
How similar are amorphous calcium carbonate and calcium phosphate? A comparative study of amorphous phase formation conditions†
I.
Buljan Meić
a,
J.
Kontrec
a,
D.
Domazet Jurašin
 b,
A.
Selmani
b,
B.
Njegić Džakula
a,
N.
Maltar-Strmečki
b,
D. M.
Lyons
c,
M.
Plodinec
d,
M.
Čeh
e,
A.
Gajović
d,
M. Dutour
Sikirić
*b and
D.
Kralj
*a
b,
A.
Selmani
b,
B.
Njegić Džakula
a,
N.
Maltar-Strmečki
b,
D. M.
Lyons
c,
M.
Plodinec
d,
M.
Čeh
e,
A.
Gajović
d,
M. Dutour
Sikirić
*b and
D.
Kralj
*a
aDivision of Materials Chemistry, Ruđer Bošković Institute, Bijenička c. 54, 10 000 Zagreb, Croatia. E-mail: kralj@irb.hr; Fax: +385 1 468 0098; Tel: +385 1 468 0207
bDivision of Physical Chemistry, Ruđer Bošković Institute, Bijenička c. 54, 10 000 Zagreb, Croatia. E-mail: sikiric@irb.hr; Fax: +385 1 468 0245; Tel: +385 1 456 0941
cCenter for Marine Research, Ruđer Bošković Institute, Giordano Paliaga 5, 52210 Rovinj, Croatia
dDivision of Materials Physics, Ruđer Bošković Institute, Bijenička c. 54, 10 000 Zagreb, Croatia
eDepartment of Nanostructured Materials, Jožef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia
First published on 28th November 2017
Abstract
Amorphous calcium carbonate (ACC) and calcium phosphate (ACP) increasingly attract attention as initial solid phases in vertebrate and invertebrate hard tissue formation, as well as in materials science as a possible new synthetic route for advanced materials preparation. Although much is known about these two amorphous phases and similarities in the mechanisms of their formation are recognized, no attempt has been made to investigate their formation under defined and comparable initial experimental conditions, viz. supersaturation, the ratio of constituent ions, ionic strength and the presence of relevant inorganic additives. In this paper, the formation of ACC and ACP in three model precipitation systems of increased chemical complexity was investigated: (a) systems containing constituent ions, (b) systems containing additional co-ions, and (c) systems with higher ionic strength and addition of Mg2+. The results have shown that ACP is more stable and was formed at lower relative supersaturations in comparison to ACC. The precipitation domain of both phases expanded with increasing complexity of precipitation systems, with the ACP precipitation domains always being larger than those of ACC. In addition to stability, the presence of inorganic ions, especially Mg2+, influences the composition of both amorphous phases. The obtained results indicate that general similarity between ACC and ACP exists, but it could also be concluded that the similar chemical environment in which they form not necessarily leads to similar structural properties.
1. Introduction
For a number of years, amorphous solid phases have attracted attention due to their frequent occurrence in biological systems as well as special functions they perform and/or mechanisms of their formation which open new synthetic pathways for advanced functional materials.1,2 Amorphous biominerals are of special interest considering that many of them have an important role in the formation of hard tissues whose properties still cannot be matched by man-made materials. It is estimated that among known biominerals approximately one fifth are amorphous (amorphous being defined as a material with no discernible X-ray diffraction pattern).1 Usually, they serve as temporary storage granules or they are molded into complex structures.1–3 The most abundantly available amorphous biominerals are amorphous calcium carbonate (ACC) and amorphous calcium phosphate (ACP). Although they are found in different organisms, ACC in invertebrates and ACP in vertebrates, they perform similar roles. Moreover, recent investigations of non-classical pathways of mineralization pointed to the similarity in mechanisms of their formation in vitro.4Amorphous calcium carbonate (ACC), biogenic or synthetic, is an important (meta)stable phase or transient precursor to crystalline calcium carbonate (CaCO3) minerals.5 Depending on the preparation method or its biogenic source, ACC varies in composition, stability and structure.5,6 Biogenic ACC has been identified in different organisms not only as an unstable precursor to crystalline calcite7,8 and aragonite9 but also as a stable structural component persistent throughout the life of the organism.10 Organisms use organic macromolecules, magnesium, phosphate and structural water to induce ACC formation and to control its stability.11–13 Water content plays a major role in the biogenic and synthetic structure of ACC and according to the literature it appears in different amounts, from 0.0–1.58 mol H2O per mol CaCO3, and structural forms, i.e. mobile or structural H2O.14–17 Depending on water content, two different forms of ACC, anhydrous (transient) and hydrous (stable), are classified in biogenic systems.5,7 ACC formed by the synthetic route typically crystallizes very rapidly in aqueous solution and its lifetime is associated with hydrate content since it transforms into a stable polymorph by a dissolution–(re)precipitation process parallel to loss of all water content.17 Advanced characterization methods enabled some new findings regarding the ACC structure, specifically the degree of distinct short-range ordering. In biogenic ACC, this ordering depends on genetic control and macromolecules which are present,5,18 while ACC prepared in the laboratory under certain formation conditions points as well to short-range protostructures similar to crystalline CaCO3 polymorphs, so called proto-vaterite, proto-calcite19 and proto-aragonite.20 However, when ACC is precipitated from a highly supersaturated solution and without additives, no protostructure is detected.14,17
Amorphous calcium phosphate (ACP) is the first solid phase formed during precipitation of calcium phosphate solid phases (CaPs) from basic and neutral solutions.21,22 In the 1960s, Posner proposed the spherical cluster Ca9(PO4)6, later named Posner's cluster, as ACP's basic structural unit.23 Although the existence of Posner's clusters was accepted, only recent development of experimental techniques (cryogenic transmission electron microscopy, atomic force microscopy and dynamic light scattering) enabled the revelation of their formation under different experimental conditions.24–27 However, it should be noted that the similarity of the sizes of prenucleation clusters (PNCs) observed in different systems and Posner's clusters should not be taken as proof of their chemical and structural identity.26 A large number of Posner's clusters are randomly close packed in spherical structures 30–100 nm in diameter, which in turn aggregate into chain-like aggregates.28,29In vitro, when ACP is in contact with the mother liquor, it readily transforms into more stable crystalline phases, namely octacalcium phosphate [OCP, Ca8(HPO4)2(PO4)4·5H2O], calcium hydrogen phosphate dihydrate (DCPD, CaHPO4·2H2O), calcium-deficient apatite [CaDHA, Ca10−x(HPO4)x(PO4)6−x(OH)2−x, 0 < x < 1] and/or hydroxyapatite [HAP, Ca10(PO4)6(OH)2].29–32 It has been believed for a long time that ACP has a similar precursor role in organisms. However, it was only after Olszta and Gower's study33 has shown that, in vitro, amorphous liquid-phase mineral precursors facilitate intrafibrillar mineralization of type-I collagen that the search for ACP in vivo has started. Mahamid et al.34 demonstrated the existence of ACP in newly formed zebrafish fin bones, which crystallizes during maturation of the bone. Akiva et al.35 investigated mineral deposition in the caudal fin bone of larval zebrafish and showed that mineral particles located between the bones and at some distance, both in close association with blood vessels, consist of ACP and OCP-like phases. In addition, enamel formation also proceeds through ACP which transforms into apatitic crystals.36 In trabecular bone, 100–300 nm ACP regions exist in its disordered phase.37 ACP was found not only in vertebrates, but also in small amounts in certain invertebrates' tissues, such as lobster cuticle, which mainly consists of calcite and ACC.38
The stability of both ACP and ACC still remains one of the key questions for understanding biomineralization processes and the application of biomimetics in materials science. Different ions (e.g. magnesium),39–42 small molecules (e.g. citrate)43 and macromolecules44,45 are known to stabilize them, but the detailed mechanism of their action is still not known. Despite frequent ACP and ACC comparison in the literature and attempts to draw parallels in their properties and behavior,46 no attempts have been made to prepare both phases under comparable initial experimental conditions, i.e. supersaturation, the ratio of constituent ions, ionic strength and the presence of relevant inorganic additives, and compare the conditions of their formation. Our study is an additional attempt in this direction in which, to the best of our knowledge, for the first time precipitation of amorphous calcium carbonate and calcium phosphate has been investigated under experimental conditions which are as similar as possible.
In our previous work, the properties of stable and metastable CaCO3 and CaP solid phases, formed under comparable initial experimental conditions after one hour aging time in systems of various complexities, were compared.47 The existence of metastable calcium carbonate and calcium phosphate phases in much broader reactant concentration domains than physiological ones and the fact that their stabilization could be additionally achieved by rather simple mechanisms prompted us to extend this strategy to the comparison of their amorphous phases. Formation of ACC and ACP was investigated in three precipitation systems of different chemical complexities; (a) a system containing constituent ions, (b) a system containing additional co-ions and (c) a system with higher ionic strength and addition of Mg2+ which simulates the inorganic chemical environment in living organisms.47 The precipitation domains of both phases in the investigated precipitation systems were determined, as well as the influence of the imposed chemical complexity of the system on the properties of the obtained amorphous precipitate.
2. Experimental
2.1. Materials and solution preparation
Analytical grade chemicals, calcium hydroxide (Ca(OH)2), calcium chloride (CaCl2), carbonic acid (H2CO3), phosphoric acid (H3PO4), sodium carbonate (Na2CO3), sodium hydrogen phosphate (Na2HPO4), sodium chloride (NaCl) and magnesium chloride (MgCl2), all obtained from Sigma Aldrich, Germany, and Milli-Q water (Millipore) were used in all experiments.Stock solutions were prepared by using chemicals which had been dried overnight in a desiccator over silica gel or by diluting concentrated acids to the required concentration. The calcium hydroxide stock solution was prepared by adding an excess of calcium hydroxide into water. The suspension was then filtered through a 0.22 μm membrane filter and the saturated solution was kept under nitrogen. The exact concentration was determined by potentiometric titration using a standard HCl solution (c = 0.10 mol dm−3) and the total calcium content was additionally determined by ion chromatography (model ICS 1100). The carbonic acid stock solutions were prepared by bubbling a high-grade carbon dioxide stream into water until the apparent constancy of the measured pH was obtained. The exact concentrations of freshly prepared stock solutions were determined by potentiometric titration using a standard NaOH solution (c = 0.10 mol dm−3).
2.2. Precipitation systems and characterization
ACP and ACC formation was investigated in systems of different chemical complexities as used in our previous study:47• simple (only constituent ions):
| Ca(OH)2 – H2CO3 and Ca(OH)2 – H3PO4 |
• complex (constituent ions and corresponding inert counterions):
| CaCl2 – Na2CO3 and CaCl2 – Na2HPO4 |
• physiological (constituent ions with addition of sodium chloride and magnesium ions):
CaCl2 – Na2CO3 – 0.15 M NaCl – MgCl2, Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mg = 1:2 and :Mg = 1:2 and |
| CaCl2 – Na2HPO4 – 0.15 M NaCl – MgCl2, Ca:Mg = 1:2. |
In each of the precipitation systems, CaPs and CaCO3 precipitation was investigated within a wide range of initial reactant concentrations:
| 1.0 × 10−5 mol dm−3 < ci(Ca)tot; ci(CO3)tot or ci(PO4)tot < 1.0 × 10−2 mol dm−3. |
The concentration range is broader than concentration domains that correspond to physiological systems (plasma, artificial saliva, extrapallial fluid) or seawater. The highest value of the concentrations was determined by the solubility of Ca(OH)2 and CO2. However, in both physiological systems, the concentrations were extended to 4.0 × 10−2 mol dm−3, in order to be able to compare the relative supersaturations to those obtained in the simple systems. Detailed information about the experimental procedure is presented in our previous work.47 In short, in all cases, precipitation was initiated by pouring carbonate or phosphate solution into an equal volume of calcium or calcium/magnesium solution. During precipitation, the systems were magnetically stirred. All experiments were performed at 25 °C, and the initial pH was not adjusted in any precipitation system. The samples were isolated by filtering the entire volume of the suspension through a 0.22 μm membrane filter. Since the volumes were relatively small with respect to the filtration funnel, the separation process was typically completed after few seconds. Considering the susceptibility of ACC to transformation, ACC samples were filtered immediately after mixing in CaCO3 systems, while in the case of CaPs, samples were isolated after 10 min reaction time. ACP samples were washed with ethanol, dried overnight under vacuum and ACC samples were dried using lyophilization. The dried ACP samples were kept at 4 °C and ACC samples at −18 °C. All these procedures were performed in order to prevent their possible transformation into the metastable and stable polymorphs.
The composition and structure of the obtained precipitates were determined by FT-IR spectroscopy (FTIR spectrometer equipped with an attenuated total reflection module, Tensor II, Bruker), powder X-ray diffraction (Rigaku Ultima IV diffractometer in Bragg–Brentano geometry using CuKα radiation), thermogravimetric measurement (Mettler TG 50 thermobalance with a TC 10 TA processor, 10 K min−1), DSC (model Perkin-Elmer Pyris Diamond) and electron paramagnetic resonance spectroscopy (EPR, Varian E-109 X-band spectrometer equipped with a Bruker ER 041 XG microwave bridge and a Bruker ER 4111 VT variable-temperature unit with a flow of N2 gas). For EPR analysis, samples were irradiated, in the presence of air, using a 60Co gamma ray source of the Ruđer Bošković Institute, at a dose rate from 9.28 Gy s−1 up to a cumulative dose of 25 kGy. Routine dosimetry was performed with 5 mm-diameter alanine dosimeters (Bruker Instruments, Germany) using a Varian E-109 spectrometer equipped with a Bruker ER 041 XG microwave bridge for readout. The actual doses were within 2% of the target dose. A manganese standard reference, Mn2+ in MnO, was used to calibrate the magnetic field of the EPR spectrometer. The morphology of the obtained precipitates was observed by FE-SEM (JEOL JSM-7000F microscope) and transmission electron microscopy (TEM, Zeiss TEM 902A microscope and JEOL JEM-2100, 80 keV). The chemical composition was determined by ion chromatography (model ICS 1100).
| S-1(ACC) = [c(Ca2+)·c(CO32−)·γ22)/KspACC0]1/2 − 1 | (1) |
| S-1(ACP) = [c(Ca2+)3·c(PO43−)2·γ23γ32)/KspACP0]1/5 − 1 | (2) |
The EPR spectra were simulated with a custom-built program in MATLAB (The MathWorks Inc., Natick, Massachusetts, USA) using the EasySpin program package.49
3. Results and discussion
3.1. Precipitation domains of ACC and ACP formation
It is considered that the main parameters which cause the nucleation of an amorphous phase in a precipitation process are the initial high supersaturation and large unit cell of the crystalline form. Specifically, in the case of calcium phosphates and carbonates, highly hydrated calcium and possibly hydrogen carbonate/phosphate ions may contribute to amorphous phase formation.50 The composition of precipitates formed after 1 hour (ref. 47) prompted us to determine the precipitation diagrams of ACP and ACC for each of the investigated systems. It is assumed that the construction of precipitation diagrams, using initial reactant concentrations, yields a range of basic information about the system in question and therefore represents a first step for more complex investigations. The precipitation diagrams of amorphous phases within the investigated concentration regions are shown in Fig. 1. The lines denote solubility boundaries (blue for ACC and grey for ACP) while shaded areas denote ACP and ACC precipitation domains. | ||
| Fig. 1 Precipitation diagrams of amorphous calcium carbonate (ACC) and amorphous calcium phosphate (ACP): simple (a), complex (b) and physiological (c) systems obtained immediately after commencement of reaction for ACC and after 10 min aging time for ACP at 25 °C. DCPD – calcium hydrogen phosphate dihydrate. | ||
Although it seems that, according to the known lower solubility of ACP, the respective S-1 curve is positioned incorrectly (at higher values of total concentrations of reactants), it should be considered that the relative supersaturations are defined as the nth root of the quotient of activities of constituent ions and thermodynamic solubility product (eqn (1) and (2)). Therefore, diagrams in which the position of S-1 curves are shown as a function of concentrations/activities of constituent ions, more intuitively reflect the difference in solubility of calcium and carbonate amorphous phases, i.e. lower solubility of ACP (please see Fig. SI 2†). The shapes of ACC and ACP solubility boundaries in the simple system are markedly different from those in the complex and physiological precipitation systems. It was shown before that the asymmetry indicates the formation of specific ion pairs51 or complexes in solution or increased solubility of solid phases at higher concentrations of acids. On the other hand, the apparently symmetrical shape of solubility boundaries with respect to the equivalence line in the complex and physiological systems reveals the absence of any significant ion pairing or complex formation.
The precipitation domains of ACC and ACP are markedly different in all investigated precipitation systems. In the simple system, ACC precipitation occurred in a very narrow domain of calcium and carbonate concentrations (0.007 mol dm−3 ≤ c(H2CO3) and c(Ca(OH)2) ≤ 0.01 mol dm−3, 20 < (S-1)calcite < 33, initial pH range: 9.5–11.7). The ACC precipitation domain in the complex system is broader than that in the simple system including higher concentrations of CaCl2 and Na2CO3 (18 < (S-1)calcite < 33, initial pH range: 10.4–10.9). As expected,3,41 addition of magnesium ions into the physiological precipitation system significantly influenced the ACC precipitation region, which is broader than the domains obtained in the other two systems. In addition, it was found that in solution with high initial concentrations of calcium and carbonate (initial supersaturation, 16 < (S-1)calcite < 33, initial pH range: 10.3–10.6), amorphous precursors precede the formation of crystalline metastable polymorphs, monohydrocalcite and aragonite.47 Additionally, in a certain concentration region (0.007 < c(Ca2+)/mol dm−3 < 0.023 and 0.015 < c(CO32−)/mol dm−3 < 0.023) and at c(CO32−) ≥ c(Ca2+), ACC remains stable even after 60 min of reaction (initial pH = 10.2–10.8). During the precipitation process in this system, minimal or even no pH change has been observed.
The ACP precipitation domain significantly increases with the increasing complexity of precipitation systems as well and extends to much lower supersaturations than those for ACC. In the simple system, ACP forms in very narrow reactant concentration region (5 < (S-1) < 6) in which the initial pH varied from 7.9–12.0, while in the complex system, the precipitation domain is significantly larger (1 < (S-1) < 2.6) and corresponds to an initial pH range of 7.0–8.4. In this system, ACP was not detected only at the highest investigated reactant concentrations. Instead, the formation of a DCPD and CaDHA mixture observed after 60 min (ref. 47) indicated that ACP could be the first step in precipitation. This was confirmed by the shape of the pH curves in which, before the steep drop in pH, a region of slower pH change was observed. Although the precipitate was analyzed after 5 minutes, a mixture of DCPD and CaDHA (Fig. SI 3†) was also obtained.
In the physiological system, the ACP formation domain extends over the entire investigated reactant concentration region (0.4 < (S-1) < 4.0, initial pH = 5.4–8.2) in which a precipitate was detected. Furthermore, at the highest reactant concentrations, ACP was obtained in a mixture with DCPD. The stabilizing effect of Mg2+ ions is well known. Moreover, Ding et al. recently showed that adsorbed Mg2+ ions are more efficient in inhibiting the ACP transformation than incorporated ones.40 Interestingly, no influence of Mg2+ on DCPD formation has been observed.39 Unlike simple and complex systems, in the physiological system, ACP was detected over the entire precipitation domain even after 60 min of ageing.47
The obtained results indicated that ACP forms over a broader reactant concentration and initial pH region and at lower reactant concentrations than ACC, as well as that ACP is more stable than ACC when in contact with solution.
3.2. Composition, structure and morphology of ACC and ACP in systems of different complexities
A number of studies have shown that the composition, structure and morphology of both ACP and ACC can considerably vary depending on the formation conditions.3,5,13,17,31 It was even proposed that their structure can be similar to the basic structure of different calcium phosphate and calcium carbonate phases into which they transform.2,48,52,53 In the case of ACP, two different phases were detected, differing in their solubility and morphology (usually noted as ACP1 and ACP2).31 Synthetic ACC classification depends on the preparation method and variation in thermal stability, water content, particle size and morphology, as well as in short-range order.16,20,48,54In order to determine the influence of the system complexity, the representative precipitates formed in different precipitation systems were subjected to detailed physico-chemical analysis.
Simple system. In this system, very high turbidity in parallel with a simultaneous drop in pH (Fig. SI 5a†) was noted, indicating the formation of a precursor amorphous phase in the system with very high initial supersaturation. The mineralogical composition of precursors, i.e. particles precipitated immediately after mixing of reactants, was primarily identified by FT-IR and XRD (Fig. 2a). The FTIR spectra of the initial phase (ACCsimple) were characterized by a broad band between 2800 and 3600 cm−1 (O–H stretching) and a sharp band at 1641 cm−1 (O–H bending), both of which correspond to structural water within ACC.41 In addition, ACC was also identified by the presence of a doublet on the main asymmetric ν3 CO3 band (at 1485 and 1434 cm−1). Besides the presence of water and ν3 doublet, atomic disorder of the amorphous CaCO3 is generally proven by the apparent absence of the ν4 symmetric vibration at 713 cm−1 for calcite and 745 cm−1 for vaterite.4
 | ||
| Fig. 2 FTIR spectra (a and f), PXRD patterns (b and g), SEM micrographs (c1, c2 and h), TEM micrographs (d1 and i1), selected area electron diffraction (SAED; d2* and i2) and TGA results (e and j) with DSC curves (insets*) of representative amorphous calcium carbonate precipitates formed in simple (c(Ca(OH)2) = c(H2CO3) = 0.01 mol dm−3, a–e, ACCsimple) and complex (c(CaCl2) = c(Na2CO3) = 0.01 mol dm−3, f–j, ACCcomplex) precipitation systems, isolated at the beginning of reactions, at 25 °C. The red arrow in (c2) points to calcite. *The enlarged SAED pattern in (d2) and DSC insets in (e) and (j) are properly presented in ESI† Fig. SI 4 and SI 7. | ||
However, detailed analysis of spectroscopic bands in ACCsimple revealed that crystallization had already started. The bands at 872 cm−1 (ν2) and 710 cm−1 (ν4) belong to a crystalline polymorph, while out-of-plane bending for ACC is observed at about 862–868 cm−1 and ν4 at about 700 cm−1.55 The FTIR spectrum of a stable precipitate, formed after 60 min of precipitation in the same system, is characteristic for the mixed phase, containing calcite and vaterite, as presented in ref. 47.
The powder X-ray diffraction (PXRD) pattern supported the results of IR analysis and Fig. 2b confirms that the initial precipitate consisted of a poorly ordered material, with two broad amorphous maxima at approximately 30° and 45° 2θ which are characteristic of ACC, while the crystalline polymorphs were not detected.
In contrast, the SEM analysis revealed that recrystallization had already started, showing spherical amorphous particles of about 100 nm, together with few larger calcite crystals (Fig. 2c1 and c2 and SI 7†). The product obtained after one hour contained both regular rhombohedral calcite and cauliflower-like vaterite.47 The TEM micrograph (Fig. 2d1) revealed irregular elongated particles of about 30–100 nm which are, according to the SAED pattern (Fig. 2d2 and SI 4†), partially crystallized and belong to the dominant calcite phase with the presence of a small amount of the vaterite phase.
Complex system. Similar to the simple precipitation system, high turbidity and a simultaneous drop in pH were noted (Fig. SI 5b†).47 The precipitate isolated immediately after the mixing of reactants is ACC as identified by FT-IR and XRD (Fig. 2f and g). Detailed FTIR spectral analysis revealed that as with the simple model system, crystallization of CaCO3 polymorphs had just started, as confirmed by the ν2 band at 873 cm−1 and ν4 bands at 711 and 744 cm−1, while the ACC phase was confirmed by the presence of water and a ν3 doublet at 1479 and 1423 cm−1.
SEM and TEM analyses (Fig. 2h, i1 and i2) revealed that samples formed in the complex system consisted of regular spherical ACC particles of about 50–100 nm and SAED patterns showed no presence of crystalline domains. This obtained amorphous precursor also transforms into a mixture of stable calcite and metastable vaterite,47 as shown previously.
FTIR/ATR analyses confirmed the presence of water components in amorphous samples in the simple and complex model systems, but gave no insight into the structure and amount of hydrated components, therefore thermogravimetric (TG) and differential scanning calorimetry (DSC) analyses were performed. It was found that in the simple and complex systems, initial dehydration started at temperatures of about 46 °C and 81 °C (Fig. 2e and j). The literature data suggest that in this stage of dehydration, loss of surface-bound water is the first step of transformation.12 The second loss of mass for ACCsimple as well as for ACCcomplex, recorded in thermogravimetric curves at about 170–185 °C, corresponds to loss of structural water and points to the crystallization of a stable polymorph (Fig. 2e and j). It was estimated that the water content in ACCsimple as well as in ACCcomplex was about 17% and the molar ratio of water to calcium carbonate was about 1.1. FTIR/ATR analyses showed that the solid phase formed after thermal transformations of samples is calcite, while DSC analysis of the same samples showed an exothermic peak at 182 °C (simple system) and at 162 °C (complex system), which is similar to results observed by Tobler et al.56 Thus, they showed that ACC prepared in the precipitation system in which NaOH was not added transformed into calcite at 162 °C, while the addition of NaOH caused the increase of the transformation temperature. Similar behaviour was observed by Schmidt et al.,15 who concluded that the dehydration and recrystallization behaviour of ACC strongly correlates with the preparation conditions. According to the presented results, we can conclude that the precursors formed in the simple and complex systems are similar in composition and structure. They are transient phases and therefore very unstable in solution, as well as after thermal treatment.3,15
Physiological system. It was shown previously that in highly supersaturated (S-1calcite > 30) Na2CO3–CaCl2–MgCl2–NaCl systems,47 metastable monohydrocalcite and aragonite precipitated, while at a specific pH and Ca2+/CO32− ratio and at lower supersaturations (16 < (S-1)calcite < 20), amorphous particles were stable even after 60 min of reaction.
According to abundant literature data considering biogenic or synthetic ACC formation in the presence of magnesium, i.e. Mg-ACC,57–59 it was expected that Mg2+ ions will exert a major influence on stabilization, thermogravimetric parameters and the structure of amorphous phases formed in physiological systems. Indeed, the precursors formed in a highly supersaturated, Mg-bearing environment (Mg/Ca = 2) showed longer stability, when in contact with solution, than those formed in the Mg-free environment of the other two systems.
Different ACC samples formed in physiological systems have been classified with respect to solid phases into which they transformed after 60 minutes of ageing in solution.47 Thus, ACCphysI transforms into aragonite, ACCphysII into monohydrocalcite (MHC), while ACCphysIII remains amorphous. The amount of amorphous phases precipitated in the physiological system is much larger in comparison to the respective simple or complex concentration domain, which is certainly a consequence of their stabilization caused by the presence of Mg2+.
Unlike ACCphysI and ACCphysII, particles formed at medium supersaturation and c(Na2CO3) > c(CaCl2) remain amorphous even after 60 min of reaction,47 and correspond to ACCphysIII. Since the amount of sample at the beginning of the reaction was low, the amorphous particles aged for 1 hour were collected and analyzed. Consequently, information about possible differences between the properties of unstable and stable amorphous calcium carbonate could be obtained.
Fig. 3 shows the results of detailed characterization of samples formed in the physiological system. According to FTIR/ATR analyses, ACCphysI and ACCphysII are completely amorphous and differ just slightly in the out-of-plane bending, ν2 band. PXRD patterns (Fig. 3b and g) confirmed the amorphous structure, but also pointed to the presence of NaCl (peaks around 30 and 45°) which is to be expected since the samples were not properly washed in order to prevent possible transformation into a crystalline phase. In the case of ACCphysIII, which was stable even after 60 min and thoroughly washed, the PXRD pattern revealed a purely amorphous structure without any trace of NaCl (Fig. 3l). The FTIR spectra of precipitates formed after 60 min of ACC transformation showed bands characteristic for monohydrocalcite and aragonite.47
 | ||
| Fig. 3 FTIR spectra (a, f and k), PXRD patterns (b, g and l), SEM micrographs (c, h and m), TEM micrographs including selected area electron diffraction (SAED) (d, i and n) and TGA results (e, j and o) with DSC curves (insets*) of representative amorphous calcium carbonate precipitates formed in physiological (ACCphysI (c(CaCl2) = c(Na2CO3) = 0.023 mol dm−3, c(MgCl2) = 0.046 mol dm−3, a–e), ACCphysII (c(CaCl2) = 0.015 mol dm−3, c(Na2CO3) = 0.023 mol dm−3, c(MgCl2) = 0.046 mol dm−3, f–j) and ACCphysIII (c(CaCl2) = c(Na2CO3) = 0.01 mol dm−3, c(MgCl2) = 0.046 mol dm−3, k–o)) precipitation systems, isolated at the beginning of reactions, at 25 °C. *Enlarged TGA results and DSC insets in (e), (j) and (o) are properly presented in ESI† Fig. SI 8. | ||
The SEM micrograph of the ACCphysIII sample (Fig. 3m) showed spherical particles with smooth surfaces that are larger (around 250–400 nm) and less agglomerated than those in the case of ACCphysI and ACCphysII (Fig. 3c and h). TEM micrographs and the corresponding SAED patterns, presented in Fig. 3d, i and n, also confirmed the amorphous structure of the particles formed in the physiological system (Fig. 3c, h and m).
In comparison to ACCsimple and ACCcomplex, thermal transformation of the respective samples formed in physiological systems was gradual and finished at much higher temperatures. The peaks at 46, 81 and 97 °C for ACCphysI and at 46, 81 and 109 °C for ACCphysII (Fig. 3e and j) pointed to a major loss of water content of about 15–16% (adsorbed surface water and interior pore water). The FTIR analysis of solids obtained after thermal transformation at 200 °C confirmed the existence of ACC and the presence of some remaining water. However, after heating the samples to about 500 °C, an additional mass loss of approximately 2.5% (structural water) was detected and the resulting solid phase was found to be calcite. It was estimated that the water content in ACCphysI as well as in ACCphysII was about 18.5% and the molar ratio of water to calcium carbonate was about 1.2.
DSC analyses (Fig. 3e and j and SI 8†) of ACCphysI, as well as of ACCphysII, showed an exothermic peak corresponding to crystallization of calcite, which was shifted to 378 °C (ACCsimple, θ = 182 °C and ACCcomplex, θ = 162 °C). For ACCphysIII, θ = 214 °C is lower. A likely explanation for such unexpected thermal properties may be a long aging time (t = 60 min). However, data from FTIR/ATR (Fig. 3k) as well as TG analyses (Fig. 3o) showed that the molar ratio of water to calcium carbonate was lower and is about 0.9. In order to additionally explore the TG and DSC data of ACCphysIII, in the case of what appeared to be the most stable precursor, the ACCphysIII sample, the aging time was prolonged to 70 min of reaction, before transformation into aragonite was observed. These results could indicate that this sample already started to dehydrate after 60 min of reaction, followed by structural reorganization and formation of aragonite by the 70th minute. We suggest that these results are in agreement with the dissolution–(re)precipitation process of ACC transformation in solution proposed in the literature.14,17
The Mg content of amorphous samples formed under different initial conditions in the physiological system was determined using ion chromatography. It was found that ACCphysI and ACCphysII contain about 21 mol% magnesium, while ACCphysIII contains a lower amount of magnesium compared to the other two less stable samples (about 14 mol%). Unlike ACCphysI and ACCphysII which probably contain some traces of magnesium originating from solution (no washing, frozen samples), ACCphysIII was properly washed before freezing. Consequently, the magnesium content is lower.
In summary, the addition of magnesium ions into precipitation systems caused the formation of ACC phases that are thermally more stable, while their stability in solution was increased as well in comparison to respective non-Mg stabilized ACC phases. The explanation for such behaviour may be related to structural differences and high dehydration energy of magnesium ions, which, when incorporated within the structure of ACC, prevent the loss of water molecules that is a necessary step during recrystallization.57 Another likely explanation is related to shortening of the Mg–O bond which caused the stabilization of ACC.60 On the other hand, the presence of Mg2+ in solution may stabilize ACC by inhibiting the nucleation and/or growth of more stable CaCO3 modification into which ACC transforms during the solution-mediated process. A similar explanation has been proposed for preferential formation of aragonite in seawater, which is even more supersaturated with respect to calcite. In Mg-rich environments, calcite nucleation is inhibited by preferential adsorption of Mg2+ onto calcite-like nuclei that are in competition with aragonite. However, the preferential adsorption on calcite is explained by its structure being highly similar to the structure of magnesite, MgCO3, which is also a slightly soluble salt.
Simple system. The FTIR spectrum (Fig. 4a) of the precipitate obtained after 10 minutes in the simple system (ACPsimple) contained phosphate bands (asymmetric stretching mode of PO43− at 1048 cm−1, HPO42− band at 871 cm−1, and bending mode of PO43− at 555 cm−1) and water bands (broad band at 3700–2600 cm−1 and band at 1654 cm−1) characteristic for amorphous calcium phosphate.53 The absence of splitting of phosphate bands at about 1100 and 600 cm−1 further confirmed the formation of ACP.2,53 However, the PXRD peaks at 2θ 25.88° and 31.84°, characteristic for CaDHA,2 indicated that under the given conditions the transformation of ACP had already started (Fig. 4b). The TEM micrograph showed that the precipitate consists of aggregates of small spherical particles, with an average size of 17 nm, characteristic of ACP (Fig. 4c).28,29,61 SAED showed that some particles were amorphous while others were crystalline, thus confirming the PXRD results and pointing to the tendency for transformation (Fig. 4d). Our previous study has confirmed that after 60 minutes, in this system, ACP transformation into CaDHA was complete.47
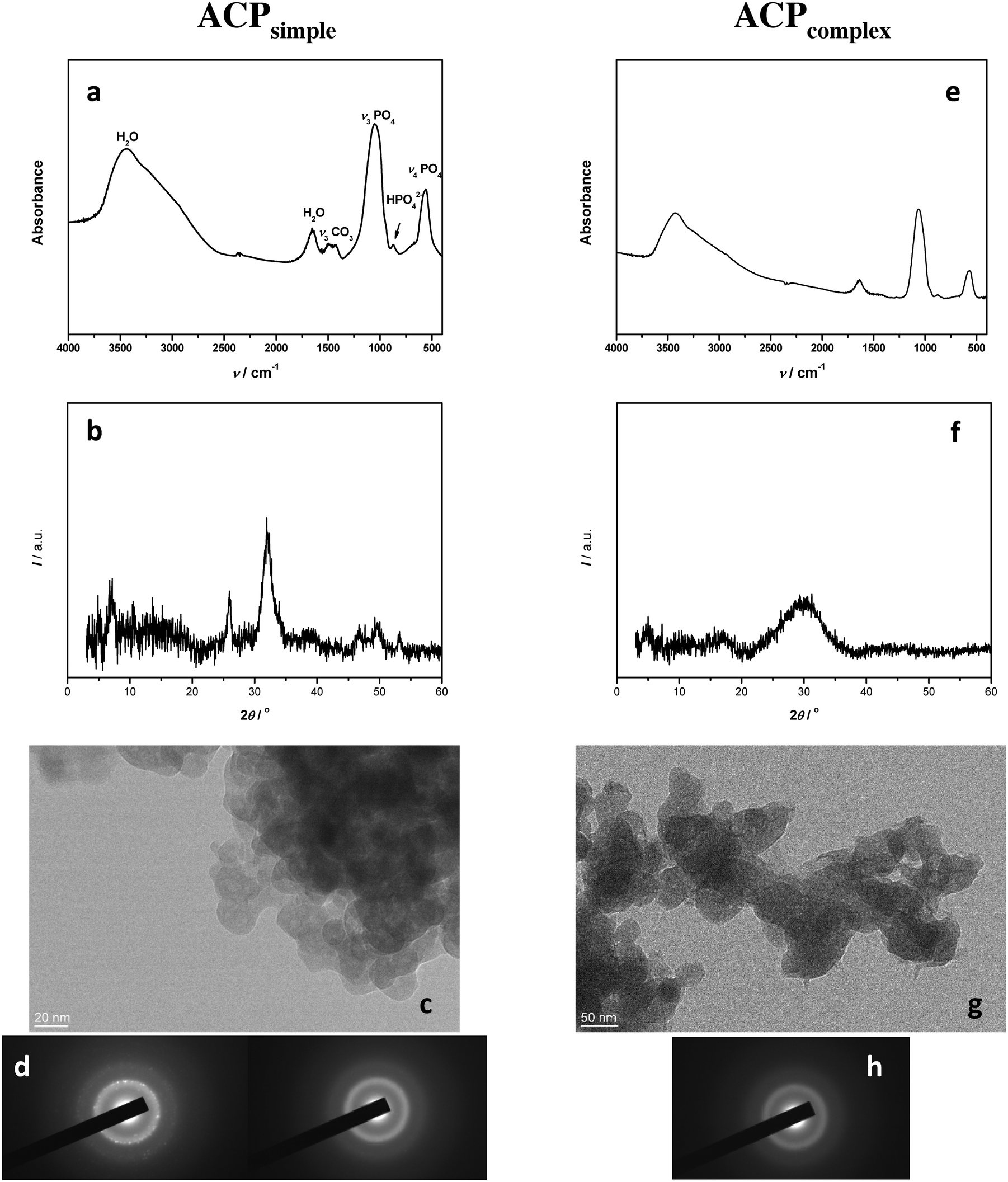 | ||
| Fig. 4 FTIR spectra (a and e), PXRD patterns (b and f), TEM micrographs (c and g) and selected area electron diffraction (SAED, d, h) of representative amorphous calcium phosphate precipitates formed in simple (c(Ca(OH)2) = 0.003 mol dm−3, c(H3PO4) = 0.002 mol dm−3, a–d, ACPsimple) and complex (c(CaCl2) = 0.005 mol dm−3, c(Na2HPO4) = 0.002 mol dm−3, e–h, ACPcomplex) precipitation systems after 10 minutes aging time at 25 °C. | ||
Complex system. The FTIR spectrum and PXRD pattern of the precipitate obtained after 10 minutes in the complex system (Fig. 4e and f, ACPcomplex) confirmed the formation of an amorphous precipitate. No splitting of bands at about 1100 and 600 cm−1 was observed and the PXRD pattern showed only a broad amorphous maximum in the 26–34° 2θ region characteristic for ACP.2 The TEM micrograph (Fig. 4g) showed that ACPcomplex also precipitated in the form of chain-like aggregates of spherical particles. However, the spherical particles in the complex system were larger, with an average size 51 nm, than the ones obtained under comparable conditions in the simple system and somewhat more elliptic, indicating the influence of the neutral electrolyte on the ACP properties. The formation of ACP was confirmed with SAED (Fig. 4h), indicating a slower rate of ACP transformation in the complex system. Similar to the simple system, after 60 minutes, ACP transformed into CaDHA.47
Physiological system. Unlike in the simple and complex systems, in the physiological system after 10 min reaction time, ACP was detected both at medium (ACPphysI) and at high supersaturations (ACPphysII). In both cases, the FTIR spectra (Fig. 5a and e) and PXRD patterns (Fig. 5b and f) were characteristic of ACP.2,53 However, TEM micrographs (Fig. 5c, g and i) revealed the differences between these two samples. Similar to the simple and complex precipitation systems, ACPphysI precipitated in the form of chain-like aggregates of spherical particles with an average size of 31 nm. In contrast, TEM micrographs and SAED (Fig. 5g–j and SI 10†) revealed that at higher supersaturations (ACPphysII), a small amount of large, though not well developed, thin plate-like crystals appeared together with ACP. Since they were not detected by FTIR and PXRD, it can be concluded that less than 5% of such crystalline phases existed in the mixture. It was shown previously that after 60 minutes in this concentration region, a mixture of DCPD and ACP precipitated,47 so the observed large crystals are most probably DCPD in the early stage of formation.
 | ||
| Fig. 5 FTIR spectra (a and e), PXRD patterns (b and f), TEM micrographs (c, g, i), selected area electron diffraction (SAED, d, h, j) of representative amorphous calcium phosphate precipitates formed in physiological precipitation system at medium supersaturation (c(CaCl2) = 0.005 mol dm−3, c(Na2HPO4) = 0.002 mol dm−3, c(MgCl2) = 0.01 mol dm−3, a–d, ACPphysI) and high supersaturation (c(CaCl2) = c(Na2HPO4) = 0.01 mol dm−3, c(MgCl2) = 0.02 mol dm−3, e–j, ACPphysII) after 10 minutes aging time at 25 °C. | ||
Differences between ACP precipitates obtained in different precipitation systems were also reflected in their thermal behaviour (Fig. 6). For ACP samples formed in the simple and physiological systems, the largest loss of mass occurs in the temperature range from room temperature to about 135 °C, followed by an almost continuous loss of mass until 650 °C. Similar behaviour was observed by Kanazawa et al.62 Thus, the first mass loss may be ascribed to the loosely bound water molecules adsorbed on the surface of ACP particles, while the second one is usually ascribed to more strongly bound inter-cluster water molecules.2,53,63,64 Both mass losses were larger for ACP formed in the simple system, totalling to the mass loss of 19.15% for ACPsimple and 15.67% for ACPcomplex.
 | ||
| Fig. 6 TGA curves of ACP precipitates formed in simple (ACPsimple), complex (ACPcomplex) and physiological systems (ACPphysI and ACPphysII) after 10 minutes aging time at 25 °C. | ||
Indeed, somewhat different behaviour was observed for ACP formed in the physiological system. Both ACPphysI and ACPphysII contained larger amounts of water than ACPsimple and ACPcomplex (i.e. w = 28.29% and w = 30.02%, respectively). The first loss of mass was also larger than that for ACP formed in the simple and complex systems. However, unlike the ACPsimple and ACPcomplex systems, the first mass loss was not followed by a continuous mass loss. A discontinuity in the rate of loss (marked with the arrow) in the temperature range 150–350 °C for ACPphysI and 150–415 °C for ACPphysII was detected. It was shown that such discontinuity in TGA curves depends on solution composition and becomes more expressed with lowering of the Ca/P ratio.64
The results of structural analyses indicate somewhat different arrangement of ions and composition of precipitates obtained in the respective precipitation systems. The values of the Ca/P ratio determined for different samples confirm this observation. Thus, the Ca/P ratio decreased with increasing chemical complexity of the precipitation system: Ca/P ratios of 1.64, 1.59, 1.55 and 1.21 were obtained for ACPsimple, ACPcomplex, ACPphysI and ACPphysII, respectively. The values are consistent with literature data which indicated that ACP with a Ca/P ratio varying from 1.0–2.2 can be formed.2,53 The markedly different Ca/P ratio of ACPphysII can be a result of the presence of DCPD (Ca/P ratio = 1) in the mixture, as well as the fact that at lower pH, HPO42− ions can be incorporated into the structure instead of PO43−, thus resulting in a lower Ca/P ratio.53
In order to further verify the difference in ion arrangement, EPR analysis was performed. Fig. 7 and SI 11† show the experimentally obtained EPR spectra and corresponding simulations of γ-irradiated ACPsimple, ACPcomplex, ACPphysI and ACPphysII.
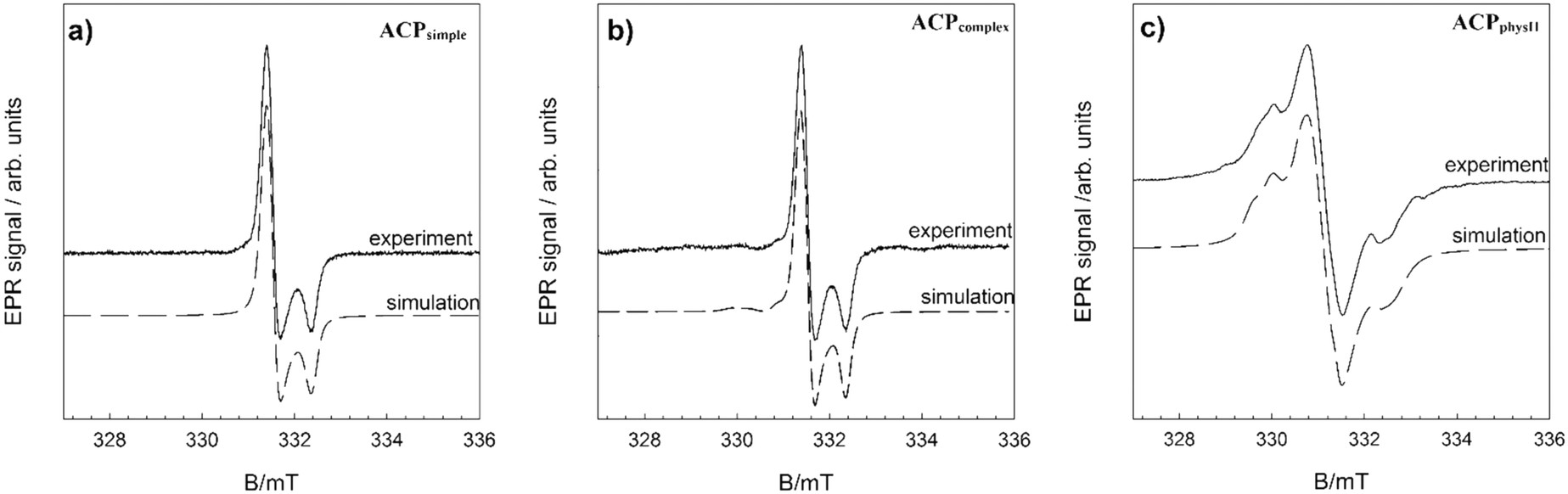 | ||
| Fig. 7 EPR spectra and corresponding simulations of γ-irradiated ACP precipitate formed in a) simple (ACPsimple), b) complex (ACPcomplex) and c) physiological systems (ACPphysII) after 10 minutes aging time at 25 °C. | ||
As expected, the spectra of γ-irradiated ACPs obtained in different precipitation systems vary substantially, in accordance with previous studies.65 The EPR spectrum (Fig. 7a) of the precipitate obtained after 10 minutes in the simple system (ACPsimple) represents the contribution of CO2− radicals with orthorhombic symmetry characteristic for A-type carbonate apatite samples cooled in a CO2 flow.65 In the EPR spectrum of γ-irradiated ACPcomplex, shown in Fig. 7b, signals from species with orthorhombic symmetry are present, with the main contribution from CO2− and the minor presence of CO3− and CO− radicals, typical for a disordered (amorphous) structural state.66 Samples ACPphysI and ACPphysII exhibit complex EPR spectra, similar in terms of spectral features, but with differences in signal intensity (Fig. SI 11†). Specifically, the EPR signal intensity, i.e. the concentration of radicals detected in ACPphysI, is approximately 6 times smaller than that in ACPphysII. Differences in radical concentration for such amorphous samples could be explained by the location of radicals in the lattice and differences in the short-range order arrangement of atoms of these samples. The absence of crystalline structure in ACPphysI leads to less stable radicals as compared to the ACPphysII sample in which, in addition to ACP, a small amount of crystalline DCPD has been detected (Fig. 5e–j). This leads to lower intensity due to the rapid recombination of the primarily unstable radicals formed upon γ-irradiation.67 The identified species and the spin Hamiltonian parameters obtained from simulation of the EPR spectra are listed in Table 1.65–67
| Sample | Radicals | g x | g y | g z | Weight/% |
|---|---|---|---|---|---|
| ACPsimple | CO2−orthorombic | 2.002![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) |
1.997 |
2.002![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) |
100.00 |
| ACPcomplex | CO2−orthorombic | 2.003 |
1.997![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) |
2.001![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) |
92.56 |
| CO− | 2.006 |
2.003![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) |
2.001 |
3.43 | |
| CO3− | 2.006![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) |
2.008 |
2.011 |
4.01 | |
| ACPphysII | CO2−orthorombic | 2.002 |
1.996 |
2.001 |
16.25 |
| CO2−axial | 2.001![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) |
2.001 |
1.993 |
25.96 | |
| CO3− | 2.004 |
2.007![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) |
2.011 |
14.26 | |
| CO− | 2.006 |
2.003 |
2.001 |
40.78 | |
| CO33− | 2.004![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) |
2.003 |
2.001![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) |
2.75 |
The existence of different types or concentrations of carbonate radicals, incorporated in the structure of various ACP samples, points to the difference in their short-range order.
It has long been known that the difference in the reactant concentrations, their ratio, and pH and the presence of inorganic ions will result in formation of ACCs and ACPs of different composition.2,5,6,53 Both ACC and ACP contain a significant amount of water, which has an important role in formation and stabilization.2,53,69 In ACC, water appears in different structural forms, i.e. mobile or structural H2O.14,15 In ACP, water is either located in inter-cluster space or more loosely bound on the surface of ACP associates. It is assumed that water present in inter-cluster space enables association of Posner's clusters into spherical particles.2,53 However, no direct relationship between the water content and stability of either amorphous phases has been established.2,53,69
We were also not able to prove such a relationship. The water content of precipitated amorphous phases did change due to increased chemical complexity of the precipitation systems. And again, this effect was much more expressed for ACP. ACCsimple and ACCcomplex contained somewhat less water than ACCphysI and ACCphysII. They also showed dehydration and crystallization behaviour different from the more gradual water loss continuing to higher crystallization temperature observed in ACCphysI and ACCphysII samples (Fig. 2e, j and 4e, j). Similar differences in water content and structure between ACP precipitates obtained in the simple and complex precipitation systems, on the one side, and physiological system, on the other side, were obtained (Fig. 6). However, while traces of crystalline phases were observed in both ACCsimple and ACCcomplex, they were detected only in ACPsimple, which, according to TGA, has higher water content than ACPcomplex (Fig. 6).
The change in ACP composition was further confirmed by the decrease of the Ca/P ratio (from 1.64 to 1.55 and 1.21) with increasing chemical complexity of the systems. It is known that depending on the reactant concentration and ratio, as well as pH, ACP's Ca/P ratio can vary from 1.0–2.2.2,53 A large range of Ca/P ratios found in ACP was taken as proof of the possibility that amorphous phases of different CaPs can exist.2,53 This line of thinking was also motivated by Gebauer's study of ACCs in which he has shown that ACCs with different short-range order, resembling the short-range order of calcite or vaterite, exist. This led to the introduction of protocrystalline structuring notion, i.e. protocalcite and protovaterite.19 Such a notion is rarely used in the calcium phosphate literature.2 The reason could lie in the fact that OCP and CaDHA, metastable phases through which ACP transforms into HA, contain parts of the structure similar to that of HA.
The EPR spectra of amorphous calcium phosphate phases obtained in different precipitation systems indicate that they have different short-range order (Fig. 7 and Table 1). Interestingly, regardless of this difference, both ACPsimple and ACPcomplex transformed after 60 minutes into CaDHA.47 Moreover, CaDHA's Ca/P ratio in both cases was 1.5, the average size of particles was similar (9 and 10 nm, respectively) and only a slight difference in morphology was observed.47 This implicates that, at least in the case of calcium phosphates, amorphous phases with different short-range order could transform into the same crystalline form. In contrast, ACCsimple and ACCcomplex both transform into a vaterite and calcite mix, while amorphous particles formed in the physiological system transform into aragonite and MHC or remain amorphous like in the case of ACCphysIII. Unlike in the case of ACP, we can't make any certain conclusion about the short-range order of the investigated ACC samples, relying only on the information gained from the chosen characterization methods. Also, it should be noted that ACCs synthesized without additives and at higher supersaturation, like in the case of ACCsimple and ACCcomplex, usually do not show any order that is related to crystalline polymorphs14,70 and we can only assume that differences in their composition probably influence their solution and thermal stability, as well as transformation path.
In addition, the morphology and size of both ACC and ACP particles also varied to different extents with changing the complexity of the precipitation system. ACCsimple differs in morphology compared to samples precipitated in complex and physiological systems, containing irregular elongated particles (probably as a consequence of initial crystallization) as seen in Fig. 2c and d. Also, amorphous particles formed in the ACCphysIII system are evidently larger than those formed in lower complexity model systems. Unlike ACC, the morphology of ACPcomplex is the one which differs from those of ACPsimple and ACPphys (Fig. 3 and 4). In the complex system, the largest, somewhat elliptical particles were obtained, contrary to smaller, spherical particles obtained in the simple and physiological systems.
The obtained results indicate that a general similarity between ACC and ACP exists, but it could also be concluded that the similar chemical environment in which they form not necessarily leads to similar structural properties.
Conclusion
Amorphous calcium carbonate and calcium phosphate phases have been precipitated under similar conditions, in three chemically different environments, in order to compare the relevant experimental parameters responsible for their properties.The results of kinetic, structural, morphological and chemical analyses indicated that the precipitation domains of ACC and ACP significantly differ. Thus, ACP precipitated at lower reactant concentrations in comparison to ACC. In addition, both ACC and ACP precipitation domains extend in the presence of simple reactant co-ions (Na+, Cl−), the effect being more evident for ACP. The presence of Mg2+ ions stabilized both amorphous phases that could be detected even after 1 hour of aging in solution.
The morphology and size of both ACC and ACP particles vary to different extents with changing the complexity of the precipitation system. In addition, the structure and composition of ACC and ACP formed in simple and complex systems are markedly different from those obtained in the physiological system.
Although a general similarity between respective ACC and ACP phases can be observed, the similar chemical environment in which they form not necessarily leads to similar structural properties. In addition, the obtained results show that tailoring the ACC and ACP properties can be achieved by rather simple methods. This can in turn be a starting point for developing bottom-up approaches in (bio)materials synthesis and for understanding the amorphous phase formation in general.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the Croatian Science Foundation under project IP-2013-11-5055. The authors are indebted to Mrs. Nevenka Nekić for technical assistance. The authors would like to thank the Laboratory for Radiation Chemistry and Dosimetry, Ruđer Bošković Institute, Zagreb, Croatia, for allowing the use of the 60Co-gamma irradiation source.Notes and references
- H. A. Lowenstam and S. Weiner, On Biomineralization, Oxford University Press, New York, 1989 Search PubMed.
- S. V. Dorozhkin, Acta Biomater., 2010, 6, 4457–4475 CrossRef CAS PubMed.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161–1164 CrossRef CAS PubMed.
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergström and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15(12), 959–970 CrossRef CAS.
- S. Roger Qiu and C. A. Orme, Chem. Rev., 2008, 108, 4784–4822 CrossRef PubMed.
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. London, Ser. B, 1997, 264, 461–465 CrossRef CAS.
- E. Beniash, L. Addadi and S. Weiner, J. Struct. Biol., 1999, 125, 50–62 CrossRef CAS PubMed.
- I. M. Weiss, N. Tuross, L. Addadi and S. Weiner, J. Exp. Zool., 2002, 293, 478–491 CrossRef CAS PubMed.
- E. Foran, S. Weiner and M. Fine, Sparus aurata, Sci. Rep., 2013, 3, 1–5 Search PubMed.
- C. E. Killian, R. A. Metzler, Y. U. T. Gong, I. C. Olson, J. Aizenberg, Y. Polit, F. H. Wilt, A. Scholl, A. Young, A. Doran, M. Kunz, N. Tamura, S. N. Coppersmith and P. U. P. A. Gilbert, J. Am. Chem. Soc., 2009, 131, 18404–18409 CrossRef CAS PubMed.
- P. Raiteri and J. D. Gale, J. Am. Chem. Soc., 2010, 132(49), 17623–17634 CrossRef CAS PubMed.
- S. Weiner, Y. Levi-Kalisman, S. Raz and L. Addadi, Connect. Tissue Res., 2003, 44(Suppl 1), 214–218 CrossRef CAS PubMed.
- F. M. Michel, J. MacDonald, J. Feng, B. L. Phillips, L. Ehm, C. Tarabrella, J. B. Parise and R. J. Reeder, Chem. Mater., 2008, 20(14), 4720–4728 CrossRef CAS.
- M. P. Schmidt, A. J. Ilott, B. L. Phillips and R. J. Reeder, Cryst. Growth Des., 2014, 14, 938–951 CAS.
- A. V. Radha, T. Z. Forbes, C. E. Killian, P. U. P. A. Gilbert and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(38), 16438–16443 CrossRef CAS PubMed.
- C. Rodriguez-Navarro, K. Kudłacz, Ö. Cizerc and E. Ruiz-Agudoa, CrystEngComm, 2015, 17, 58–72 RSC.
- J. H. E. Cartwright, A. G. Checa, J. D. Gale, D. Gebauer and C. I. Sainz-Diaz, Angew. Chem., Int. Ed., 2012, 51, 11960–11970 CrossRef CAS PubMed.
- D. Gebauer, P. N. Gunawidjaja, J. Y. P. Ko, Z. Bacsik, B. Aziz, L. Liu, Y. Hu, L. Bergström, C. Wai Tai, T.-K. Sham, M. Edén and N. Hedin, Angew. Chem., Int. Ed., 2010, 49, 8889–8891 CrossRef CAS PubMed.
- M. Farhadi-Khouzani, D. M. Chevrier, P. Zhang, N. Hedin and D. Gebauer, Angew. Chem., Int. Ed., 2016, 55, 1–5 CrossRef PubMed.
- L. Wang and G. H. Nancollas, Chem. Rev., 2008, 108, 4628–4669 CrossRef CAS PubMed.
- E. D. Eans, I. H. Gillesen and A. S. Posner, Nature, 1965, 208, 365–367 CrossRef.
- A. S. Posner and F. Betts, Acc. Chem. Res., 1975, 8, 273–281 CrossRef CAS.
- K. Onuma and A. Ito, Chem. Mater., 1998, 10, 3346–3351 CrossRef CAS.
- A. Oyane, K. Onuma, T. Kokubo and A. Ito, J. Phys. Chem. B, 1999, 103, 8230–8235 CrossRef CAS.
- A. Dey, P. H. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1010–1014 CrossRef CAS PubMed.
- L. Wang, S. Li, E. Ruiz-Agudo, C. V. Putnis and A. Putnis, CrystEngComm, 2012, 14, 6252–6256 RSC.
- Lj. Brečević, V. Hlady and H. Füredi-Milhofer, Colloids Surf., 1987, 28, 301–313 CrossRef.
- B.-Y. Ofir, R. Govrin-Lippman, N. Garti and H. Füredi-Milhofer, Cryst. Growth Des., 2004, 4, 177–183 Search PubMed.
- Lj. Brečević and H. Füredi-Milhofer, Calcif. Tissue Res., 1972, 10, 82–90 CrossRef.
- J. Christoffersen, M. R. Christoffersen, W. Kibalczyc and F. A. Andersen, J. Cryst. Growth, 1989, 94, 767–777 CrossRef CAS.
- S. Jiang, W. Jin, Y.-N. Wang, H. Pan, Z. Sun and R. Tang, RSC Adv., 2017, 7, 25497–25503 RSC.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y.-Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef.
- J. Mahamid, A. Sharir, L. Addadi and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12748–12753 CrossRef CAS PubMed.
- A. Akiva, G. Malkison, A. Masic, M. Kerschnitzki, M. Bennet, P. Fratzl, L. Addadi, S. Weiner and K. Yaniv, Bone, 2015, 75, 192–200 CrossRef CAS PubMed.
- E. Beniash, R. A. Metzler, R. S. K. Lam and P. U. P. A. Gilbert, J. Struct. Biol., 2009, 166, 133–143 CrossRef CAS PubMed.
- O. A. Tertuliano and J. R. Greer, Nat. Mater., 2016, 15, 1195–1202 CrossRef CAS PubMed.
- Y. Levi-Kalisman, S. Raz, S. Weiner, L. Addadi and I. Sagi, Adv. Funct. Mater., 2002, 12, 43–48 CrossRef CAS.
- M. H. Salimi, J. C. Heughebaert and G. H. Nancollas, Langmuir, 1985, 1, 119–122 CrossRef CAS.
- H. Ding, H. Pan, X. Xu and R. Tang, Cryst. Growth Des., 2014, 14, 763–769 CAS.
- E. Loste, R. M. Wilsonb, R. Seshadric and F. C. Meldrum, J. Cryst. Growth, 2003, 254, 206–218 CrossRef CAS.
- X. Yang, B. Xie, L. Wang, Y. Qin, Z. J. Henneman and G. H. Nancollas, CrystEngComm, 2011, 13, 1153–1158 RSC.
- Y. Chen, W. Gu, H. Pan, S. Jiang and R. Tang, CrystEngComm, 2014, 16, 1864–1867 RSC.
- Y. B. Li and W. J. Weng, J. Mater. Sci.: Mater. Med., 2007, 18, 2300–2308 Search PubMed.
- S. E. Wolf, J. Leiterer, V. Pipich, R. Barrea, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2011, 133, 12642–12649 CrossRef CAS PubMed.
- K. Bleek and A. Taubert, Acta Biomater., 2013, 9, 6283–6762 CrossRef CAS PubMed.
- I. Buljan Meić, J. Kontrec, D. Domazet Jurašin, B. Njegić Džakula, L. Štajner, D. M. Lyons, M. Dutour Sikirić and D. Kralj, Cryst. Growth Des., 2017, 17(3), 1103–1117 Search PubMed.
- M. Kellermeier, A. Picker, A. Kempter, H. Cölfen and D. Gebauer, Adv. Mater., 2014, 26, 752–757 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- H. Aoki, Science and medical applications of hydroxyapatite, Takayama Press System Center Co., Tokyo, 1991 Search PubMed.
- B. Težak, Croat. Chem. Acta, 1968, 40, 63–78 Search PubMed.
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- C. Combes and C. Rey, Acta Biomater., 2010, 6, 3362–3378 CrossRef CAS PubMed.
- Lj. Brečevic and A. E. Nielsen, J. Cryst. Growth, 1989, 98, 504–510 CrossRef.
- R. Gueta, A. Natan, L. Addadi, S. Weiner, K. Refson and L. Kronik, Angew. Chem., Int. Ed., 2007, 46, 291–294 CrossRef CAS PubMed.
- D. J. Tobler, J. D. Rodriguez-Blanco, H. O. Sørensen, S. L. S. Stipp and K. Dideriksen, Cryst. Growth Des., 2016, 16(8), 4500–4508 CAS.
- J. D. Rodriguez-Blanco, S. Shaw, P. Bots, T. Roncal-Herrero and L. G. Benning, Geochim. Cosmochim. Acta, 2014, 127, 204–220 CrossRef CAS.
- Z. Zhang, Y. Xie, X. Xun, H. Pan and R. Tang, J. Cryst. Growth, 2012, 343, 62–67 CrossRef CAS.
- Y.-Y. Wang, Q.-Z. Yao, G.-T. Zhou and S.-Q. Fu, Eur. J. Mineral., 2015, 27, 717–772 CrossRef CAS.
- Y. Politi, D. R. Batchelor, P. Zaslansky, B. F. Chmelka, J. C. Weaver, I. Sagi, S. Weiner and L. Addadi, Chem. Mater., 2010, 22, 161–166 CrossRef CAS.
- A. Selmani, I. Coha, K. Magdić, B. Čolović, V. Jokanović, S. Šegota, S. Gajović, A. Gajović, D. Jurašin and M. Dutour Sikirić, CrystEngComm, 2015, 17, 8529–8548 RSC.
- T. Kanazawa, T. Umegaki and N. Uchiyama, J. Chem. Technol. Biotechnol., 1982, 32, 399–406 CrossRef CAS.
- J. M. Sedlak and R. A. Beebe, J. Colloid Interface Sci., 1974, 47, 483–489 CrossRef CAS.
- Y. Kojima, K. Sakama, T. Toyama, T. Yause and Y. Arai, Phosphorus Res. Bull., 1994, 4, 47–52 CrossRef CAS.
- D. U. Schramm and A. M. Rossi, Appl. Radiat. Isot., 2000, 52, 1085–1091 CrossRef CAS PubMed.
- F. Callens, P. Moens and R. Verbeeck, Calcif. Tissue Int., 1995, 56, 543–548 CrossRef CAS PubMed.
- D. V. Rokhmistriv, O. T. Nikolov, O. A. Gorobchenko and K. I. Loza, Appl. Radiat. Isot., 2012, 70, 2621–2626 CrossRef PubMed.
- Lj. Brečević and D. Kralj, Croat. Chem. Acta, 2007, 80(3–4), 467–484 Search PubMed.
- A. Fernadez-Martinez, H. Lopez-Martinet and D. Wang, Structural characterization and the occurrence of polyamorphism in amorphous calcium carbonate, ed. A. E. S. van Driessche, M. Kellermeier, L. G. Benning and D. Gebauer, New Perspective on mineral nucleation and growth, From solution precursors to solid materials, Springer, Cham, 2017, ch. 4 Search PubMed.
- M. F. Khouzani, D. M. Chevrier, P. Güttlein, K. Hauser, P. Zhang, N. Hedin and D. Gebauer, CrystEngComm, 2015, 17, 4842–4849 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ce01693j |
| This journal is © The Royal Society of Chemistry 2018 |