causes a decrease of SASA. This decrease is one of the factors of the compaction of tau.” Moreover, they wrote that: “The increase in temperature from 293 K to 333 K influences the transient population of secondary structures, and consequently its overall fold. There is a second effect of the change in the secondary structures: namely a change in L, the contour length of tau. When residues shift from a random coil to local structures like α-helices, 3-helices, PP II helices, and β-turns, the contour length decreases. This change of L is a component of the total compaction phenomenon.” What is lacking in the analysis by Battisti and colleagues is an indication of the actual driving force that provides the stabilization of such secondary structure elements on increasing the temperature, and how it is related to the claimed enhancement of hydrophobic interaction.
In the present comment, I would like to provide a reliable explanation of the molecular origin of the compaction phenomenon by assigning a precise meaning to the expression hydrophobic interaction. The latter refers to an “effective” attraction between nonpolar moieties in water; its actual driving force is the gain in translational entropy of water molecules associated with the decrease in solvent-excluded volume due to protein compaction.6,7 Water molecules push the polypeptide chain to populate compact conformations in order to gain translational entropy for the increase in the accessible configurational space. The solvent-excluded volume of a conformation is proportional to the SASA of the conformation itself; from the theoretical point of view, this is taken into account by the creation of a cavity suitable to host such conformation in the liquid.8 Cavity creation, at constant temperature and pressure, leads to an increase in liquid volume corresponding to the partial molar volume of the cavity, but, nevertheless, causes a geometric constraint for liquid molecules whose centre can at most lie on the cavity SASA, if the cavity itself has to exist. Of course, this “geometric” description contrasts with the old description based on the imagined presence of icebergs surrounding nonpolar moieties in water.9 The old picture is untenable simply because it has not received support by direct structural information recorded by means of different experimental probes.10,11
The “geometric” description can be tested by considering cavities possessing the same van der Waals volume, but a different shape so that their SASA is different. Such a situation is not far from reality because the change in volume associated with denaturation is a very small quantity,12 and so the polypeptide chain populates conformations possessing the same molecular volume and different SASA values. To provide rough quantitative estimates, I have considered a spherical cavity of 10 Å radius and four prolate spherocylindrical cavities of different radii and cylindrical lengths, all possessing the same van der Waals volume (the precise measures are reported in Table 1). For these simple geometries, classic scaled particle theory,13 SPT, provides analytical formulae to calculate the Gibbs energy cost of cavity creation in a liquid, ΔGc. The latter is the right measure of the solvent-excluded volume effect, being a purely entropic quantity.14,15 To perform SPT calculations, the experimental density of liquid water over the 20–60 °C temperature range and 1 atm has been used,16 and the effective diameter of water molecules has been fixed to σ(H2O) = 2.8 Å,17 and considered to be temperature-independent. The selected σ(H2O) value is reliable because it is close to the location of the first peak in the oxygen–oxygen radial distribution function of water.18
| a (Å) | l (Å) | SASA (Å2) | ΔGc 20 °C | ΔGc 30 °C | ΔGc 40 °C | ΔGc 50 °C | ΔGc 60 °C | |
|---|---|---|---|---|---|---|---|---|
| 1 | 10 | — | 1633 | 482 | 495 | 508 | 519 | 529 |
| 2 | 5.5 | 36.7 | 2189 | 637 | 655 | 671 | 685 | 699 |
| 3 | 5.0 | 46.7 | 2393 | 691 | 710 | 728 | 744 | 758 |
| 4 | 4.5 | 59.8 | 2654 | 759 | 779 | 799 | 816 | 832 |
| 5 | 4.0 | 78.0 | 3013 | 849 | 872 | 894 | 913 | 931 |
The ΔGc values calculated by means of classic SPT formulae in water over the 20–60 °C temperature range are reported in Table 1 and are shown in Fig. 1. As expected, they increase in magnitude with an increase in the cavity SASA,6–8 which measures the solvent-excluded volume effect caused by each cavity (i.e., by the object to be hosted in the cavity). Moreover, these ΔGc values increase in magnitude with an increase in temperature, regardless of the cavity shape.17 This temperature dependence is a special feature of water because in other liquids the ΔGc values decrease upon increasing the temperature,17 and it is not due to classic SPT, since it also emerged from the results of computer simulations using reliable models of the relevant liquids.19 Water is special because its density remains practically constant over the 0–100 °C temperature range16 due to the strength of H-bonds with respect to the random thermal energy. According to statistical mechanics,20 the fundamental formula to calculate ΔGc is:
ΔGc = −RT·ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) pins pins | (1) |
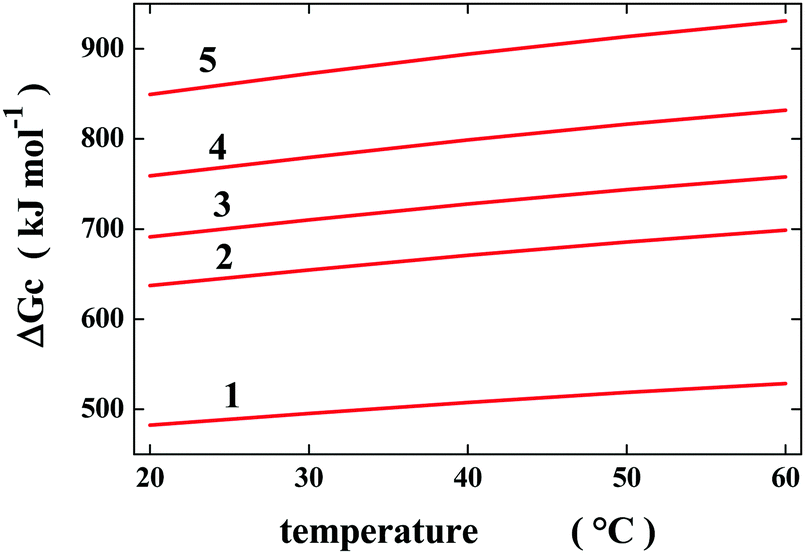 | ||
| Fig. 1 Temperature dependence of the Gibbs energy cost to create water cavities possessing the geometric measures reported in Table 1. Classic SPT calculations have been performed using the experimental density of water and σ(H2O) = 2.8 Å, considered to be temperature-independent. | ||
 | ||
| Fig. 2 Temperature dependence of the Gibbs energy decrease associated with the change from the most elongated prolate spherocylindrical cavity (numbered 5) to the other prolate spherocylindrical ones, ΔΔGc = ΔGc(i) − ΔGc(5); see Table 1 and the text for further details. | ||
It is important to underscore that Rg and SASA are quantities of different nature: the latter accounts for all the details of a structure, whereas the first is coarse-grained, capturing an overall feature of a structure. Indeed the “structures” D1(20 °C) and D2(60 °C), obtained by means of MD simulations and the ensemble optimization method, possess Rg = 69.1 Å and 57.0 Å, and SASA = 39800 Å2 and 37300 Å2, respectively (see Tables 2 and 4 in ref. 4). These two “structures” show a decrease in Rg amounting to 17.4% and a decrease in SASA amounting to 6.3%.
On the other hand, it should be recognized that the formation of an α-helix starting from a fully elongated chain is associated with a large decrease in solvent-excluded volume (i.e., a significant decrease in the contour length of the chain, using the polymer language adopted by Battisti and colleagues4), which can be measured by the large SASA decrease. Therefore, as originally pointed out by Snir and Kamien,25 α-helix formation is driven by the gain in translational entropy of water molecules. In other words, water molecules force the polypeptide chains to populate compact conformations characterized by small SASA values, and this can also be realized by forming secondary structural elements. The simulation results of Battisti and colleagues nicely match the present “geometric” scenario. A significant decrease in the total SASA of protein tau was obtained on passing from 20 to 60 °C (see the numbers listed in the second column of Table 4 of ref. 4). Note that it is not correct to take into account solely the nonpolar SASA, because the solvent-excluded volume of a conformation cannot be partitioned, being a “geometric” property of the whole polypeptide chain.8
According to my theoretical approach,7,8,17,26 the conformational stability of polypeptide chains (i.e., looking at a huge ensemble of molecules) is mainly governed by three contributions: (a) the one associated with the gain (upon collapse/folding) or loss (upon swelling/unfolding) of the translational entropy of water molecules depending on the solvent-excluded volume of the chain; (b) the one associated with the gain (upon swelling/unfolding) or loss (upon collapse/unfolding) of the conformational entropy of the chain; (c) the one associated with the trade-off between intra-chain energetic interactions and the intermolecular ones with water molecules for the denatured state and the native state ensembles (in this matter, the H-bonds play a fundamental role). The coupling between these three contributions rules the folding cooperativity. The balance of the three contributions is delicate and, in fact, the native state is only marginally stable.27 Therefore, the finding that the denatured state ensemble of several proteins, at room temperature in water, seems to be well described by the random walk model28 should not come as a surprise.
In conclusion, I have tried to provide a clear explanation of the molecular origin of hydrophobic interaction, of its strengthening on increasing the temperature, and of its relationship with the formation of secondary structural elements. This analysis should be useful to arrive at a more complete rationalization of the interesting results of Battisti and colleagues.
Conflicts of interest
There are no conflicts of interest to declare.References
- R. Wuttke, H. Hofmann, D. Nettels, M. B. Borgia, J. Mittal, R. B. Best and B. Schuler, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5213–5218 CrossRef CAS PubMed.
- D. Nettels, S. Müller-Späth, F. Küster, H. Hofmann, D. Haenni, S. Rüegger, L. Reymond, A. Hoffmann, J. Kubelka, B. Heinz, K. Gast, R. B. Best and B. Schuler, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20740–20745 CrossRef CAS PubMed.
- M. Aznauryan, D. Nettels, A. Holla, H. Hofmann and B. Schuler, J. Am. Chem. Soc., 2013, 135, 14040–14043 CrossRef CAS PubMed.
- A. Battisti, G. Ciasca, A. Grottesi and A. Tenenbaum, Phys. Chem. Chem. Phys., 2017, 19, 8435–8446 RSC.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- G. Graziano, J. Phys. Chem. B, 2009, 113, 11232–11239 CrossRef CAS PubMed.
- G. Graziano, Phys. Chem. Chem. Phys., 2010, 12, 14245–14252 RSC.
- A. Merlino, N. Pontillo and G. Graziano, Phys. Chem. Chem. Phys., 2017, 19, 751–756 RSC.
- W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed. Engl., 1993, 32, 1545–1579 CrossRef.
- P. Buchanan, N. Aldiwan, A. K. Soper, J. L. Creek and C. A. Koh, Chem. Phys. Lett., 2005, 415, 89–93 CrossRef CAS.
- G. Graziano, J. Phys. Chem. B, 2014, 118, 2598–2599 CrossRef CAS PubMed.
- T. V. Chalikian, Annu. Rev. Biophys. Biomol. Struct., 2003, 32, 207–235 CrossRef CAS PubMed.
- R. A. Pierotti, Chem. Rev., 1976, 76, 717–726 CrossRef CAS.
- B. Lee, J. Chem. Phys., 1985, 83, 2421–2425 CrossRef CAS.
- G. Graziano, J. Phys. Chem. B, 2006, 110, 11421–11426 CrossRef CAS PubMed.
- G. S. Kell, J. Chem. Eng. Data, 1975, 20, 97–105 CrossRef CAS.
- G. Graziano, Phys. Chem. Chem. Phys., 2014, 16, 21755–21767 RSC.
- J. M. Sorenson, G. Hura, R. M. Glaeser and T. Head-Gordon, J. Chem. Phys., 2000, 113, 9149–9161 CrossRef CAS.
- H. S. Ashbaugh and L. R. Pratt, J. Phys. Chem. B, 2007, 111, 9330–9336 CrossRef CAS PubMed.
- H. Reiss, Adv. Chem. Phys., 1965, 9, 1–84 CrossRef.
- B. Guillot and Y. Guissani, J. Chem. Phys., 1993, 99, 8075–8094 CrossRef CAS.
- B. Widom, J. Phys. Chem., 1982, 86, 869–872 CrossRef CAS.
- G. Graziano, Phys. Chem. Chem. Phys., 1999, 1, 3567–3576 RSC.
- D. Ben-Amotz and R. Underwood, Acc. Chem. Res., 2008, 41, 957–967 CrossRef CAS PubMed.
- Y. Snir and R. D. Kamien, Science, 2005, 307, 1067 CrossRef CAS PubMed.
- A. Pica and G. Graziano, Phys. Chem. Chem. Phys., 2016, 18, 14426–14433 RSC.
- D. C. Rees and A. D. Robertson, Protein Sci., 2001, 10, 1187–1194 CrossRef CAS PubMed.
- J. A. Riback, M. A. Bowman, A. M. Zmyslowski, C. R. Knoverek, J. M. Jumper, J. R. Hinshaw, E. B. Kaye, K. F. Freed, P. L. Clark and T. R. Sosnick, Science, 2017, 358, 238–241 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2018 |