
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature-induced polymorphism in methyl stearate†
X.
Liu
 *a,
A. S.
Gibbs
c,
G. S.
Nichol
a,
C. C.
Tang
b,
K. S.
Knight
c,
P. J.
Dowding
d,
I.
More
d and
C. R.
Pulham
*a
*a,
A. S.
Gibbs
c,
G. S.
Nichol
a,
C. C.
Tang
b,
K. S.
Knight
c,
P. J.
Dowding
d,
I.
More
d and
C. R.
Pulham
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: Xiaojiao.Liu@ed.ac.uk
bDiamond Light Source Ltd, Harwell Science & Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
cHRPD, ISIS Facility, Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire OX11 0LA, UK
dInfineum UK Ltd, Milton Hill Business and Technology Centre, Abingdon, OX13 6BB, UK
First published on 2nd August 2018
Abstract
The crystallisation of methyl stearate under a range of crystallisation conditions has been studied and three new polymorphs have been identified and structurally characterised. Form III (monoclinic, space group Cc, Z = 8) was obtained at room temperature by slow evaporation of a saturated solution in CS2. Form IV (monoclinic, space group C2/c, Z = 4) was obtained by slow cooling of the melt. Both structures were characterised by single crystal X-ray diffraction. Form V (monoclinic, space group Cc, Z = 4) was obtained from the melt by rapid cooling. X-ray and neutron powder diffraction methods were employed in the determination of this structure. Form V shows highly anisotropic thermal expansion, with expansion along the crystallographic b-axis being substantially greater than along the other two axes.
1. Introduction
Combustion of fossil fuels accounts for 98% of global carbon dioxide emissions1 and so there is an urgent requirement to reduce CO2 emissions by reducing consumption of these fuels. Hence there is growing interest in the use of biofuels, such as biodiesel, that are obtained from sustainable, biomass-based sources. In 2003, the EU set a target for the use of biodiesel of 5.75% of total fuel consumption by 2010, whilst the current EU target for 2020 is 10%.2Biodiesel mainly consists of a mixture of fatty acid methyl esters (FAMEs) that are derived from vegetable oils or animal fats (feedstocks) by transesterification reactions.3 Whilst biodiesel has some advantages as a fuel, it is well known that it shows poor performance at low temperature.4 One disadvantage is its poor cold-flow engine performance. For example, in field studies, FAMEs derived from soybean oil began to solidify when the environmental temperatures approached 0 °C.5 This critical temperature is substantially higher than that of petroleum-based diesels. When the environmental temperature reaches the crystallisation temperature of diesel or biodiesel, high-molecular weight components begin to nucleate and crystallise. If the fuel remains cold for long periods of time, these crystals can grow large enough in size to block engine filters and injectors, causing very serious start-up and operational problems for vehicles. Mitigation strategies have been demonstrated through modification of the composition of biodiesel by reducing the ratio of saturated/unsaturated FAMEs6,7 in order to enhance the cold-flow performance. However, unsaturated FAMEs are susceptible to oxidation even under ambient conditions and indeed it has been shown that polyunsaturated fatty acids are very prone to autoxidation.8–11 The degradation of biodiesel not only significantly decreases the chain length of the acid groups, leading to lower cetane number values,12,13 but can also produce organic acids and polymeric materials that can potentially cause damage to engines. Given the growing importance of biodiesel and its associated problems at low temperatures, it is therefore crucial to understand its crystallisation behaviour in order to design efficient additives that will improve its cold-flow performance. However, a major challenge of such studies is presented by the nature of biodiesel – it is a complex mixture of multiple components with variable compositions.13,14 A better understanding of the crystallisation behaviour of single components of biodiesel is needed, hence the focus of this paper: the low-temperature crystallisation behaviour of one of the main components of biodiesel, namely methyl stearate (C19H38O2).
Methyl stearate is a fully saturated methyl ester with a 17-carbon linear chain (Fig. 1) and a melting point of 311 K.
 | ||
| Fig. 1 Molecular structure of methyl stearate. | ||
The crystal structure of methyl stearate has intrigued crystallographers for decades on account of the problems in growing suitable quality single crystals for X-ray diffraction. These difficulties arise from the nature of materials with long hydro-carbon chains and reflect the tendency to form thin, plate-shaped crystals with thicknesses of only a few microns.15 The first crystal structure of methyl stearate was published in 1960, and was solved in the monoclinic crystal system (A2/a) with unit cell parameters a = 5.61 Å, b = 7.33 Å, c = 106.6 Å, β = 116.47° (form I).16 The paper reported that single crystals were grown from CS2 solution at −15 °C to −12 °C, and X-ray diffraction data were collected at room temperature. In 1970, MacGillavry grew single crystals and recorded X-ray diffraction data under the same crystallisation and data collection conditions, and reported a new orthorhombic (Pnab) structure with unit cell parameters a = 5.613(5) Å, b = 7.354(5) Å, c = 95.417(4) Å (form II).17 Recently, a group from the University of Leeds studied the morphology and growth of methyl stearate as a function of crystallisation conditions and rationalised the observed morphology on the basis of a monoclinic form (space group C2) with unit cell parameters a = 5.60 Å, b = 7.39 Å, c = 47.96 Å, β = 91.15°. However, no further structural details were reported.18
Form II contains one molecule in the asymmetric unit. There are four molecular layers in the unit cell, packed in a tilted manner and forming roof-like “ripples” along the c-axis (Fig. 2a). The folding angle between the least squares planes though the methyl stearate molecules is approximately 150°. The ester groups (head) of the methyl stearate in one layer are close to the ester groups of methyl stearate molecules in the neighbouring layer, thereby packing in “head-to-head” bilayers. In the crystallographic bc plane (Fig. 2b), the chain direction is perpendicular to the lamella layers, and parallel with the c-axis. Inter-chain interactions dominate packing behaviour.
 | ||
| Fig. 2 Packing plot of form II of methyl stearate. (a) View parallel to the crystallographic b-axis; (b) view parallel to the crystallographic c-axis. | ||
Given the uncertainties in the literature about these polymorphs and the crystallisation conditions under which they are formed, our aim was to explore the crystallisation of methyl stearate under a wide range of conditions.
2. Experimental
High-purity methyl stearate (>99%) was obtained from Tokyo Chemical Industry. The following experiments were conducted without further purification of the sample. Initial attempts focussed on repeating the experimental methods described in ref. 16 and 17. Although it proved possible to grow very thin crystals of methyl stearate in CS2 at low temperatures, it proved impossible to manipulate and transfer the crystals to the diffractometer without melting or dissolution. Instead, experiments were subsequently conducted that involved (i) crystallisation from solution at ambient temperature, and (ii) crystallisation from the melt under both slow and rapid cooling conditions. Neutron powder diffraction studies were also conducted on the fully deuterated isotopomer of methyl stearate in order to corroborate the structure of the polymorph characterised by powder X-ray diffraction and to locate hydrogen positions.2.1. Form III and form IV
Form III crystallised as colourless, thin, plate-like crystals from a saturated solution of CS2 (Sigma Aldrich) at room temperature by slow evaporation. A suitable specimen was selected and mounted on a Rigaku Oxford Diffraction SuperNova diffractometer (Cu-Kα radiation, λ = 1.5418 Å). The crystal was protected from melting during transfer to the diffractometer by cooling in dry ice and it was subsequently maintained at 120 K during data collection using an Oxford Cryostream Plus.Form IV was obtained from a finely ground, polycrystalline sample of methyl stearate loaded into a borosilicate glass capillary of 0.5 mm diameter. This was attached to a goniometer head and mounted on a Rigaku Oxford Diffraction SuperNova diffractometer (Mo-Kα radiation, λ = 0.7107 Å). The sample was melted by warming to 323 K and the temperature was held for 10 min. It was then cooled at a rate of 5 K h−1 to 283 K to induce nucleation and crystallisation of a single crystal, followed by cooling to 120 K at a cooling rate of 360 K h−1. Single crystal diffraction data were recorded at 120 K.
Unit cell parameters of form III and form IV (Table 1) were obtained using the CrysAlisPro program.19 Olex2 software20 was employed for structure determination. Structures were solved with the ShelXT21 program using Intrinsic Phasing methods. Refinements were performed by ShelXL22 program using least squares minimisation.
| Form I16 | Form II17 | Form III | Form IV | Form V | |
|---|---|---|---|---|---|
| Space group | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| A2/a | Pnab | Cc | C2/c | Cc | |
| a (Å) | 5.610 | 5.613(6) | 95.583(8) | 47.795(13) | 49.013(1) |
| b (Å) | 7.330 | 7.354(5) | 7.291(8) | 7.113(4) | 7.213(1) |
| c (Å) | 106.600 | 95.147(4) | 5.586(4) | 5.532(4) | 5.556(1) |
| β (°) | 116.78 | 90 | 92.57(1) | 90.48(3) | 77.536(2) |
| V (Å3) | 3913.37 | 3927.48(3) | 3889.6(5) | 1880.49(5) | 1917.78(7) |
| Z | 8 | 8 | 4 | 8 | 4 |
| Z′ | 1 | 1 | 2 | 0.5 | 1 |
| R | R 1 = 0.1543 | R 1 = 0.1160 | R wp = 0.11076 | ||
| wR2 = 0.4416 | wR2 = 0.3129 | R exp = 0.0240 | |||
| Collection temperature (K) | ∼293 | ∼293 | 120 | 120 | 193 |
2.2. Form V
A 2θ range from 1–29° was used for indexing diffraction patterns (Table 1) using Topas 4.1.25 The space group was determined as Cc. The following unit cell parameters were obtained at 193 K: a = 49.012(8) Å, b = 7.212(2) Å, c = 5.555(10) Å, β = 77.527(6)°; good values of Rwp = 9.324%, Rexp = 2.262% and GoF = 4.121 were obtained. Initially, the structure solution indicated that the 17-carbon chain in methyl stearate was partly disordered and so in order to obtain reliable structural coordinates from the XRPD pattern, distance- and angle-restrained refinements with non-hydrogen atoms were performed simultaneously using Rietveld refinement. All hydrogen atoms were added in calculated positions. The final fit of the Rietveld refinements is shown in Fig. 3: the difference plot, combined with the figures of merit (Table 1), confirm the excellent fit obtained.
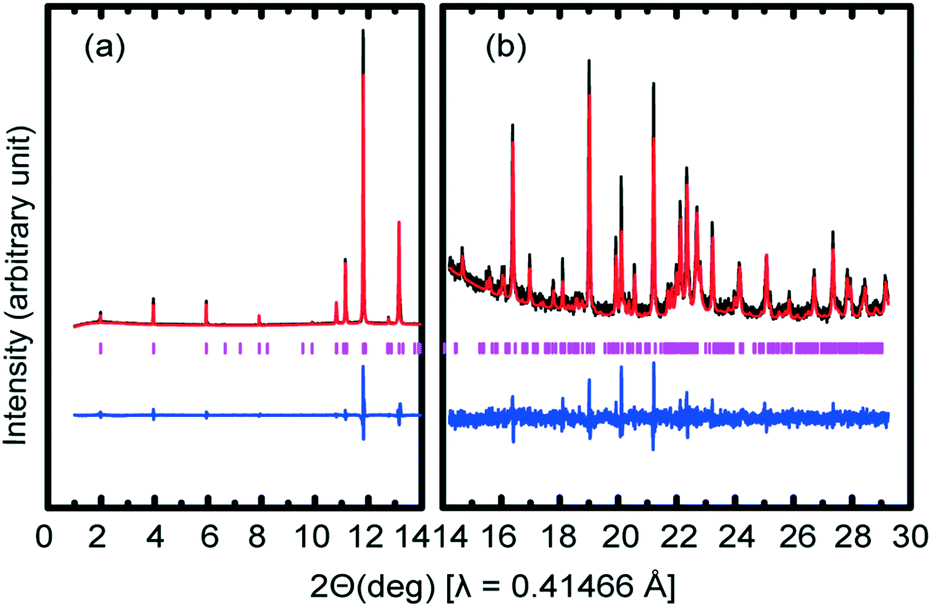 | ||
| Fig. 3 Rietveld refinement of synchrotron XRPD data of form V of methyl stearate collected at 193 K on beamline I11. The experimental (black), calculated profile (red) and difference (blue). Vertical tick marks (pink) show calculated peak positions. The arbitrary unit of (a) is 24 times of the unit of (b). | ||
2.2.2.1. General sample preparation. Unless otherwise indicated, all reagents were purchased from Sigma Aldrich and used without further purification. All reactions were performed in oven-dried glassware.
Preparation of the NaHSO4·SiO2 catalyst26. 10 g of SiO2 (column chromatographic grade, 60 Å, 200–400 mesh) was added to a solution of 4.14 g (0.03 mole) of NaHSO4 in 20 mL of water. After stirring for 15 min at room temperature, the mixture was gently heated on a hot plate (50 °C) with stirring, until a free-flowing white solid was obtained. The solid was then dried in an oven at 120 °C for 48 h prior to use.
Preparation of perdeuterated methyl stearate. Deuterated stearic acid was kindly provided by the ISIS Deuteration Facility. 5 g of deuterated stearic acid was added to 300 mL methanol-d4 (≥99.8 atom %D) in a 500 mL round-bottomed flask with 5.5 g of NaHSO4·SiO2 prepared as described above. The flask was sealed with Parafilm M and the contents stirred at 45 °C. This was based on a procedure described in ref. 27, but conducted at the higher temperature of 45 °C in order to increase the reaction rate. The reaction (Scheme 1) was monitored by 13C NMR (recorded in CHCl3) until the peak of the acid group of deuterated stearic acid (δ = 182 ppm) was no longer visible. The mixture was cooled and filtered under vacuum, and the recovered catalyst was washed with deuterated methanol (10 mL). The combined solution was concentrated in vacuo to obtain the required product as a white solid.
 | ||
| Scheme 1 Synthesis of perdeuterated methyl stearate. | ||
2.2.2.2. Data collection and analysis. High-resolution neutron powder diffraction data of perdeuterated methyl stearate were collected on the high resolution powder diffractometer (HRPD), at the ISIS Neutron Facility, STFC-Rutherford Appleton Laboratory, UK. A polycrystalline sample was prepared following the preparation method of the sample used for synchrotron XRPD studies as described above. 2 g of the sample was gently packed into a vanadium slab can before being cooled to 4.2 K in situ on the diffractometer. Data were collected simultaneously using the backscattering detector banks, the 90° banks, and the low-angle banks (2θ = 30°) over the time-of-flight range 30–130 ms. Data were then collected upon warming the sample from 4.2 K to 300 K and then cooling to 10 K for 20 min intervals, holding the sample at each required temperature for 10 min in order to allow thermal equilibration before data collection. Long data collections (8 h) were conducted at 4.2 K, 193 K, and 300 K. On account of the large crystallographic unit cell of methyl stearate, only the diffraction patterns recorded by the backscattering detector (centred around 168°) and the complementary detector at 90° were employed in the structural analysis.
Rietveld refinement of the crystal structure was conducted using the GSAS program27 and used as its starting point the structure of form V of methyl stearate solved from XRPD data. Bond length and bond angle restraints were imposed on the ester group of methyl stearate. The final profile fits of calculated patterns compared to recorded diffraction patterns are shown in Fig. 4. The crystallographic data and details of the Rietveld refinement are given in Table 1.
 | ||
| Fig. 4 Rietveld refinement plot of neutron powder data of form IV of methyl stearate collected at 193 K on the HRPD instrument. The experimental (black), calculated profile (red) and difference (blue). Vertical tick marks (pink) show calculated peak positions. (a) A plot of pattern recorded by the backscattering detector (bank 1); (b) a plot of pattern recorded by the complementary detector at 90° (bank 2). | ||
3. Results and discussion
3.1. Form III
The crystal structure of form III of methyl stearate contains two independent molecules (A and B) in the asymmetric unit (Fig. 5). The two molecules differ essentially in the conformation of the ester groups. The CI–O1–C2–O2 torsion angle is −5.8° in the molecule A, while the CIA–O1A–C2A–O2A torsion angle is 3.3° in the molecule B. The bond angle O2–C2–O1 in molecule A is 120.9°, as expected for the central sp2 hybridised carbon atom. However, the corresponding bond angle in molecule B (O2A–C2A–O1A) refines as 112.3°, suggesting some degree of packing disorder. | ||
| Fig. 5 The two independent molecules in the asymmetric unit of the crystal structure of form III (view along the b-axis). | ||
There are four molecular layers showing tilted packing in the unit cell (Fig. 6a). Unlike in form II, the “head-to-head” bilayers in form III that arise from the two independent asymmetric methyl stearate molecules form oblique layers with the same tilt angles with the crystallographic bc plane (1 0 0). The repeat bilayer thickness (C19A–C19 distance) is 44.93 Å. The thicknesses of the single layer A (C1–C19 distance) and B (C1A–C19A distance) are 21.23 Å and 21.26 Å, respectively. The gap between layer A and layer B is 2.44 Å (C1–C1A distance), while the distance between two bilayers is 2.81 Å. The tilt angle of the least squares line through molecule A with respect to the bilayer normal (where the bilayer plane is defined by the least squares plane of C19 atoms) is 55.6°, whereas the corresponding angle for molecule B is 56.2°. In the crystallographic ab plane (Fig. 6b), the chain direction is perpendicular to the lamella layers, parallel with the a-axis. The packing behaviour is also dominated by inter-chain interactions.
 | ||
| Fig. 6 Packing plot of form III of methyl stearate. (a) View along the b-axis, shown without hydrogen atoms (distances between each plane are labelled, the planes of the boundaries of bilayers are shown in pink while those of single layers are shown in blue); (b) view along the c-axis, hydrogen atoms are shown in calculated positions. | ||
3.2. Form IV
The crystal structure was refined in space group C2/c with a crystallographically-imposed inversion centre located at the centre point of the C10–C10 bond. The asymmetric unit thus consists of half a molecule of methyl stearate with disorder of the ester head group caused by inversion symmetry.Chains of molecules of methyl stearate pack in homologous, oblique layers, parallel to the crystallographic bc plane. (Fig. 7a) The least-squares line through the molecule forms a 42° tilt angle with the crystallographic ab plane. In the ab plane (Fig. 7b), the chain direction is perpendicular to the lamella layers, parallel with the a-axis. The packing behaviour is also dominated by inter-chain interactions.
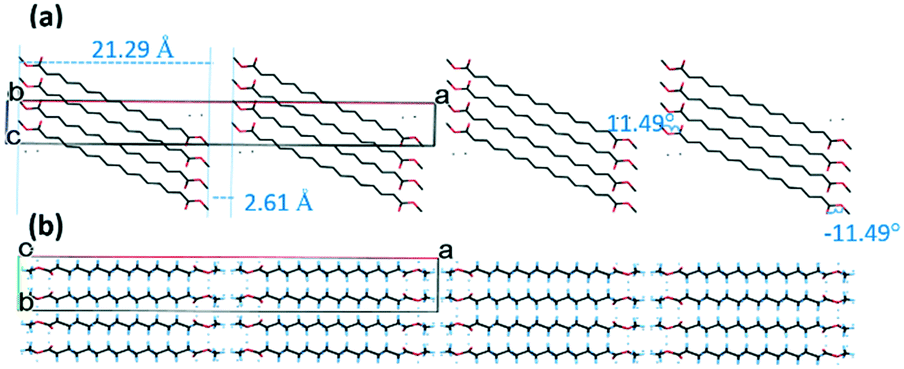 | ||
| Fig. 7 Packing plot of methyl stearate form IV. (a) View along the b-axis, shown without hydrogen atoms, torsion angles and distances between least-square planes are labelled; (b) view along the c-axis, shown with partially disordered hydrogen atoms. | ||
3.3. Form V
These show excellent agreement with the XRPD studies (Table 2) thereby confirming that this is indeed the correct structure. The small differences in unit-cell volumes between perdeuterated and hydrogenous samples reflect the well-known geometric isotope effect (GIE)28–30 in which E–D bond lengths are slightly shorter that E–H bonds. This geometrical H/D isotope effect is also particularly striking for materials that are dominated by hydrogen-bonding interactions that stabilise crystallographic packing.31 Although clearly in the case of methyl stearate there is no hydrogen bonding, and hence the origin of the smaller unit-cell volume for the perdeuterated compound is simply the shorter C–D bond length compared to the C–H bond length.
| Parameters | X-ray (synchrotron) | Neutron |
|---|---|---|
| a (Å) | 49.013(1) | 48.947(2) |
| b (Å) | 7.213(1) | 7.198(1) |
| c (Å) | 5.556(1) | 5.547(1) |
| β (°) | 77.536(2) | 77.453(1) |
| V (Å3) | 1917.78(7) | 1907.60(3) |
| Space group | Cc | Cc |
| Z | 4 | 4 |
| R | R wp = 0.11076 | R wp = 0.0169 |
| R exp = 0.0240 | R p = 0.0179 | |
| T (K) | 193 | 193 |
The crystal structure of form V of methyl stearate has one molecule in the asymmetric unit. The packing behaviour is similar to that of form III. However, there are two molecular layers in the unit cell. Molecules of methyl stearate pack in oblique chain layers, parallel to the crystallographic bc plane. (Fig. 8a) The least squares line though the molecule forms a 13.98° tilt angle with crystallographic ab plane. A change of packing behaviour of methyl stearate has been clearly observed. Unlike the “head to head” bilayer packing in form II and III, a “head to tail” monolayer structure was observed in form V of methyl stearate. In the crystallographic bc plane, (Fig. 8b) the chain direction is perpendicular to the lamella layers, parallel with the c-axis. The packing behaviour is also dominated by inter-chain interactions.
 | ||
| Fig. 8 Packing plot for form V of methyl stearate. (a) View parallel to the b-axis, shown without hydrogen atoms; (b) view parallel to the c-axis, shown with deuterium positions. | ||
Unit cell parameters over a range of temperatures of hydrogenous and deuterated samples of form V of methyl stearate are tabulated in the ESI† (Tables S1 and S2). The relative thermal expansions of the unit cell parameters are shown in Fig. 9a (deuterated) and Fig. 9b (hydrogenous), and highlight the highly anisotropic thermal expansion of methyl stearate. For the monoclinic crystal system, one of the principal axes of the thermal expansion coincides with the crystallographic b-axis.32 The other two principal axes are in the (0 1 0) plane. Fig. 9 shows clearly that the unit cell b-axis of form V increases significantly more with temperature than do the c-axis and the β angle. The crystallographic a-axis remains almost constant over the entire temperature range. A likely explanation for this is that the molecules of methyl stearate lie along the a-axis, and so the length of the a-axis is dominated by C–C covalent bonds within the hydrocarbon chains. These are relatively rigid and so would not be expected to be strongly affected by temperature changes. In contrast, along the b-axis there are only very weak intermolecular interactions between chains of molecules and so thermal expansion is substantially greater along this direction.
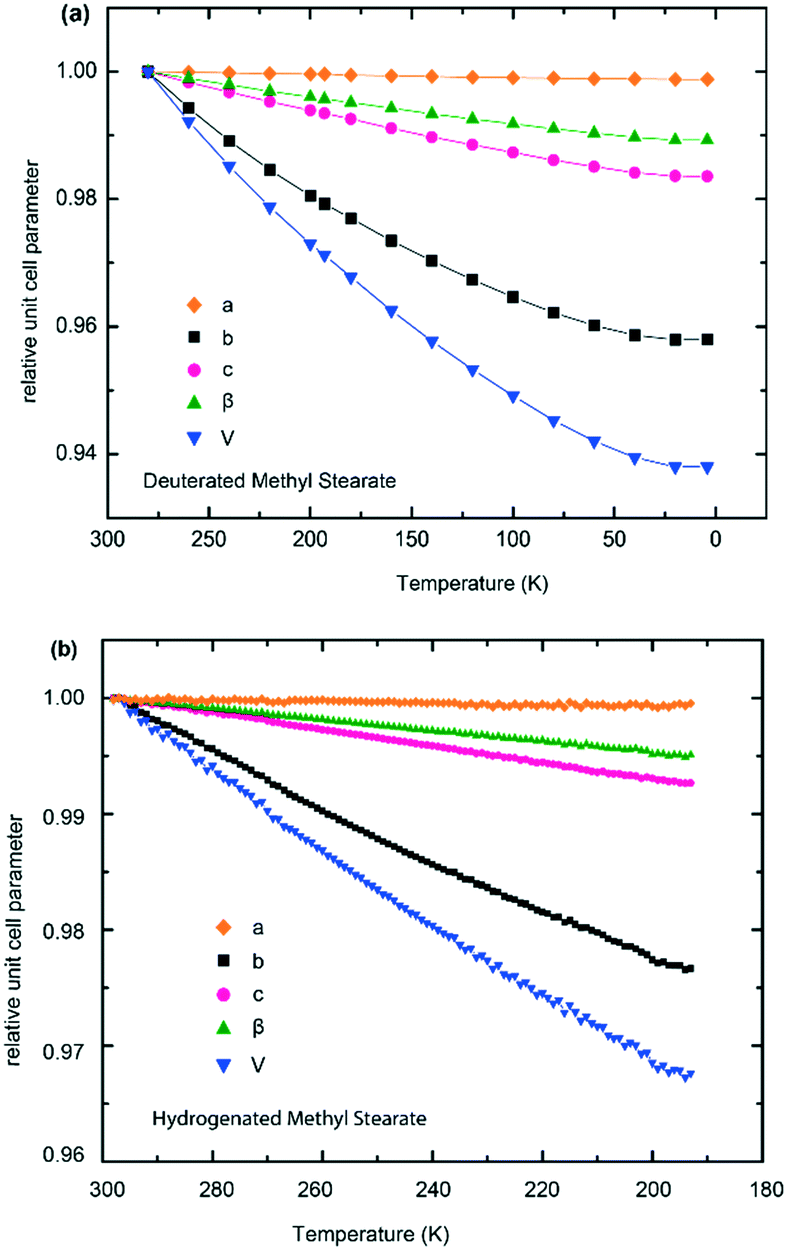 | ||
| Fig. 9 Multiplots of the relative unit cell expansion of form V of methyl stearate. (a) Deuterated methyl stearate over the temperature range of 4.2 K to 280 K (neutron data); (b) hydrogenous methyl stearate over the temperature range of 193 K to 298 K (synchrotron X-ray data). | ||
The temperature dependence of the unit cell volume of deuterated methyl stearate in the temperature range of 4.2 K to 280 K was fitted to a Debye–Einstein model33 (eqn (1) and (2))
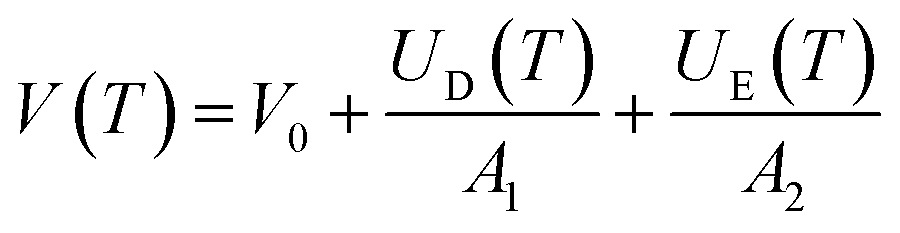 | (1) |
 | (2) |
The fit (shown in Fig. 10) resulted in the values θD = 189 ± 14 K, θE = 1287 ± 186 K, V0 = 1842.4 ± 0.4 Å3, A1 = 4.17 ± 0.03 × 104 J cm−3, A2 = 1.78 ± 1.04 × 103 J cm−3. The Debye temperature obtained (≈189 K) is consistent with those of other low-dimensional organic compounds.34,35 The relatively large errors in the fitted parameters for the Einstein component of the model can largely be accounted for by the restricted experimental temperature range in relation to the Einstein temperature. No anomalies in the thermal expansion behaviour were detected within the temperature resolution of these experiments.
 | ||
| Fig. 10 Volume thermal expansion coefficient of form V of deuterated methyl stearate fitted to a Debye–Einstein model. | ||
It was possible to compare the different thermal expansion coefficients of the lattice parameters induced by deuteration over the temperature range 193–280 K. The thermal expansion coefficients of both axial  and volumetric
and volumetric  are calculated and tabulated in Table 3. This shows that the thermal expansion behaviour of hydrogenous and deuterated methyl stearate is very similar over this temperature range. It is interesting to note that thermal expansion coefficients of all the unit cell parameters of the deuterated methyl stearate are slightly higher than those for the hydrogenous sample. This suggests that the intermolecular interactions in deuterated methyl stearate are slightly weaker than those of the hydrogenous sample.
are calculated and tabulated in Table 3. This shows that the thermal expansion behaviour of hydrogenous and deuterated methyl stearate is very similar over this temperature range. It is interesting to note that thermal expansion coefficients of all the unit cell parameters of the deuterated methyl stearate are slightly higher than those for the hydrogenous sample. This suggests that the intermolecular interactions in deuterated methyl stearate are slightly weaker than those of the hydrogenous sample.
| Sample type | α a (× 10−6 K−1) | α b (× 10−4 K−1) | α c (× 10−5 K−1) | α β (× 10−5 K−1) | α V (× 10−4 K−1) |
|---|---|---|---|---|---|
| Hydrogenous | 2.91(4) | 2.28(1) | 7.02(3) | 4.54(4) | 3.16(4) |
| Deuterated | 4.29(1) | 2.50(1) | 7.39(1) | 4.90(6) | 3.40(7) |
It was possible to perform Rietveld refinements for form VI based on the high quality NPD data. The structure of form V of deuterated methyl stearate at 193 K, solved from neutron powder diffraction, was employed as a model. Comparison of Rietveld refinements performed on the data collected at 280 K and 300 K (Fig. 11) highlights the phase change. In particular, it is notable that a number of reflections (such as Bragg peaks at d-spacings of 1.27 Å, 2.22 Å, and 2.39 Å) shown in the patterns collected at 300 K, could not be fitted by the model structure of form V, or indeed any of the known forms. Unfortunately, on account of the large unit cell volumes of the polymorphs of methyl stearate, combining with the limited d-spacing range of the data recorded from HRPD instrument, it was not possible to determine the unit cell parameters of form VI. Moreover, this transition was not observed in the PXRD pattern of the hydrogenous sample at the maximum temperature of 298 K. This strongly suggests that there may be an effect of deuteration, either on the formation of form VI or on the transition temperature. An alternative explanation is that this phase transition is associated with a rearrangement of some of the hydrogen (deuterium) atoms. This would appear clearly in the neutron experiment, which is very sensitive to scattering from deuterium atoms, but might not appear in the X-ray experiment on account of the only weak contribution to the scattering from hydrogen atoms.
 | ||
| Fig. 11 Rietveld refinements of the neutron powder patterns collected at 280 K and 300 K, using form V of deuterated methyl stearate as model structure, show mismatched peaks induced by the phase transition. In both cases the experimental profile (black), calculated profile (red) and difference (blue). Vertical tick marks (pink) show calculated peak positions. Blue arrows indicate mismatching reflections. (a) a plot of pattern recorded by the backscattering detector (bank 1); (b) a plot of pattern recorded by the complementary detector at 90° (bank 2). | ||
Forms II and III were both obtained from a CS2 solution and show some degree of similarity in molecular packing. Methyl stearate molecules in these two forms all pack in “head to head” (and “tail to tail”) oblique bilayers, with four molecular layers in the unit cell. The most significant difference between the structures of form II and III is the tilt angle between molecules inside the bilayer. In form II, which crystallised under low temperature, methyl stearate molecules stack in roof-like “waves” with an approximately open 120° angle between the least squares line though the methyl stearate moieties. By contrast, in form III, which was crystallised at room temperature, the open angle between the methyl groups of adjacent layers is 180°. Both form IV and V were crystallised from the melt, but using different cooling rates. They both exhibit tilted monolayers, with two molecular layers in the unit cell, while as forms II and III, show bilayer packing behaviour leading to a doubled crystallographic axis along with direction of the hydrocarbon chain. A similar difference in packing has also been reported for long-chain alkanes;15,38 it was suggested that this difference could be induced by the presence of impurities at a level of 2% being required for the formation of bilayers. In this case, the solvent could possibly act as an impurity inducing the layer doubling, although we saw no evidence of incorporation of CS2 in these studies. Another possibility of the difference of the packing behaviour may be induced by the conformation of the material its polarity. Methyl stearate contains hydrophobic hydrocarbon chain and a more hydrophilic ester group. It is therefore more likely to form lipid bilayers in solution due to the hydrophobic interactions. The structure obtained from CS2 solution therefore shows “head to head” packing. In contrast, this cannot happen on crystallisation from the melt. This type of “head to head” packing of methyl stearate could give some into the design of additives for biodiesel. Given that biodiesel contains FAME compounds dissolved in hydrocarbons, it is therefore likely that crystallites formed under low-temperature conditions will feature packing that involves the formation of “head-to-head” bilayers.
Different cooling rates of the melt clearly influence the outcome of crystallisation. Form IV, which was crystallised by slow cooling, shows a highly disordered structure, in contrast to form V which was crystallised by rapid cooling. This is perhaps a somewhat surprising result as one would normally expect rapid quenching of a melt to result in structures that are more disordered. The disordered packing behaviour of form IV suggests that it is less stable than form V.
The packing behaviour in all of the polymorphs is dominated by inter-chain interactions, which appear to be weaker for deuterated methyl stearate compared to the hydrogenous sample.39 This is clearly shown in the significant difference in thermal expansion coefficients of the two isotopomers over a similar temperature range and is perhaps a possible explanation as to why the phase transition observed in the deuterated sample from form V to VI was not observed in the hydrogenous sample over the same temperature range. Finally, we should comment on the almost identical lattice parameters of form IV identified in this study and the monoclinic form reported in ref. 18. The only difference is in the assignment of space group (C2/c for form IV compared with C2 in the previous report). Given the consistency between the structures of form IV determined by both X-ray and neutron techniques, there is a high degree of confidence that the space group is indeed C2/c rather than C2.
4. Conclusions
This research has shown that the crystallisation behaviour of methyl stearate is strongly affected by temperature. A new polymorph, form III, was successfully obtained by growing a single crystal from a saturated CS2 solution at room temperature and data collection was conducted at 120 K. Form IV was obtained by crystallisation from the melt followed by slow cooling. Structural characterisation using single crystal X-ray diffraction showed disordered packing of the molecules in this form. The crystal structure of form V, obtained by quench cooling from the melt, was solved and refined using a combination of synchrotron XRPD and high-resolution neutron powder diffraction. The thermal expansion behaviour of this form was also investigated. Form V of methyl stearate shows highly anisotropic thermal expansion, with significantly greater expansion along the crystallographic b-axis compared to the a- and c-axes. The deuterated sample showed a higher volume thermal expansivity. The crystal structure of form III shows “head to head (ester group)” bilayer packing behaviour with similar unit cell parameters as published for form II. Differences are shown in packing angles of molecules between layers. Forms IV and V show similar monolayer packing behaviours with similar unit cell parameters but unlike form III, form V are shown “head to tail (end of chain group)” packing, while the length of crystallographic axis of form V along hydrocarbon chain accommodation only shows half of those of form III. In contrast to form V, form IV structure has a crystallographic inversion centre located in the centre of the molecule and the whole structure exhibits whole molecule disorder. Furthermore, a phase transition from form V to form VI (280–300 K) was observed in the neutron diffraction experiments for a fully deuterated sample, but no evidence for this transition was observed from a hydrogenous sample in a corresponding synchrotron XRPD study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors grateful acknowledge Infineum Ltd. for funding this research. We thank Diamond Light Source for access to beamline I11 (NT12259-1) that contributed to the results presented here. We gratefully acknowledge the Science and Technology Facilities Council (STFC) for access to neutron beamtime at ISIS, and also for the provision of sample preparation, HRPD facilities. Many thanks to Prof Ken Lewtas, Dr Sumit Konar, Dr Adam Michalchuk, Dr Katrina Warrack, Dr Boris Zakharov, and Dr Peter Hutchins for helpful discussions. X. Liu is grateful for an Edinburgh Global Research Scholarship and PhD studentship funding from Infineum Ltd.References
- J. G. J. Olivier, M. Muntean and J. A. H. W. Peters, PBL Netherlands Environ. Assess. Agency Eur. Comm. Jt. Res. Cent., 2015, pp. 1–78 Search PubMed.
- I. B. Fridleifsson, Renewable Sustainable Energy Rev., 2001, 5, 299–312 CrossRef.
- N. N. A. N. Yusuf, S. K. Kamarudin and Z. Yaakub, Energy Convers. Manage., 2011, 52, 2741–2751 CrossRef.
- C. R. Krishna, K. Thomassen, C. Brown, T. A. Butcher, M. Anjom and D. Mahajan, Ind. Eng. Chem. Res., 2007, 46, 8846–8851 CrossRef.
- R. O. Dunn, Prog. Energy Combust. Sci., 2009, 35, 481–489 CrossRef.
- P. Saxena, S. Jawale and M. H. Joshipura, Procedia Eng., 2013, 51, 395–402 CrossRef.
- P. Saxena, S. Jawale and M. H. Joshipura, Procedia Eng., 2013, 51, 395–402 CrossRef.
- M. B. Dantas, A. R. Albuquerque, A. K. Barros, M. G. Rodrigues Filho, N. R. Antoniosi Filho, F. S. M. Sinfrônio, R. Rosenhaim, L. E. B. Soledade, I. M. G. Santos and A. G. Souza, Fuel, 2011, 90, 773–778 CrossRef.
- G. Knothe, Fuel Process. Technol., 2007, 88, 669–677 CrossRef.
- S. T. Ahmed, M. M. Islam, A. B. M. R. Bostami, H. S. Mun, Y. J. Kim and C. J. Yang, Food Chem., 2015, 188, 481–488 CrossRef PubMed.
- B. Bozan and F. Temelli, Bioresour. Technol., 2008, 99, 6354–6359 CrossRef PubMed.
- A. I. Bamgboye and A. C. Hansen, Int. Agrophys., 2008, 22, 21–29 Search PubMed.
- I. Kralova and J. Sjöblom, J. Dispersion Sci. Technol., 2010, 31, 409–425 CrossRef.
- S. K. Hoekman, A. Broch, C. Robbins, E. Ceniceros and M. Natarajan, Renewable Sustainable Energy Rev., 2012, 16, 143–169 CrossRef.
- S. R. Craig, G. P. Hastie, K. J. Roberts and J. N. Sherwood, J. Mater. Chem., 1994, 4, 977–981 RSC.
- E. von Sydow and S. Aleby, Acta Crystallogr., 1960, 13, 487–492 CrossRef.
- C. H. MacGillavry and M. Wolthuis-Spuy, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 645–648 CrossRef.
- D. M. Camacho, K. J. Roberts, F. Muller, D. Thomas, I. More and K. Lewtas, Cryst. Growth Des., 2017, 17, 563–575 CrossRef.
- Rigaku Oxford Diffraction, Technol. UK Ltd, Yarnton, Oxford, UK, 2014 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- S. P. Thompson, J. E. Parker, J. Potter, T. P. Hill, A. Birt, T. M. Cobb, F. Yuan and C. C. Tang, Rev. Sci. Instrum., 2009, 80, 75107 CrossRef PubMed.
- S. P. Thompson, J. E. Parker, J. Marchal, J. Potter, A. Birt, F. Yuan, R. D. Fearn, A. R. Lennie, S. R. Street and C. C. Tang, J. Synchrotron Radiat., 2011, 18, 637–648 CrossRef PubMed.
- A. A. Coelho, TOPAS-Academic V7, http://www.topasacademic.net, 2017 Search PubMed.
- K. V. Srinivas, I. Mahender and B. Das, Synlett, 2003, 2003, 2419–2421 Search PubMed.
- A. C. Larson and R. B. Von Dreele, Los Alamos Natl. Lab. Rep. LAUR, 2004, vol. 748, pp. 86–748 Search PubMed.
- K. Ando, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 172104 CrossRef.
- M. Ichikawa, J. Mol. Struct., 2000, 552, 63–70 CrossRef.
- T. Udagawa and M. Tachikawa, J. Mol. Struct.: THEOCHEM, 2009, 912, 63–66 CrossRef.
- T. Udagawa, T. Ishimoto, H. Tokiwa, M. Tachikawa and U. Nagashima, J. Phys. Chem. A, 2006, 110, 7279–7285 CrossRef PubMed.
- K. Wang, J. Zhang, J. Wang, H. Zhang, Z. Wang, W. Yu, X. Wang, Q. Lu, M. Ba and R. I. Boughton, J. Appl. Phys., 2005, 98, 46101 CrossRef.
- A. Fortes, E. Suard and K. Knight, Science, 2011, 331, 742–746 CrossRef PubMed.
- W. C. Germs, K. Guo, R. A. J. Janssen and M. Kemerink, Phys. Rev. Lett., 2012, 109 Search PubMed.
- J. Odin, J. C. Lasjaunias, K. Biljaković, P. Monceau and K. Bechgaard, Solid State Commun., 1994, 91, 523–527 CrossRef.
- E. von Sydow, The normal fatty acids in solid state, Stockholm, 1956 Search PubMed.
- F. Framcis and S. H. Piper, J. Am. Chem. Soc., 1939, 61, 577–581 CrossRef.
- P. Teare, Acta Crystallogr., 1959, 12, 294–300 CrossRef.
- M. Ichikawa, Nippon Kessho Gakkaishi, 1998, 40, 10–18 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1589573, 1811171–1811173. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01055b |
| This journal is © The Royal Society of Chemistry 2018 |