
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
X-ray Raman scattering: a new in situ probe of molecular structure during nucleation and crystallization from liquid solutions†
Laila H.
Al-Madhagi
 ab,
Sin-Yuen
Chang
b,
Mahalingam
Balasubramanian
c,
Anna B.
Kroner
b,
Elizabeth J.
Shotton
b,
Elizabeth A.
Willneff
d,
Bhoopesh
Mishra
ae and
Sven L. M.
Schroeder
*ab
ab,
Sin-Yuen
Chang
b,
Mahalingam
Balasubramanian
c,
Anna B.
Kroner
b,
Elizabeth J.
Shotton
b,
Elizabeth A.
Willneff
d,
Bhoopesh
Mishra
ae and
Sven L. M.
Schroeder
*ab
aSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK. E-mail: S.L.M.Schroeder@leeds.ac.uk
bDiamond Light Source Ltd., Didcot, Oxfordshire OX11 0DE, UK
cAdvanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, USA
dSchool of Design, The University of Leeds, Leeds LS2 9JT, UK
eDepartment of Physics, Illinois Institute of Technology, Chicago, IL 60616, USA
First published on 28th September 2018
Abstract
X-ray Raman scattering (XRS) has been used for in situ probing of solute molecule speciation in solution during cooling crystallization. The C and N K-edges of aqueous imidazole were measured as a function of temperature to monitor the transition from the undersaturated state through supersaturation to crystallization. A new jacketed-vessel crystallizer with internal flow was used, which enables thermal control and minimizes radiation damage. We have demonstrated that the C and N K-edges of imidazole are sensitive to changes in local bonding. In line with this, an abrupt change in the N K-edge fine structure indicates the onset of desolvation and crystallization from the supersaturated solution. In contrast, negligible changes are observed in the C and N K-edge spectra acquired during cooling, indicating that the average solvation structure around imidazole molecules does not change significantly while traversing the thermodynamically metastable supersaturated zone. To the best of our knowledge this is the first time X-ray Raman scattering has been used for studying molecular speciation in organic aqueous solutions during crystallization. Time-dependent density functional theory (TD-DFT) calculations of the near-edge spectra were performed using implicit, explicit and combined solvation models to elucidate the likely binding sites of the water molecules. An explicit solvation model with one water molecule coordinating each nitrogen moiety in the imidazole ring accurately reproduces the peak positions and intensities of the XRS spectra of aqueous imidazole solution.
Introduction
Understanding the nucleation of organic solutes from solutions is crucial to control the physical and chemical properties of crystalline products, with relevance for the chemical and the pharmaceutical industries. Current nucleation theory does not allow predictive design of crystallization processes to target crystal properties. More detailed information about molecular transformations and self-assembly processes during nucleation and crystallization is required to progress this research field.1–4Nucleation of crystals from solution must be associated with the formation of solute–solute bonds at the expense of solute–solvent interactions. Classical nucleation theory (CNT) describes this process by postulating a prenucleation equilibrium between solvated solute molecules and small clusters of solute molecules that are held together by solute–solute interactions. CNT further assumes that the balance between the cohesive energy gain in the nascent crystal volume and destabilization by the formation of the crystal/solution interface determines a critical cluster size, above which further crystal growth becomes the thermodynamically spontaneous process. Clusters with subcritical size form and re-dissolve again in the prenucleation equilibrium. One would therefore expect that at high supersaturation an unknown mole fraction of the solute molecules exists in prenucleation clusters1 in which solute–solvent bonds are replaced by solute–solute interactions. This process is possibly mediated by the solvent molecules in the solvation shells surrounding the solute molecules.
A number of spectroscopic and scattering techniques have been used to probe the interactions of solute molecules in solution generally, and specifically during nucleation. Some of these techniques are: nuclear magnetic resonance (NMR),5,6 optical microscopy,7 Raman and Fourier transform infra-red (FTIR) spectroscopy,8 UV-vis spectroscopy9 and small- and wide-angle X-ray scattering (SAXS and WAXS).10 However, with these techniques there has been limited success in providing molecular-level information about the supersaturated state from which nucleation occurs. Neutron diffraction and accompanying structure simulations can provide an excellent molecular representation of solution structures,11,12 but the measurements are slow and hence not suitable for capturing the structural evolution around solute molecules during nucleation and crystallization.
Near-edge X-ray absorption fine structure (NEXAFS) spectroscopy probes local structure via excitation of core level electrons (usually 1s) to unoccupied states in the valence region of molecules. The technique has recently been shown to be able to detect the formation of prenucleation equilibrium during the reactive interfacial nucleation of metal nanoparticles.13–15 The involvement of valence levels makes NEXAFS also highly sensitive to chemical and physical changes around organic molecules in solution and in the solid state. Differences due to crystal polymorphism,16 solute–solvent interactions,17–19 protonation and hydrogen bonding18,20,21 have been reported.
However, NEXAFS of organic molecules is a soft X-ray synchrotron radiation technique requiring ultra-high vacuum (UHV) beamlines, with most available spectrometers housed in UHV chambers as well. NEXAFS measurements on liquids thus require sample handling in a vacuum chamber. This imposes constraints on experimental control that, while probably addressable through novel instrument designs, have so far prevented studies of solution structure under controlled supersaturation conditions. Most commonly used designs are liquid microjets that expand into a vacuum chamber.22–25 In a liquid microjet system, evaporative cooling and adiabaticity of the sudden pressure drop prevent control of the temperature, which is required to maintain well-defined supersaturation conditions. Moreover, turbulence and interfaces in the microjet nozzles act as nucleation sites, often resulting in blockage by growth of solid deposits from supersaturated solutions.
X-ray Raman scattering (XRS) is a non-resonant X-ray scattering technique that is an alternative to NEXAFS for probing unoccupied valence density of states. XRS probes not only dipole but also higher-order transitions, depending on the momentum transfer involved. The momentum transfer is related to the angle between the incident beam and the detection angle for the scattered photons. At low momentum transfer (vide infra) the spectra are dominated by dipole transitions. XRS utilizes hard X-rays; hence it is effectively an alternative technique for measuring NEXAFS that avoids the experimental challenges associated with the use of soft X-rays. The deep penetration depth (∼2 mm at 10 keV) of hard X-rays makes XRS a bulk-sensitive technique, with the added benefit of significantly inhibited self-absorption distortions of the spectra.26 The use of hard X-rays also allows the design of ambient-pressure environmental cells with precise temperature and pressure control. It has been shown previously that these characteristics make the technique suitable for studying molecular structure in the bulk of liquid water and aqueous solutions,26,27 including comparisons of structure in liquid water and ice28,29 and exploring the effect of isotope substitution on local structure.30 More recently, XRS has been applied to examine the effects of organic solutes on the structure of water.31 Here, we show that XRS can be used as an in situ technique for characterizing solute speciation during cooling crystallization, from the undersaturated state through the metastable zone with high supersaturation until the onset of crystallization.
Imidazole was chosen as the test system for the in situ XRS measurements because it has high solubility in water (∼11 mol L−1 at 25 °C (ref. 32)), which ensures good signal-to-noise ratios. Moreover, imidazole is of high biological,33–36 pharmaceutical37–39 and industrial40,41 importance. For imidazole in the solid state, X-ray42,43 and neutron44 diffraction analysis revealed a motif of hydrogen-bonded molecule chains in one direction, with 3D-cohesion involving π–π interactions between the chains. For aqueous solutions, previous studies by UV spectroscopy9 and energy dispersive X-ray diffraction45 indicated self-association of the solute molecules by π–π stacking. However, molecular dynamics (MD) simulations illustrated that chain-like hydrogen-bonding is more likely than such stacked structures, both in aqueous solutions at different concentrations46 and in liquid imidazole.47 MD simulations also indicated that the local structure around the imidazole molecules does not change strongly as a function of concentration.46 A recent NEXAFS study of imidazole solutions17 supported the MD simulation results, in that the N K-edge absorption spectra exhibited only minor changes in the concentration range from 0.5 to 8.2 M. The XRS study reported here complements this NEXAFS study17 and extends the scope of characterization to the supersaturated state.
Experimental
Crystallizer design
Commonly used hard X-ray sample environments for liquid samples include microfluidic cells,48 closed-loop flow cells utilizing peristaltic pumps49,50 and liquid jets.51 Since cryogenic cooling is not an option for reducing radiation damage of the sample when studying liquids, circulating or flushing samples is required to reduce the contact time between the sample and the X-ray beam. The use of microfluidic cells is one way of achieving this. However, their stock volume is small, which is convenient for precious or hazardous samples, but may not be sufficient for diluting the influence of radiation-induced chemical changes. Closed-loop flow cells often use higher stock volumes. However, maintaining the equilibrium temperature, and thus the metastable state of the supersaturated system, is challenging across a combination of a stock solution reservoir, flow tubing and the region where X-ray probing takes place, making these cells less attractive for crystallization studies.We have therefore designed a different type of crystallizer (Fig. 1) built around a jacketed vessel of the type commonly used in laboratory-scale crystallization studies. The design minimizes the potential for radiation damage to the sample through circulation of a large stock volume (∼250 ml) and provides the required temperature control, i.e. the thermodynamic driving force for crystallization. It is therefore possible to follow the structural evolution of a solution from undersaturation through supersaturation to nucleation and crystal growth in a classic cooling crystallization experiment.
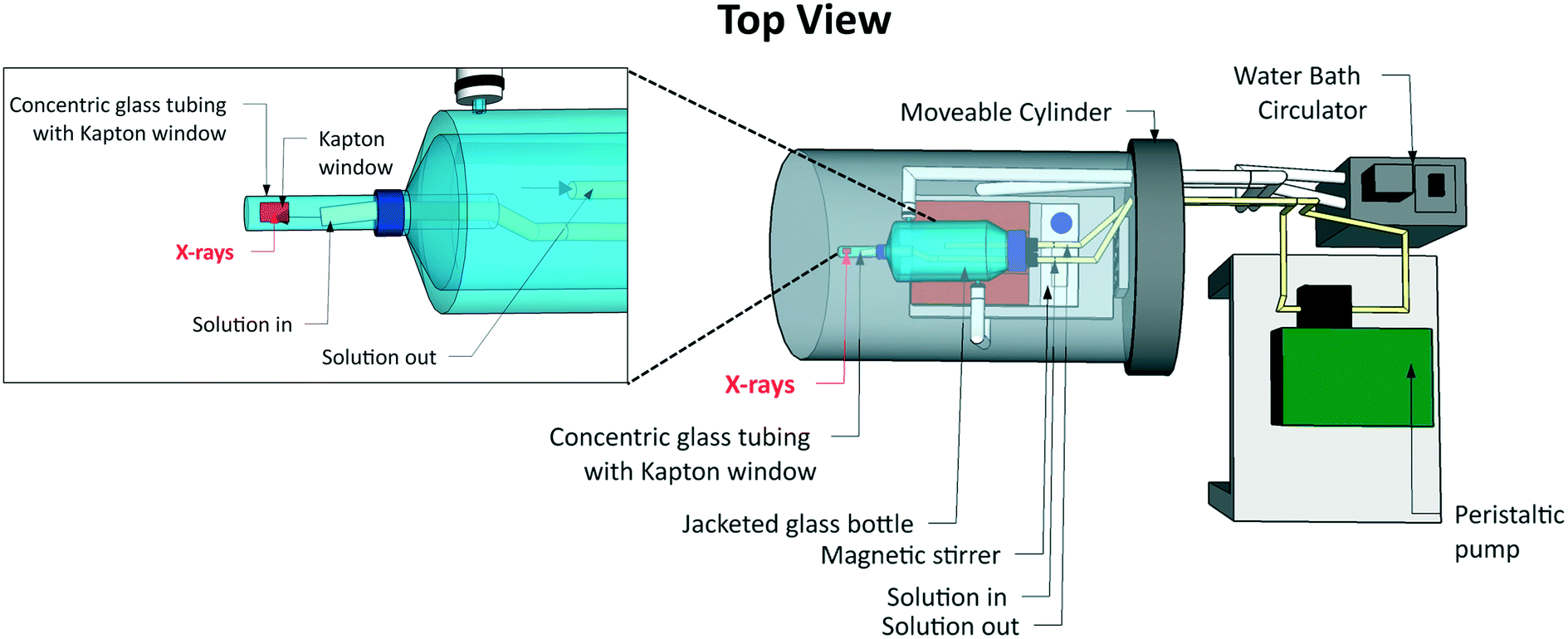 | ||
| Fig. 1 The flow cell crystallizer used for XRS measurements of solution phase samples. The shaded grey area is encapsulated in He gas. | ||
At the heart of the crystallizer is a standard jacketed glass vessel. The temperature of the crystallizing solution is controlled through the temperature of the fluid in the jacket. We used water as the temperature control fluid, with a water bath circulator to control its temperature. This jacketed vessel was modified by fitting a system of two concentric tubes to its bottom. The outer cylindrical glass tube has a closed end and was attached to the jacketed vessel using a screw cap connection. It has a Kapton window to allow probing of the sample volume directly underneath using X-rays. The Kapton window was fixed over a 10 mm wide cut-out slot covering a 180° arc. The solution flows continuously past this window and is recycled back into the main bulk of the cell via an inner concentric tube using a peristaltic pump at a flow rate of up to ∼450 ml min−1 to minimize radiation damage. The inner concentric tube diameter is 3.2 mm, which is sufficiently large to minimise turbulence that may induce nucleation. In addition to internal solution circulation, magnetic stirring was used to ensure uniform temperature distribution in the stock solution within the jacketed vessel and to mimic stirring in a real-world crystallizer.
Cooling crystallization studies
Imidazole (Sigma Aldrich, puriss >99.5%) was used without further purification. We noticed that some supplied batches contained small amounts of insoluble matter; such batches were not used for these experiments. Deionized water was used for the preparation of the 10.1 M imidazole solution. The solution had a pH value of 11 at room temperature. For the cooling crystallization study, the C and N K-edge spectra of a 10.1 M aqueous solution of imidazole were measured at 35, 20 and 13 °C. The solution was also probed after precipitation of crystals had taken place.Crystalline imidazole powder was also measured as a reference. The powder sample was pressed into a 13 mm diameter pellet with a thickness of ∼2 mm. The pellet was mounted on a spinning platform and measured with an off-centred beam to minimize radiation damage.
X-ray Raman scattering
Non-resonant inelastic X-ray scattering measures the double differential cross section d2σ/dΩdω (ref. 52) which contains a Thomson scattering cross section and a dynamic structure factor S(q,ω) (dσ/dω)Th, viz. | (1) |
At low momentum transfer (q), XRS and XAS probe the same transition matrix element for core level electrons and have similar directional sensitivities.52 The dynamic structure factor for XRS in the low q range (the dipole limit) is presented in eqn (2) and is analogous to the X-ray absorption coefficient.52
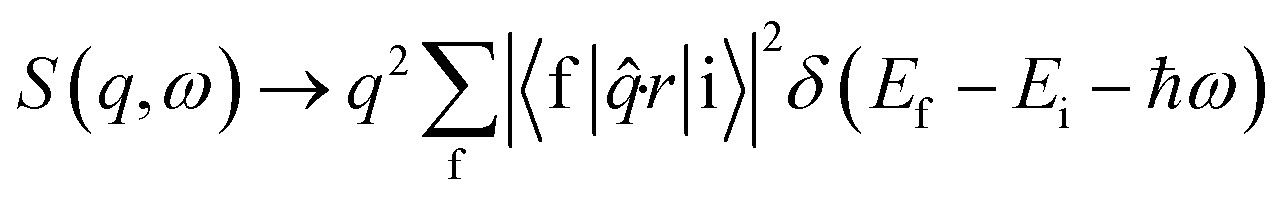 | (2) |
XRS measurements were performed at sector 20-ID of the Advanced Photon Source (APS), Chicago, USA, using the low-energy resolution inelastic X-ray scattering (LERIX) spectrometer.53,54 20-ID is an undulator beamline equipped with liquid nitrogen cooled Si(111) and Si(311) monochromators. The Si(311) monochromator was used as it achieves high spectral resolution which is needed for this study. The combined instrumental resolution of the monochromator and the analyzer crystal was ∼0.5 eV. The monochromatized beam was focused using a toroidal mirror to a spot size of 600 μm (horizontal) and 400 μm (vertical). The scattered radiation was analyzed with a combination of Si diced and undiced spherically bent crystal analyzers (SBCA). The SBCAs were tuned to use the Si(555) reflection, and the scattered radiation was detected using NaI scintillators coupled to custom-built amplifier and discriminator electronics. All data were collected using the inverse scanning mode, where the incident photon energy was scanned and the analyzer energy was fixed at 9891 eV. Data were collected from 18 of the 19 crystal spectrometers. The data presented in this study were taken from q6 diced crystal analyzer which is one of the low momentum transfer (q) analyzers with the best signal to noise ratio. This way, the data were collected at a scattering angle of 54° with q value of ∼4.6 Å−1. The monochromator drift was small (±0.137 eV) and it was monitored through frequent measurement of the elastic scattering peak. The monochromator was detuned by 15% to suppress the contribution from the harmonic component of the beam.
For the liquid samples, typical measurement times at each temperature were 2–3 hours. Over this period, several spectra were collected and checked for consistency before averaging. It is worth noting that no evidence of radiation damage was picked up in the collected spectra (individual spectra are provided in the ESI,† Fig. S1–S9). The LERIX sample chamber was flushed with He gas to reduce background scattering from air. For data analysis, the incident energy was subtracted from the XRS data to obtain near-edge spectra which were then subsequently background subtracted and normalized to the edge step height using Athena software.55
Computational methods
Excited state density functional theory (DFT) calculations were performed to generate theoretical C K-edge and N K-edge X-ray absorption (XA) spectra of imidazole in the gas, solution and solid phases. All calculations were performed using ORCA version 4.0.2, a quantum chemistry software package.56 Calculations were carried out using the B3LYP functional57 and the 6-31G* basis set for the monomeric imidazole molecule, while a minimally augmented def2-TZVP basis set58 was used for simulating the XA spectra of solvated and solid state imidazole. To speed up the calculations, the D3 dispersion correction59 was applied to all calculations. RIJCOSX approximation60 with def2/J auxiliary set was applied to the calculations for solvated and solid state imidazole.The structure of the monomeric molecule used to represent imidazole in the gas phase was constructed in Avogadro version 1.1.1, a molecular builder and visualization tool with ORCA support.61 The intramolecular structure was initially optimized in Avogadro using the UFF force field and then further refined using ground state geometry optimization calculations in ORCA. Frequency calculations were run on the optimized structure to ensure that the structure corresponded to the global energy minimum. Finally, time dependent DFT (TD-DFT) calculations were performed on the optimized structure to generate the theoretical near-edge spectra.
To simulate the water solvation shell around the imidazole molecule, both implicit and explicit solvation models were used. In models 2–4, water molecules were explicitly added around the imidazole molecule. The positions of the water molecules shown in Fig. 2 were optimized in ORCA. For comparison, model 1 used an implicit solvation model, the conductor-like polarizable continuum model (CPCM).62 Model 5 used a hybrid explicit/implicit model which added CPCM to the structure from model 4 where two water molecules bind to the protonated and non-protonated nitrogen atoms in the imidazole ring. The construction of all the solvation models as well as the geometry optimization and frequency calculations were performed using the same procedure as described above for the monomeric imidazole.
 | ||
| Fig. 2 The structural models representing imidazole in (i) the aqueous solution and (ii) solid phase. The two imidazole molecules used in the solid phase TD-DFT calculations are circled. | ||
To perform excited state simulations (TD-DFT) of solid phase imidazole, a cluster of eight imidazole molecules (Fig. 2) was taken from the crystal structure of imidazole.44 The calculations were performed for the two circled imidazole molecules (Fig. 2(ii)) in the eight molecule cluster. These two molecules are involved in hydrogen bonding as well as edge-face and π–π stacking with parallel displacement as in the crystal structure. Hence calculations on these molecules should account for the effects of the different types of imidazole–imidazole interactions that exist in the crystal structure.
It should be noted that the excited state DFT calculations presented here only model electronic states below the continuum threshold. In addition, relative changes in the core level potential energies calculated using TD-DFT are more accurate than absolute values,63,64 thus we introduced an empirical 1s core level energy offset of ∼10.5 eV (±0.4) for the C K-edge and ∼12.3 eV (±0.5) for the N K-edge when comparing calculated and experimental near-edge spectra.
Results and discussion
In Fig. 3, the C and N K-edge spectra of solid and aqueous imidazole measured using X-ray Raman scattering (XRS) are compared to the gas phase imidazole spectrum measured using inner shell electron energy loss spectroscopy (ISEELS).65 At low momentum transfer, ISEELS66 and XRS52 probe dipole transitions and hence are analogous to X-ray absorption (XA) spectra. The strong resonances in the near-edge region of the C and N K-edge spectra correspond to transitions from the 1s core level of the absorbing atom to the unoccupied valence orbitals with π* character. These are followed by the ionization potentials (IP) and broad σ* shape resonances corresponding to multiple scattering at the potential barrier to the bound neighbouring atoms. The energy difference between the centroid of the σ* shape resonances and the IP (usually termed δ) provides a highly sensitive measure of the bond length.66,67 The sensitivity of δ values to bond length variations as low as 0.001 Å have been previously illustrated for p-aminobenzoic acid68 and saccharides.69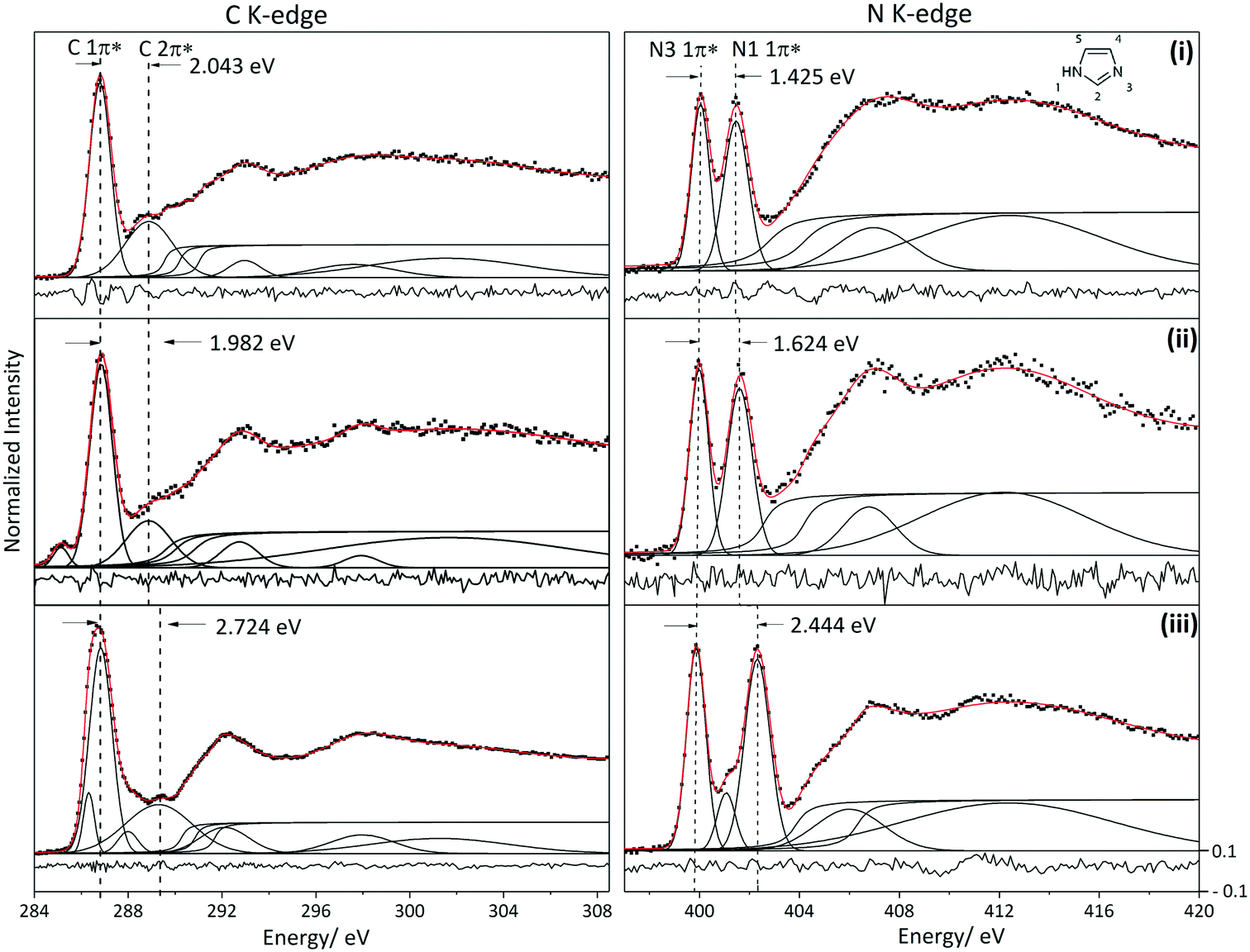 | ||
| Fig. 3 Fitted C and N K-edge spectra of the different phases of imidazole. Residual is shown at the bottom of each spectrum. (i) XRS spectra of solid state imidazole, (ii) XRS spectra of aqueous solution of imidazole measured at 35 °C and (iii) ISEELS spectra of imidazole in the gaseous phase.65 Note that the peak at ∼285.1 eV in the solution C K-edge is from the Kapton window. | ||
Non-linear least-squares fitting using Fityk software70 was utilized for the deconvolution of the near edge K-edge spectra (Fig. 3). Gaussian functions were used to model the π* and σ* shape resonances.66 The full width at half maximum (FWHM) of the fitted functions is mainly determined by life-time broadening, with similar values expected for the ionization potential (IP) step functions and the bound state π* resonances just below the IPs. The FWHMs of the Gaussian peaks modelling the π* resonances vary by ∼1 eV for the C K-edge and ∼0.3 eV for the N K-edge. The C K-edge near-edge peaks are broader because several features contribute to each π* resonance; this is illustrated later in the paper using TD-DFT calculations in which we assigned the contributions from the different carbon atoms to each π* peak. The FWHMs of the σ* shape resonances increase with energy due to lifetime broadening.66
Arctan functions were used to model the IPs.66 Considering that imidazole has three different carbon moieties and two different nitrogen moieties, three and two arctan functions were thus used to model the C K-edge and N K-edge IPs respectively. Since the ratio of the different carbon moieties in the imidazole molecule is equal, the width and height of the arctan functions in the C K-edge spectra were constrained to be equal; a similar procedure was followed for the N K-edge. The energy difference between the arctan functions (ΔIP) in the N K-edge spectra was constrained to be equal to the energy difference between the two 1π* peaks (ΔE (1π*)) because it has been illustrated previously that the transitions to π* states are dominated by the core level binding energy shifts.17 Experimental core level binding energies for C K-edge and N K-edge have been reported for aqueous imidazole solution using X-ray photoelectron spectroscopy (XPS).71 The energy differences of the arctan functions in the C K-edge for the aqueous solution phase (ΔIP (C2–C5) and ΔIP (C5–C4)) were constrained according to the values reported in the above mentioned XPS studies.71 ΔIP (N1–N3) is in good agreement with the binding energy differences reported previously.71 The resulting centroids of the fitted Gaussian and arctan curves are listed alongside the values predicted by ORCA TD-DFT calculations in Tables 1 and 2.
| Gas | Solution | Solid | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Exp/eV | Calc/eV* | Exp @ 35 °C/eV | Exp @ 20 °C/eV | Exp @ 13 °C/eV | Exp after crystallization/eV | Calc/eV* | Exp/eV | Calc/eV* | |
| * The energy shift of ∼12.3 eV (±0.5) has not been applied to the calculated transitions presented here. | |||||||||
| E (N1 1s–1π*) | 402.316 | 390.098 | 401.610 | 401.652 | 401.680 | 401.509 | 389.167 | 401.467 | 388.996 |
| E (N3 1s–1π*) | 399.872 | 388.020 | 399.986 | 400.057 | 400.049 | 399.956 | 387.513 | 400.042 | 387.600 |
| ΔE (1π*) | 2.444 | 2.078 | 1.624 | 1.595 | 1.632 | 1.553 | 1.654 | 1.425 | 1.396 |
| E (N3 1s–2π*) | 401.073 | 389.180 | — | — | — | — | 388.573 | — | 388.475 |
| ΔE (N3 2π*–1π*) | 1.201 | 1.160 | — | — | — | — | 1.060 | — | 0.875 |
| IP (N1) | 406.344 | 391.339 | 404.192 | 404.186 | 404.293 | 404.625 | 390.935 | 404.216 | 391.175 |
| IP (N3) | 403.898 | 389.125 | 402.568 | 402.591 | 402.661 | 403.072 | 389.175 | 402.792 | 389.652 |
| ΔIP | 2.446 | 2.214 | 1.624 | 1.595 | 1.632 | 1.553 | 1.760 | 1.424 | 1.523 |
| 1σ* (C–N) | 406.003 | 406.787 | 406.759 | 406.616 | 406.933 | 406.977 | |||
| 2σ* (C–N) | 412.349 | 412.181 | 412.239 | 412.029 | 411.694 | 412.419 | |||
| δ1 (1σ*–IP(N3)) | 2.144 | 4.219 | 4.168 | 3.955 | 3.861 | 4.185 | |||
| δ2 (2σ*–IP(N3)) | 8.490 | 9.613 | 9.648 | 9.368 | 8.622 | 9.627 | |||
| δ3 (1σ*–IP(N1)) | — | 2.595 | 2.573 | 2.323 | 2.308 | 2.761 | |||
| δ4 (2σ*–IP(N1)) | 6.046 | 7.989 | 8.053 | 7.736 | 7.069 | 8.203 | |||
| Structural model | N1–H | N1–C2 | C2–N3 | N3–C4 | C4–C5 | C5–N1 | N3⋯HO | N1H⋯O | N1H⋯N3 |
|---|---|---|---|---|---|---|---|---|---|
| The calculated XA spectra of structural models in bold are presented in Fig. 5. | |||||||||
| Monomeric molecule (ORCA optimized) | 1.009 | 1.368 | 1.316 | 1.380 | 1.373 | 1.382 | |||
| Solution | |||||||||
| Model 1 (implicit) | 1.008 | 1.355 | 1.320 | 1.381 | 1.369 | 1.374 | |||
| Model 2 (explicit) | 1.006 | 1.359 | 1.312 | 1.376 | 1.366 | 1.377 | 1.916 | ||
| Model 3 (explicit) | 1.013 | 1.361 | 1.313 | 1.375 | 1.370 | 1.375 | 2.022 | ||
| Model 4 (explicit) | 1.014 | 1.356 | 1.315 | 1.375 | 1.368 | 1.375 | 1.896 | 1.945 | |
| Model 5 (combined) | 1.022 | 1.351 | 1.322 | 1.379 | 1.368 | 1.374 | 1.787 | 1.884 | |
| Solid44 | 1.038 | 1.337 | 1.316 | 1.368 | 1.358 | 1.362 | 1.828 | ||
N K-edge XRS
The near-edge region of the imidazole N K-edge spectra is dominated by excitations from 1s to the 1π* orbital of the two nitrogen moieties (N3 and N1–H).17,65 An additional transition into the 2π* valence orbital is observed at the N3 centre for the gas phase spectrum.65 Comparison of the N K-edge spectra of the three phases reveals a decrease in the energy difference between the two 1π* peaks, from 2.444 eV in the gas phase, to 1.624 eV for the solution and 1.425 eV for the solid (Fig. 3). The energy difference between the two 1π* transitions and IPs for the solution is closer to that of the hydrogen bonded solid than to the gas phase monomer, indicating that hydrogen bonding likely plays an important role in the solution phase of imidazole.The energy difference between the two 1π* peaks in the solid state (1.425 eV) spectra is in good agreement with previously reported total electron-yield (TEY) NEXAFS spectra for imidazole in the solid state (1.5 eV).65 Similarly, the 1π* energy difference for the solution phase sample (1.624 eV) is in agreement with previously reported X-ray excited optical luminescence (XEOL) NEXAFS of aqueous imidazole (1.7 eV).17 However, the intensities of the two π* peaks in the solution phase XRS spectrum reported here are roughly equivalent in contrast with the previously reported NEXAFS spectra measured using XEOL which has a less intense N1 1π* peak.17 We note that the relationship between core level absorption oscillator strengths and optical photon emission is complex, involving multiple excited state decay pathways that may include the transfer of energy from the absorbing atom to nearby atoms.72 This may explain the differences in relative intensities between the near-edge features measured using XRS and XEOL.
The experimental and ORCA-calculated features (Table 1) show that the variations in ΔE(1π*) and ΔIP between the three phases stem from larger changes in the energy of 1s → 1π* transitions and the IP associated with the protonated N1 moiety when compared to N3, an interpretation that is in line with previously published results combining NEXAFS and StoBe calculations.17 The energy shifts of the near-edge resonances, the IPs and δ values associated with the N1 moiety are reflected by more significant changes in the lengths of the bonds formed by N1 when compared to N3 (Table 2). As one goes from gas to solution to solid phases, there is an increase in the N1–H bond length and an associated decrease in the N1–C2 and N1–C5 bond lengths. On the other hand, the C2–N3 bond length is almost identical and the decrease in the N3–C4 bond length is less severe when compared to that associated with N1–C bond lengths. Moreover, the δ values associated with bonds involving the N1 atom are more dependent on the physical state compared with the δ values associated with N3 (Table 1). These observations indicate that the formation of hydrogen bonds with water molecules (N1–H⋯O–H/N3⋯H–O) in the solution sample and with imidazole molecules (N1–H⋯N3) in the solid sample changes the electron density at the N1 site much more than at the N3 site. A likely explanation is that the hydrogen bonds formed at the N1 centre occur through proton donation with changes in electron density located in the N–H bond. In contrast, the N3 centre acts as a proton acceptor and participates in the aromatic system, which therefore allows significant delocalization of the electron density variation across the aromatic ring resulting in a smaller core level shift due to hydrogen bonding.
C K-edge XRS
We will first define the origin of features in the C K-edge spectra before presenting the XRS spectra of imidazole in the solution and crystalline phases. For that purpose, excited state DFT calculations were performed on an isolated imidazole molecule. Based on these calculations, a schematic molecular orbital diagram was constructed to summarize the atomic and molecular orbital energies associated with the C K-edge absorption spectra (Fig. 5). Similarly to the N K-edge spectra, the near-edge region in the C K-edge spectra is expected to be dominated by transitions into the unoccupied π* states. The calculated XA spectrum for the monomer (Fig. 4) shows a sharp peak corresponding to C4, C5 and C2 1s → 1π* transitions and C4 1s → 2π* transition and a less dominant feature corresponding to C5 and C2 1s → 2π* transitions. The energy centroids of these transitions are listed in Table 3.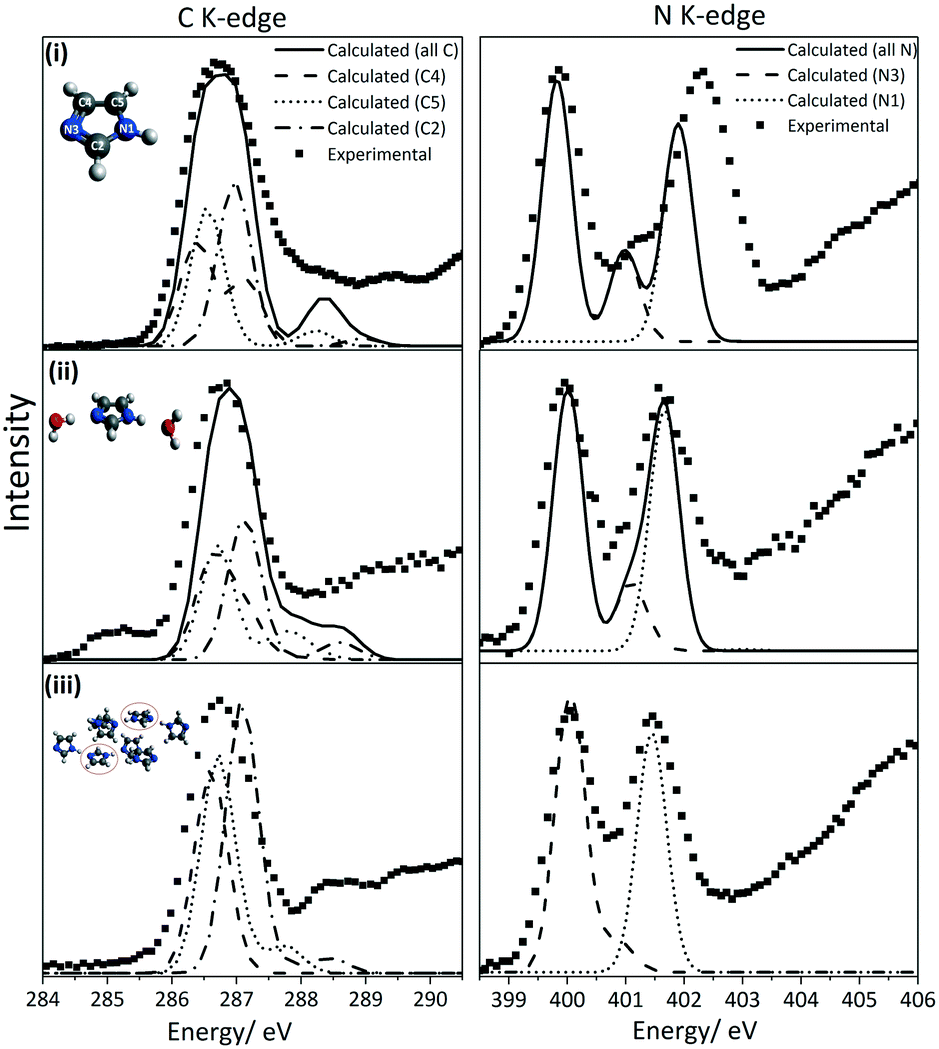 | ||
| Fig. 4 Comparison of the experimental and calculated C and N K-edge spectra. (i) ISEEL spectra of imidazole in the gaseous phase reported in reference.65 (ii) XRS spectra of the aqueous solution at 35 °C (iii) XRS spectra of solid state imidazole. For the aqueous solution sample, the TD-DFT simulated spectra used model 4 which is shown in Fig. 2. Note that the peak at ∼285.1 eV in the experimental C K-edge of the solution is from the Kapton window contribution. | ||
| Gas | Solution | Solid | ||||||
|---|---|---|---|---|---|---|---|---|
| Exp/eV | Calc/eV* | Exp @ 35 °C/eV | Exp @ 20 °C/eV | Exp @ 13 °C/eV | Calc/eV* | Exp/eV | Calc/eV* | |
| * The energy shift of ∼10.5 eV (±0.4) has not been applied to the calculated transitions presented here. | ||||||||
| E (from Kapton) | 285.106 | 285.185 | 285.170 | |||||
| E (C 1s–1π*) | 286.320 | 276.281 (C4) | 286.787 | 286.808 | 286.826 | 275.814 (C4) | 286.741 | 275.777 (C4) |
| 286.827 | 276.453 (C5) | 275.909 (C5) | 275.886 (C5) | |||||
| 276.872 (C2) | 276.323 (C2) | 276.290 (C2) | ||||||
| E (C 1s–2π*) | 286.827 | 277.049 (C4) | 288.769 | 288.860 | 288.827 | 276.298 (C4) | 288.784 | 275.922 (C4) |
| 287.989 | 278.121 (C5) | 277.095 (C5) | 276.983 (C5) | |||||
| 289.297 | 278.859 (C2) | 278.046 (C2) | 277.705 (C2) | |||||
| ΔE (C2 2π*–C 1π*) | 2.724 | 2.324 | 1.982 | 2.052 | 2.001 | 2.031 | 2.043 | 1.721 |
| IP (C2) | 291.690 | 278.267 | 290.697 | 290.697 | 290.697 | 278.238 | 290.887 | 278.606 |
| IP (C5) | 290.841 | 277.632 | 289.897 | 289.897 | 289.897 | 277.526 | 290.065 | 277.920 |
| IP (C4) | 290.266 | 277.099 | 289.497 | 289.497 | 289.497 | 277.117 | 289.424 | 277.572 |
| ΔIP (C2–C5) | 0.849 | 0.635 | 0.800 | 0.800 | 0.800 | 0.712 | 0.822 | 0.686 |
| ΔIP (C5–C4) | 0.575 | 0.533 | 0.400 | 0.400 | 0.400 | 0.409 | 0.641 | 0.348 |
| σ (C–H) | 292.103 | — | 292.566 | 292.497 | 292.612 | — | 292.786 | — |
| σ (C–C) | 297.987 | — | 297.655 | 297.768 | 297.729 | — | 297.323 | — |
| σ (C–N) | 301.266 | — | 301.302 | 300.680 | 301.191 | — | 301.206 | — |
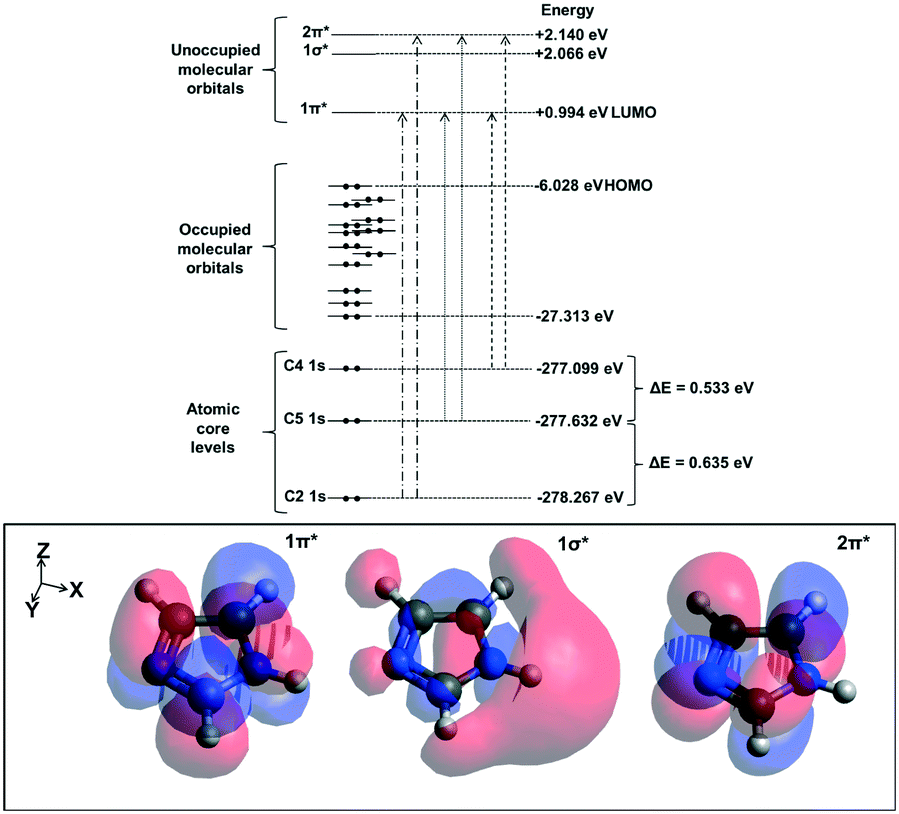 | ||
| Fig. 5 Schematic representation of the calculated atomic core levels as well as the occupied and unoccupied molecular orbitals for a monomeric imidazole molecule. The electron density of the three lowest unoccupied molecular orbitals are shown at the bottom of the figure. | ||
We now compare the experimental C K-edge spectra of imidazole in solid state and solution with the ISEELS gas phase spectrum previously reported65 (Fig. 3). The predicted near-edge features in the C K-edge corresponding to the C 1s → 1π* and C 1s → 2π* transitions are identified in the experimental spectra of imidazole in gas, solution and solid phases. The observed energy difference between the two features in the near-edge region of the solid state spectrum (2.043 eV) agrees with previously published TEY data.65 ORCA calculations for the hydrated imidazole molecule and the molecules in the crystal structure reproduced these features and enabled the identification of the contributions from the different carbon atoms (Fig. 4).
In terms of the C1s binding energies (BE), C2 is expected to have the highest 1s core level BE as it is bonded to both electronegative nitrogen moieties in the imidazole ring. The other two carbon atoms are expected to have lower BE values due to bonding to only one nitrogen neighbor, and C5 should have a slightly higher value than C4 as it is bonded to the protonated nitrogen atom (N1). This is reflected in the experimental IPs and the ORCA-calculated core level orbital energy values and the π* transition energies, which are the highest for C2 followed by C5 and C4 (Table 3). Similarly, in solid state theophylline73 and histidine74 the C1s binding energy for the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NH (equivalent to C2 in imidazole) was higher than the other carbon atoms in the imidazole ring of these molecules.
C–NH (equivalent to C2 in imidazole) was higher than the other carbon atoms in the imidazole ring of these molecules.
The energy difference between the two C K-edge peaks in the near-edge region for the solution phase is 1.982 eV, which is significantly lower than that observed in the gas phase (2.724 eV) and comparable to that of the solid phase (2.043 eV). This observation indicates that the electron density at the carbon moieties in the hydrogen bonded solution phase is similar to that in the crystalline phase where hydrogen bonding between imidazole molecules in the same plane exists alongside interplanar stacking interactions between hydrogen bonded chains. We should point out that for the solution C K-edge spectrum a fit with comparable quality can be achieved with ionisation potentials about 1 eV higher in energy (this is documented in the ESI†). However, the energy difference between the two peaks in the C K-edge spectra in that fit is similar to the energy difference reported for the gas phase spectrum, suggesting that the electron density for carbon atoms in aqueous solution is similar to that of the monomeric gas phase molecule, which appears implausible. Hence we tentatively prefer the fits presented here (Fig. 3 and 7) as they suggest similar electron density around the carbon moieties in the condensed phases (i.e. solution and solid phases). Note that the peak at 285.095 eV in the C K-edge solution phase spectrum is attributed to the contribution from the Kapton window. In contrast, the contribution of Kapton in the N K-edge spectra is minor due to the smaller atomic ratio of N in Kapton (∼0.051) as compared to C (∼0.564).
The effects of solvation
The sensitivity of the near-edge spectra to intermolecular interactions between imidazole and water is illustrated by the calculated spectra for the solvation structure models presented in Fig. 2. The structural model using two explicit water molecules, one added at each side of the imidazole ring (model 4), gave best agreement with the experimental N K-edge spectrum in terms of spectral features and relative energies of the peaks (Fig. 6 and Table 4). The energy difference between the two main 1π* peaks in the N K-edge near-edge region is greater in the XA spectra generated with explicit solvation models 2 and 3 (where water was added at one side of the imidazole ring only) than in the experimental spectrum (Fig. 6 and Table 4). This is associated with a shorter internal N1–H bond and longer hydrogen bonds between the imidazole and water molecules (Table 2). The variations between the calculated C K-edge spectra for the three explicit solvation models were minimal, indicating that the interaction of water with the N moieties results in similar effects on the C members of the aromatic ring (Fig. 6).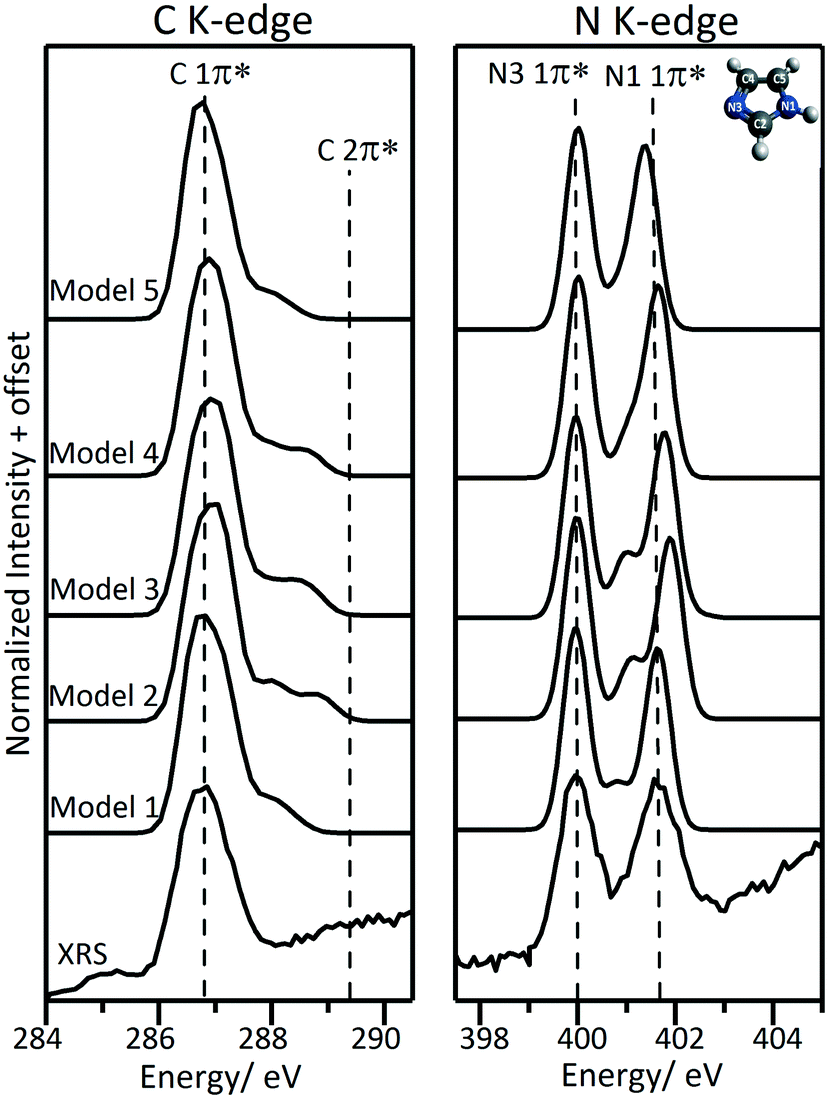 | ||
| Fig. 6 Calculated C K-edge and N K-edge XA spectra using the five solution structural models shown in Fig. 2. Note that the peak at ∼285.1 eV in the experimental C K-edge spectrum is from the Kapton window contribution. | ||
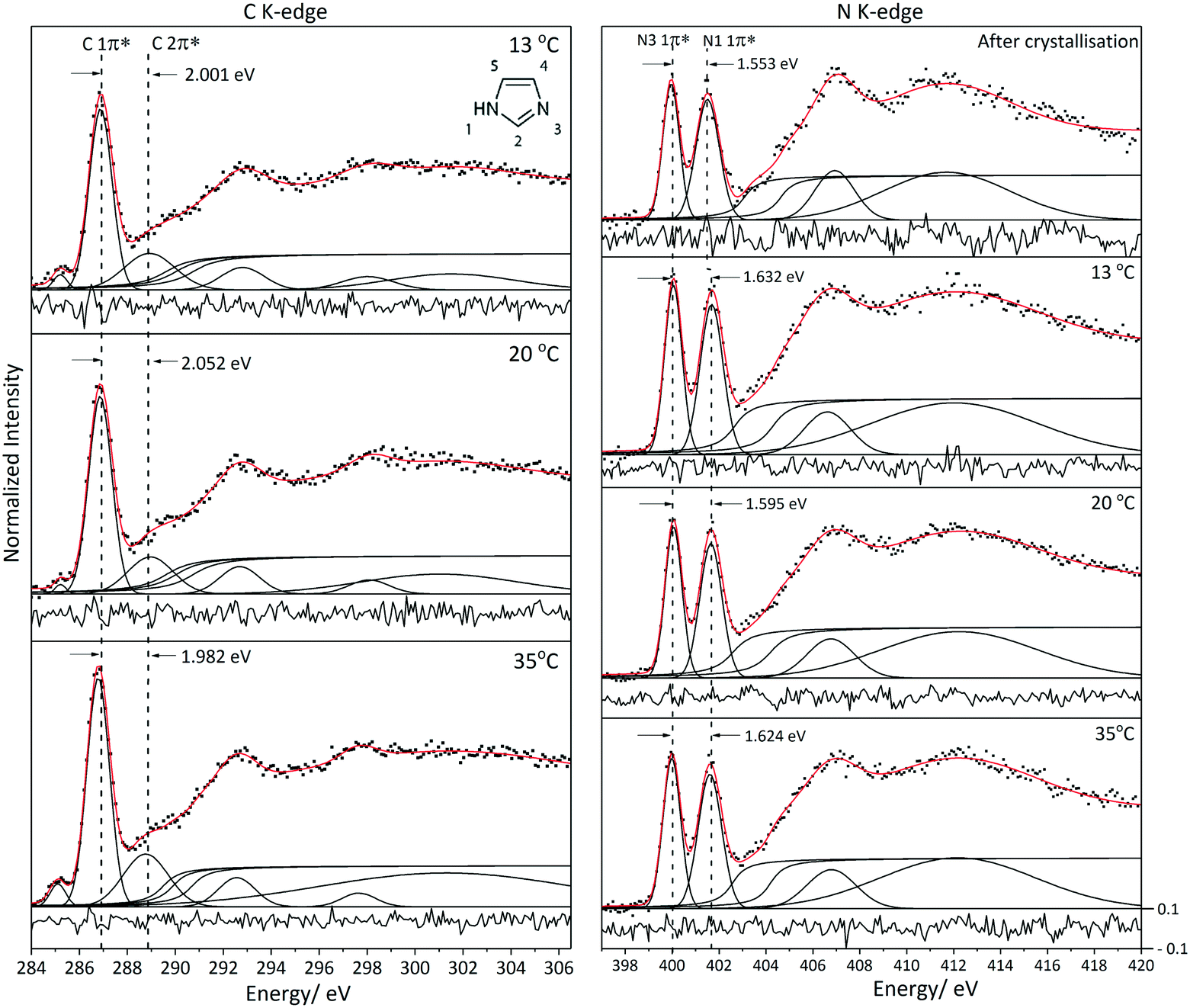 | ||
| Fig. 7 Fitted C K-edge and N K-edge XRS spectra of aqueous solution of imidazole during cooling crystallization. The residual is shown at the bottom of each spectrum. Peak positions and assignment for the C and N K-edge spectra are presented in Tables 3 and 1 respectively. Note that the peak at ∼285.1 eV in the solution C K-edge is from the Kapton window. | ||
| Structural models | ΔE (N1 1π*–N3 1π*)/eV | ΔE (N3 2π*–N3 1π*)/eV | ΔE (C2 2π*–C 1π*)/eV |
|---|---|---|---|
| XRS experimental spectrum (@ 35 °C) | 1.624 | — | 1.982 |
| Monomeric molecule (model for gas phase) | 2.214 | 1.160 | 2.324 |
| Model 1: implicit solvation model (CPCM) | 1.686 | 0.816 | 1.607 |
| Model 2: explicit solvation model (1 water molecules at the N3 side) | 1.917 | 1.113 | 2.145 |
| Model 3: explicit solvation model (1 water molecules at the N1 side) | 1.819 | 0.995 | 1.886 |
| Model 4: explicit solvation model (1 water molecules at the N3 side + 1 water molecule at the N1 side) | 1.654 | 1.060 | 2.031 |
| Model 5: combined solvation model (1 water molecules at the N3 side 1 water molecule at the N1 side + CPCM) | 1.379 | 0.866 | 1.578 |
The implicit solvation model accounts for the long range effects of the solvent, which was previously found necessary to determine the vertical and adiabatic ionization potentials of imidazole in aqueous solution.75 The implicit solvation model (model 1) gave some agreement with the experimental data; however, it shows an extra feature between the two main peaks at 400.826 eV, which is associated with the N3 1s → 2π* transition. This extra feature is only visible in the spectrum of the gas phase species (Fig. 3). The calculated energy differences between the N3 2π* and 1π* peaks are ∼1.160 eV for the monomeric molecule (analogous to gas phase system), ∼0.995–1.113 eV for the explicit solvation models (models 2–4, Table 4) and ∼0.816 eV for the implicit model (model 1). The transition from the N3 1s core to the 2π* valence orbital is shifted to lower energy in the implicit solvation model and is associated with the higher N3–C internal bond lengths when compared to the monomeric molecule and the explicit solvation models (Table 2). These changes in internal bond lengths seem to affect the C K-edge as well, with the second feature in the C K-edge shifted to lower energy compared with the experimental spectrum (Fig. 6).
A combined model where implicit solvation was used to account for the long range effects and explicit water molecules were added to account for local effects was also considered (model 5). In this case, the relative energies for the two 1π* peaks are underestimated compared with the experimental spectrum and, similarly to the implicit solvation model (model 1), the calculated transition from N3 1s → 2π* is shifted to lower energy. The N3 1s → 2π* transition was not resolved as an individual peak because the energy of the N1 1s → 1π* transition is underestimated and the two peaks overlap. The C K-edge spectrum calculated using model 5 is similar to that of the implicit solvation model (model 1).
Although the C and N K-edge spectra generated from model 4 are in good agreement with the experimental spectra, increasing the number of water molecules is expected to decrease the hydrogen bond length between water and imidazole molecules and consequently the energy split between the two 1π* peaks. This has been illustrated by previously reported micro-hydrated cluster models that were used to simulate NEXAFS spectra17 where the addition of water molecules lowers the energy differences between the two 1π* peaks and the two IPs. Moreover, it was concluded that explicit solvation to include the second coordination shell did not account for the long-range effects of the bulk solvent nor, naturally, for solute–solute interactions. Implicit solvation which accounts for long range effects of the solvent underestimates the transition to the 2π* valence orbitals.
In situ measurements in the metastable supersaturated zone
As described in the introduction, according to classical nucleation theory (CNT) the formation of crystal nuclei is believed to be preceded by self-association of solute molecules and formation of clusters with significant solute–solute interactions. Previous studies, as well as our results for the undersaturated concentrated solutions, have indicated that hydrogen bonding17,46 and π–π interactions9,45 are the dominant intermolecular interactions in aqueous imidazole solutions. TD-DFT calculations with our structural models show that the N K-edge spectra are highly sensitive to changes in hydrogen bonding at the N moieties of imidazole. Comparison of the C K-edge in the three phases (gas, solution and solid) reveals that the C K-edge is sensitive to intermolecular interactions between solute–solvent and solute–solute molecules.To capture the structural evolution of a supersaturated aqueous solution of imidazole, C and N K-edge XRS spectra were collected during cooling crystallization (Fig. 7). The spectra for the two edges were collected outside the metastable zone of a 10.1 M aqueous solution of imidazole (at 35 °C) and then in the metastable zone (at 20 °C and 13 °C). A subsequent N K-edge spectrum was significantly different from the spectra in the metastable zone, prompting visual inspection of the crystallisation vessel which indicated the presence of crystals. The photon energy difference between the two peaks in the C K-edge is ∼2.002 eV (±0.051) for the solution at different temperatures (35 °C, 20 °C and 13 °C). Similarly, the photon energy difference between the two 1π* peaks in the N K-edge spectra is ∼1.624 eV (±0.019) for the solution at different temperatures and ∼1.553 eV after crystal formation (Fig. 7). The variation in the spectral features of the C and N K-edge XRS spectra during cooling is minor and a change is observed in the N K-edge spectra once crystals have formed.
The minimal change observed in the C and N K-edge during cooling crystallization indicates that the average local coordination around the imidazole molecules does not change significantly during the cooling crystallization process. This outcome of our in situ measurements in the metastable zone is reminiscent of previous NEXAFS results for non-saturated aqueous solutions of imidazole, where minimal variation in the N K-edge spectra was detected as a function of concentration.17 They are also in line with a recent molecular dynamics simulation that suggested the solvation of the imidazole nitrogen moieties remained almost identical for different concentrations.46 This suggests that the concentration-dependent structure of aqueous imidazole can be described in terms of interactions between hydrated imidazole molecules rather than direct imidazole–imidazole interactions. By implication, desolvation of hydrated imidazole monomers appears to be the key step in nucleation of imidazole crystals. A recent simulation of NaCl nucleation from supersaturated solutions similarly suggested that the desolvation of the ions determines the attachment rate of monomers at the interface between the nuclei and the solution, rather than the diffusive flux of molecules as assumed by CNT.76
Moreover, CNT assumes that the internal structure of the critical nuclei is similar to that of the final crystals. It is interesting to note in this context, that the C and N K-edge spectra varied minimally during cooling crystallization and were distinct from those of the crystalline phase. It therefore appears that any structuring due to aggregation of monomers in the metastable zone prior to nucleation, if it exists, does not reflect the structure of the final crystalline product. This is in line with previous suggestions that the pre-nuclei differ drastically in terms of structure and composition from the bulk phase.5,8,77,78 Other “non-classical” nucleation mechanisms have been explored both theoretically and experimentally such as the two-step nucleation mechanism4 and pre-nucleation cluster mechanism.2 In two-step nucleation, formation of metastable dense liquid-like clusters is predicted to precede the nucleation event. Numerical simulation of homogenous solutions79 and DFT calculations80 showed that such liquid–liquid separation may occur near the metastable critical point. However, crystallization experiments did not provide evidence that this phase separation takes place.7 This indicates that the predicted phase separation is due to a liquid–liquid miscibility gap inside the metastable zone and should not be confused with nucleation mechanisms.1,7 Pre-nucleation clusters are thermodynamically stable associates in the solution, with a structure that may be similar or dissimilar to the final crystallized product.2 However, their presence has only been illustrated for inorganic systems.2
It is worthwhile to examine how realistic it is that pre-nucleation clusters can be detected by experimental techniques probing the overall composition of the nucleating solution. Arguably, the concentration of solute molecules in prenucleation clusters should be small in comparison with the mole fraction in the remaining bulk saturated solution. How realistic, therefore, is it that techniques such as XRS capture their presence? We can arrive at an estimate by considering that XRS was able to capture the changes in the N K-edge spectral features induced by nucleation. Let us, for the moment, make the assumption that the solution after crystal formation consists of imidazole crystals in a saturated imidazole solution (noting that this might be a simplification requiring further consideration, see below). The balance between solid state and solution state imidazole molecules is reflected by the photon energy difference between the two 1π* peaks in the N K-edge spectra. For the post-nucleation solution we determined an intermediate value between those for the crystalline phase and the undersaturated solution (at 35 °C) (Fig. 8). The observed decrease to 1.553 eV (Table 1, Fig. 8) is approximately 1/3 (∼36%) of the decrease expected if all imidazole transitioned into the crystalline form. This suggests that the imidazole molecules in the precipitated crystals were approximately 1/3 of all the imidazole molecules previously present in solution, with the remainder staying as solute molecules in a saturated solution. If nucleation of imidazole from aqueous solutions would pass through a prenucleation cluster stage it may involve up to 1/3 of the solute molecules. That no significant changes in the spectra were observed prior to the detection of crystals may therefore suggest that the fraction of solute molecules involved in cluster formation before nucleation is much lower than the fraction of molecules assembled in the crystallized product, or it may signify that imidazole–water binding remains the dominant structural motif upon cluster formation, with similar local solvation of imidazole in the undersaturated and in the supersaturated state. If prenucleation clusters are formed then imidazole–imidazole interactions do not appear to change the electronic structure of imidazole to the same extent as the hydrogen bonds to water molecules. Any self-association or cluster formation would thus involve interactions between hydrated imidazole molecules, which have not yet stripped off their inner solvation shell. In this case the N and C K-edges would not appear to be sensitive enough to detect evidence for cluster formation. The alternative explanation for our observations would be that only a very small fraction of imidazole molecules are involved in self-association and cluster formation, or perhaps that the average lifetime of clusters is so short as to result in an undetectable low concentration over time.
 | ||
| Fig. 8 The photon energy difference between the two 1π* peaks in the N K-edge spectra for solution during cooling crystallization (35 °C, 20 °C, 13 °C), after crystallization and for solid phase. | ||
It should be noted that these observations were made with solutions that contained imidazole concentrations around 10.1 mol L−1 prior to nucleation. The number of water molecules available per imidazole molecule in these solutions is significantly below four. Our results indicate an inner solvation shell of at least two hydrogen-bonded water molecules, which leaves only one or two free solvent molecules per imidazole for forming the liquid solution. This suggests that the solubility of imidazole is limited by the availability of solvent molecules. The fact that we do not observe any difference to undersaturated solutions prior to nucleation is remarkable, but also underlines the importance of local hydrogen bonding in this system.
However, returning to the point made above that assuming imidazole crystals in saturated imidazole solution may be an unrealistic simplification, the precipitated crystals that were detected after crystallization event may have a different structure and composition, for example representing unknown imidazole hydrates. This could also explain why the energy difference between the two 1π* peaks in the N K-edge spectra is at an intermediate value between that of the solid state and solution state. In this case the fraction of precipitated solid might well be higher than the estimate given above, because hydrogen bonds to water would be retained, and a lesser effect on the N K-edge would be expected than for non-hydrated imidazole. Unfortunately, examining other fitted peak parameters such as FWHMs (FWHM are provided in ESI† Table S2) does not allow us to shed further light on this issue. For example, from the N K-edge spectra of the solid and the solution in Fig. 3 one may expect slight broadening of the peak arising from the N1 nitrogen, due to an energy difference of 0.2 eV between the solid and the solution. However, the expected broadening effect would be within less than 0.1 eV, which we cannot reliably resolve with the quality of the available spectra. Additional studies will be needed to clarify these points further. Complementary work with other techniques sensitive to short-range structure, and with much faster data acquisition times (e.g. X-ray pair distribution function, XPDF, measurements) may be helpful. It may then be possible to shed more light on hydrogen and proton positions using excited state density functional theory (DFT) calculations.
Conclusions
X-ray Raman scattering (XRS) of the C and N K-edge spectra was used to follow the structural evolution of aqueous imidazole in situ during cooling crystallization. A jacketed batch crystallizer with internal flow enabled precise thermal control of the system and minimized any effects of radiation damage on the spectra. Comparison of the C and N K-edge XRS spectra of imidazole in solution and in the solid phase illustrated the sensitivity of the near-edges to changes in the local bonding of the imidazole molecules. DFT calculations confirmed the sensitivity of the C K-edge and N K-edge to imidazole–water and imidazole–imidazole hydrogen bonding. Only minimal changes were observed in the C and N K-edge spectra during cooling until an abrupt change in the N K-edge spectra indicated the onset of crystallization. The observed relative shift of the π* resonances indicated that approximately 1/3 of dissolved imidazole molecules had formed crystalline product. This also determines that the upper limit for molecules involved in prenucleation clustering in aqueous imidazole is approximately 1/3 of all solute molecules. There was no evidence for changes due to prenucleation clustering in the C and N K-edge spectra, indicating that molecules with a significantly changed inner solvation shell are at most a small mole fraction. This suggests that the nucleation of imidazole takes place from a highly concentrated solution which is dominated by local hydrogen bonding to water in the inner solvation shell around the imidazole molecules. It follows that the breakage of these solute–solvent hydrogen bonds during desolvation is likely to be the key step in the nucleation of imidazole crystals from aqueous phase.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research used resources of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory and was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357, and the Canadian Light Source and its funding partners. This research has been performed with the use of laboratory facilities at the Research Complex at Harwell, and with financial support from the Future Continuous Manufacturing and Advanced Crystallization (CMAC) Hub (EPSRC Grant EP/P006965/1). The authors would also like to acknowledge the usage of ARC3 computing facility at the University of Leeds. The authors thank Mrs Abigail Mortimer, University of York for providing the glass blowing service. LHA gratefully acknowledges the University of Leeds and Diamond Light Source for the PhD studentship. SLMS would like to thank Royal Academy of Engineering, Diamond and Infineum UK for support of the Bragg Centenary Chair. SYC is grateful to Infineum UK, AstraZeneca, Diamond Light Source for financial support. All data supporting this study are provided either in the results section of this paper or in the ESI† accompanying it.Notes and references
- R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166–2179 CrossRef CAS PubMed.
- D. Gebauer and H. Cölfen, Nano Today, 2011, 6, 564–584 CrossRef CAS.
- J. Anwar and D. Zahn, Angew. Chem., Int. Ed., 2011, 50, 1996–2013 CrossRef CAS PubMed.
- P. G. Vekilov, Cryst. Growth Des., 2010, 10, 5007–5019 CrossRef CAS PubMed.
- R. A. Chiarella, A. L. Gillon, R. C. Burton, R. J. Davey, G. Sadiq, A. Auffret, M. Cioffi and C. A. Hunter, Faraday Discuss., 2007, 136, 179–193 RSC.
- A. Spitaleri, C. A. Hunter, J. F. McCabe, M. J. Packer and S. L. Cockroft, CrystEngComm, 2004, 6, 489–493 RSC.
- P. E. Bonnett, K. J. Carpenter, S. Dawson and R. J. Davey, Chem. Commun., 2003, 698–699, 10.1039/b212062c.
- R. J. Davey, G. Dent, R. K. Mughal and S. Parveen, Cryst. Growth Des., 2006, 6, 1788–1796 CrossRef CAS.
- F. Peral and E. Gallego, J. Mol. Struct., 1997, 415, 187–196 CrossRef CAS.
- H. G. Alison, R. J. Davey, J. Garside, M. J. Quayle, G. J. T. Tiddy, D. T. Clarke and G. R. Jones, Phys. Chem. Chem. Phys., 2003, 5, 4998–5000 RSC.
- J. L. Finney and A. K. Soper, Chem. Soc. Rev., 1994, 23, 1–10 RSC.
- C. A. Koh, R. P. Wisbey, X. P. Wu, R. E. Westacott and A. K. Soper, J. Chem. Phys., 2000, 113, 6390–6397 CrossRef CAS.
- S. G. Booth, S. Y. Chang, A. Uehara, C. La Fontaine, G. Cibin, S. L. M. Schroeder and R. A. W. Dryfe, Electrochim. Acta, 2017, 235, 251–261 CrossRef CAS.
- S. Y. Chang, Y. Gründer, S. G. Booth, L. B. Molleta, A. Uehara, J. F. W. Mosselmans, G. Cibin, V. T. Pham, L. Nataf, R. A. W. Dryfe and S. L. M. Schroeder, CrystEngComm, 2016, 18, 674–682 RSC.
- S. Y. Chang, S. G. Booth, A. Uehara, J. F. W. Mosselmans, G. Cibin, V. T. Pham, L. Nataf, R. A. W. Dryfe and S. L. M. Schroeder, J. Phys.: Conf. Ser., 2016, 712, 012058 CrossRef.
- J. S. Stevens, A. Gainar, C. Jaye, D. A. Fischer and S. L. M. Schroeder, J. Phys.: Conf. Ser., 2016, 712, 012133 CrossRef.
- M. J. Thomason, C. R. Seabourne, B. M. Sattelle, G. A. Hembury, J. S. Stevens, A. J. Scott, E. F. Aziz and S. L. M. Schroeder, Faraday Discuss., 2015, 179, 269–289 RSC.
- J. S. Stevens, A. Gainar, E. Suljoti, J. Xiao, R. Golnak, E. F. Aziz and S. L. M. Schroeder, Chem. – Eur. J., 2015, 21, 7256–7263 CrossRef CAS PubMed.
- R. Golnak, J. Xiao, K. Atak, J. S. Stevens, A. Gainar, S. L. M. Schroeder and E. F. Aziz, Phys. Chem. Chem. Phys., 2015, 17, 29000–29006 RSC.
- J. S. Stevens, L. K. Newton, C. Jaye, C. A. Muryn, D. A. Fischer and S. L. M. Schroeder, Cryst. Growth Des., 2015, 15, 1776–1783 CrossRef CAS.
- F. Meyer, M. Blum, A. Benkert, D. Hauschild, Y. L. Jeyachandran, R. G. Wilks, W. Yang, M. Bar, F. Reinert, C. Heske, M. Zharnikov and L. Weinhardt, Phys. Chem. Chem. Phys., 2018, 20, 8302–8310 RSC.
- M. Faubel, S. Schlemmer and J. P. Toennies, Z. Phys. D: At., Mol. Clusters, 1988, 10, 269–277 CrossRef CAS.
- B. Winter and M. Faubel, Chem. Rev., 2006, 106, 1176–1211 CrossRef CAS PubMed.
- K. R. Wilson, B. S. Rude, T. Catalano, R. D. Schaller, J. G. Tobin, D. T. Co and R. J. Saykally, J. Phys. Chem. B, 2001, 105, 3346–3349 CrossRef CAS.
- K. R. Wilson, B. S. Rude, J. Smith, C. Cappa, D. T. Co, R. D. Schaller, M. Larsson, T. Catalano and R. J. Saykally, Rev. Sci. Instrum., 2004, 75, 725–736 CrossRef CAS.
- L. A. Naslund, J. Luning, Y. Ufuktepe, H. Ogasawara, P. Wernet, U. Bergmann, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. B, 2005, 109, 13835–13839 CrossRef PubMed.
- L. A. Naslund, D. C. Edwards, P. Wernet, U. Bergmann, H. Ogasawara, L. G. M. Pettersson, S. Myneni and A. Nilsson, J. Phys. Chem. A, 2005, 109, 5995–6002 CrossRef PubMed.
- U. Bergmann, P. Wernet, P. Glatzel, M. Cavalleri, L. G. M. Pettersson, A. Nilsson and S. P. Cramer, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 092107 CrossRef.
- J. S. Tse, D. M. Shaw, D. D. Klug, S. Patchkovskii, G. Vanko, G. Monaco and M. Krisch, Phys. Rev. Lett., 2008, 100, 095502 CrossRef PubMed.
- U. Bergmann, D. Nordlund, P. Wernet, M. Odelius, L. G. M. Pettersson and A. Nilsson, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024202 CrossRef.
- C. J. Sahle, M. A. Schroer, I. Juurinen and J. Niskanen, Phys. Chem. Chem. Phys., 2016, 18, 16518–16526 RSC.
- U. Domańska, M. K. Kozłowska and M. Rogalski, J. Chem. Eng. Data, 2002, 47, 456–466 CrossRef.
- G. Balakrishnan, A. A. Jarzecki, Q. Wu, P. M. Kozlowski, D. J. Wang and T. G. Spiro, J. Phys. Chem. B, 2012, 116, 9387–9395 CrossRef CAS PubMed.
- N. Tsud, R. G. Acres, M. Iakhnenko, D. Mazur, K. C. Prince and V. Matolin, J. Phys. Chem. B, 2013, 117, 9182–9193 CrossRef CAS PubMed.
- V. Feyer, O. Plekan, N. Tsud, V. Chab, V. Matolin and K. C. Prince, Langmuir, 2010, 26, 8606–8613 CrossRef CAS PubMed.
- V. Feyer, O. Plekan, T. Skala, V. Chab, V. Matolin and K. C. Prince, J. Phys. Chem. B, 2008, 112, 13655–13660 CrossRef CAS PubMed.
- L. De Luca, Curr. Med. Chem., 2006, 13, 1–23 CAS.
- A. R. Katritzky and A. F. Pozharskii, Handbook of heterocyclic chemistry, Pergamon Press, Amsterdam; Lausanne, New York, 2000 Search PubMed.
- G. Zhao and H. Lin, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 137–147 CrossRef CAS PubMed.
- J. Mao, H. Abushammala, H. Hettegger, T. Rosenau and M.-P. Laborie, Polymer, 2017, 9, 473 Search PubMed.
- T. R. Chen, A. C. Yeh and J. D. Chen, Tetrahedron Lett., 2005, 46, 1569–1571 CrossRef CAS.
- G. Will, Z. Kristallogr., Kristallgeom., Kristallphys., Kristallchem., 1969, 129, 211–221 CrossRef CAS.
- S. Martinez-Carrera, Acta Crystallogr., 1966, 20, 783–789 CrossRef CAS.
- B. M. Craven, R. K. McMullan, J. D. Bell and H. C. Freeman, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 2585–2589 CrossRef.
- L. Gontrani, R. Caminiti, L. Bencivenni and C. Sadun, Chem. Phys. Lett., 1999, 301, 131–137 CrossRef CAS.
- S. Y. Liem, M. S. Shaik and P. L. A. Popelier, J. Phys. Chem. B, 2011, 115, 11389–11398 CrossRef CAS PubMed.
- M. S. Shaik, S. Y. Liem, Y. N. Yuan and P. L. A. Popelier, Phys. Chem. Chem. Phys., 2010, 12, 15040–15055 RSC.
- C. J. Sahle, C. Henriquet, M. A. Schroer, I. Juurinen, J. Niskanen and M. Krisch, J. Synchrotron Radiat., 2015, 22, 1555–1558 CrossRef PubMed.
- I. Juurinen, T. Pylkkanen, K. O. Ruotsalainen, C. J. Sahle, G. Monaco, K. Hamalainen, S. Huotari and M. Hakala, J. Phys. Chem. B, 2013, 117, 16506–16511 CrossRef CAS PubMed.
- T. Pylkkanen, J. Lehtola, M. Hakala, A. Sakko, C. Monaco, S. Huotari and K. Hamalainen, J. Phys. Chem. B, 2010, 114, 13076–13083 CrossRef CAS PubMed.
- H. N. Chapman, P. Fromme, A. Barty, T. A. White, R. A. Kirian, A. Aquila, M. S. Hunter, J. Schulz, D. P. DePonte, U. Weierstall, R. B. Doak, F. R. N. C. Maia, A. V. Martin, I. Schlichting, L. Lomb, N. Coppola, R. L. Shoeman, S. W. Epp, R. Hartmann, D. Rolles, A. Rudenko, L. Foucar, N. Kimmel, G. Weidenspointner, P. Holl, M. N. Liang, M. Barthelmess, C. Caleman, S. Boutet, M. J. Bogan, J. Krzywinski, C. Bostedt, S. Bajt, L. Gumprecht, B. Rudek, B. Erk, C. Schmidt, A. Homke, C. Reich, D. Pietschner, L. Struder, G. Hauser, H. Gorke, J. Ullrich, S. Herrmann, G. Schaller, F. Schopper, H. Soltau, K. U. Kuhnel, M. Messerschmidt, J. D. Bozek, S. P. Hau-Riege, M. Frank, C. Y. Hampton, R. G. Sierra, D. Starodub, G. J. Williams, J. Hajdu, N. Timneanu, M. M. Seibert, J. Andreasson, A. Rocker, O. Jonsson, M. Svenda, S. Stern, K. Nass, R. Andritschke, C. D. Schroter, F. Krasniqi, M. Bott, K. E. Schmidt, X. Y. Wang, I. Grotjohann, J. M. Holton, T. R. M. Barends, R. Neutze, S. Marchesini, R. Fromme, S. Schorb, D. Rupp, M. Adolph, T. Gorkhover, I. Andersson, H. Hirsemann, G. Potdevin, H. Graafsma, B. Nilsson and J. C. H. Spence, Nature, 2011, 470, U73–U81 CrossRef PubMed.
- W. Schülke, Electron Dynamics by Inelastic X-Ray Scattering, Oxford Univ. Press, Oxford, 2007 edn, 2007 Search PubMed.
- T. T. Fister, G. T. Seidler, L. Wharton, A. R. Battle, T. B. Ellis, J. O. Cross, A. T. Macrander, W. T. Elam, T. A. Tyson and Q. Qian, Rev. Sci. Instrum., 2006, 77, 063901 CrossRef.
- G. T. Seidler, T. T. Fister, J. O. Cross and K. P. Nagle, AIP Conf. Proc., 2007, 882, 911–913 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- J. J. Zheng, X. F. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Aust. J. Chem., 2012, 4, 17 CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS.
- R. van Leeuwen, O. V. Gritsenko and E. J. Baerends, in Density Functional Theory I: Functionals and Effective Potentials, ed. R. F. Nalewajski, Springer Berlin Heidelberg, Berlin, Heidelberg, 1996, pp. 107–167, DOI:10.1007/3-540-61091-x_4.
- E. Apen, A. P. Hitchcock and J. L. Gland, J. Phys. Chem., 1993, 97, 6859–6866 CrossRef CAS.
- J. Stöhr, NEXAFS spectroscopy, Springer, Berlin; New York, 1st edn, 1996 Search PubMed.
- F. Sette, J. Stohr and A. P. Hitchcock, J. Chem. Phys., 1984, 81, 4906–4914 CrossRef CAS.
- J. S. Stevens, A. Gainar, E. Suljoti, J. Xiao, R. Golnak, E. F. Aziz and S. L. M. Schroeder, J. Phys.: Conf. Ser., 2016, 712 Search PubMed.
- A. Gainar, J. S. Stevens, C. Jaye, D. A. Fischer and S. L. M. Schroeder, J. Phys. Chem. B, 2015, 119, 14373–14381 CrossRef CAS PubMed.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- D. Nolting, N. Ottosson, M. Faubel, I. V. Hertel and B. Winter, J. Am. Chem. Soc., 2008, 130, 8150–8151 CrossRef CAS PubMed.
- L. Soderholm, G. K. Liu, M. R. Antonio and F. W. Lytle, J. Chem. Phys., 1998, 109, 6745–6752 CrossRef CAS.
- J. S. Stevens, S. J. Byard, C. A. Muryn and S. L. M. Schroeder, J. Phys. Chem. B, 2010, 114, 13961–13969 CrossRef CAS PubMed.
- J. S. Stevens, A. C. de Luca, M. Pelendritis, G. Terenghi, S. Downes and S. L. M. Schroeder, Surf. Interface Anal., 2013, 45, 1238–1246 CrossRef CAS.
- B. Jagoda-Cwiklik, P. Slavicek, L. Cwiklik, D. Nolting, B. Winter and P. Jungwirth, J. Phys. Chem. A, 2008, 112, 3499–3505 CrossRef CAS PubMed.
- N. E. R. Zimmermann, B. Vorselaars, D. Quigley and B. Peters, J. Am. Chem. Soc., 2015, 137, 13352–13361 CrossRef CAS PubMed.
- D. W. Oxtoby, Acc. Chem. Res., 1998, 31, 91–97 CrossRef CAS.
- R. C. Burton, E. S. Ferrari, R. J. Davey, J. L. Finney and D. T. Bowron, J. Phys. Chem. B, 2010, 114, 8807–8816 CrossRef CAS PubMed.
- P. R. tenWolde and D. Frenkel, Science, 1997, 277, 1975–1978 CrossRef CAS.
- V. Talanquer and D. W. Oxtoby, J. Chem. Phys., 1998, 109, 223–227 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ce00929e |
| This journal is © The Royal Society of Chemistry 2018 |