
Catalyst-free and visible light promoted trifluoromethylation and perfluoroalkylation of uracils and cytosines†
Yang
Huang‡
a,
Yun-Yun
Lei‡
a,
Liang
Zhao
a,
Jiwei
Gu
b,
Qiuli
Yao
a,
Ze
Wang
a,
Xiao-Fei
Li
*a,
Xingang
Zhang
 *b and
Chun-Yang
He
*a
*b and
Chun-Yang
He
*a
aGeneric Drug Research Center of Guizhou Province, School of Pharmacy, Zunyi Medical University, Zunyi, 563003, P. R. China. E-mail: hechy2002@163.com; 359238630@qq.com
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, P. R. China. E-mail: xgzhang@mail.sioc.ac.cn
First published on 22nd October 2018
Abstract
Fluoroalkylated enaminones, such as trifluridine and 5-trifluoromethyluracil, have widespread applications in pharmaceuticals and agrochemicals. Although these kinds of pharmaceutical agent often bear CF3 and perfluoroalkyl motifs in the core structure, access to such analogues typically requires multi-step synthesis. Here, we report a mild, metal-free and operationally simple strategy for the direct perfluoroalkylation of uracils, cytosines and pyridinones through a visible-light induced pathway from perfluoroalkyl iodides. This photochemical transformation features synthetic simplicity, mild reaction conditions without any photoredox catalyst, and high functional group tolerance, providing a facile route for applications in medicinal chemistry.
The incorporation of fluoroalkyl groups into (hetero)arenes can dramatically alter the physical and chemical properties of the parent molecules and results in numerous applications in pharmaceuticals, agrochemicals, and functional materials.1 Among fluorine-containing functional groups, the trifluoromethyl and perfluoroalkyl (RF) groups have a privileged role in the field of medicinal chemistry because their incorporation into small molecules often enhances metabolic stability, improves lipophilicity and increases the protein binding affinity of the drug candidates.2 Therefore, it is of great interest to develop novel and effective synthetic methods to construct these fluoroalkylated (hetero)arenes.3
The structural entity of uracils is a kind of important electron-deficient heteroarene in pharmaceuticals and life sciences.4 For example, 5-trifluoromethyluracil (5-TFU) is an important intermediate in the field of medicinal chemistry,5 and trifluridine is an antiviral drug.6 Therefore, the development of an efficient and environmentally benign method to access such structures is very desirable. Traditionally, 5-TFU is manufactured from 3,3,3-trifluoropropene as a starting material in six steps.7 Recently, some new methods by using Fe(II)/H2O2, Cu(II)/K2S2O8 or electrochemical reagents have been developed.8 However, an alternative method using commercially available reagents remains highly desirable.
Recently, metal- and organocatalyst-based photoredox catalysis reactions induced by visible light have been demonstrated as an attractive strategy for generating perfluoroalkyl free radicals under mild reaction conditions.9 As a milestone progress in the realm of visible-light induced radical fluoroalkylation, Melchiorre's group realized a noncovalent interaction initiated single-electron-transfer (SET) fluoroalkylation driven by the photochemical activity of an electron donor–acceptor (EDA) complex.10 These strategies are promising because no catalyst is needed.11 Curran and Studer developed the concept that an electron can also be treated as an efficient catalyst to realize various transformations.12 However, the connection of electrophilic perfluoroalkyl radicals with electron-deficient (hetero)arenes via this catalyst-free strategy has never been reported.
Herein, we demonstrated a metal-free, operationally simple protocol for the synthesis of 5-trifluoromethylated/perfluoroalkylated uracils via a visible light-promoted reaction with inexpensive perfluoroalkyl iodides as fluoroalkylating reagents. The reaction can also extend to cytosines and 4-pyridinones, providing a useful and facile access to some valuable molecules for application in medicinal chemistry.
We began this study by choosing uracil 1a and CF3I 2 as model substrates.13 In our previous studies, we found that the solution of CF3I in DMSO could be stored at room temperature without declining the titer, thus making the manipulating process more convenient.14 When this stable and practically manipulated trifluoromethyl iodide solution combined with uracil 1a and Na2CO3 in DMSO was irradiated by blue LEDs for 12 hours, the trifluoromethylated uracil 3a was obtained in 30% yield (Table 1, entry 1). Further optimization of inorganic bases demonstrated that Cs2CO3 was the most effective one, and the desired product 3a was isolated in 81% yield. However, organic bases, which are traditionally used as electron-donors in previous reports, did not provide any reactivity (Table 1, entries 9–11). Then, a series of reaction media were screened (Table 1, entries 12–15), and no desired product was detected when MeOH or DCE was used. Only trace yield was observed when the reaction was conducted in DMF and MeCN. Visible light with other wavelengths, such as green and yellow light, demonstrates less reactivity (Table 1, entries 16 and 17). Control experiments, carried out by performing the reaction in the dark or in the absence of a base, failed to provide the desired product, which indicates that a photochemical transformation was involved in the reaction (Table 1, entries 18 and 19).
|
|
||||
|---|---|---|---|---|
| Entry | Light source | Base | Solvent | Yieldb (%) |
| a Reaction conditions (unless otherwise specified): 1a (0.4 mmol, 1.0 equiv.), CF3I (1.2 mmol, 3.0 equiv.), base (0.8 mmol, 2.0 equiv.), solvent (3.0 mL), room temperature, 12 h. b Determined by 19F-NMR spectroscopy using fluorobenzene as an internal standard; the number within parentheses represents the yield of the isolated product. | ||||
| 1 | Blue LED | Na2CO3 | DMSO | 30 |
| 2 | Blue LED | K2CO3 | DMSO | 65 |
| 3 | Blue LED | Cs2CO3 | DMSO | 85 (81) |
| 4 | Blue LED | K3PO4 | DMSO | 56 |
| 5 | Blue LED | NaOH | DMSO | 53 |
| 6 | Blue LED | K2HPO4 | DMSO | Trace |
| 7 | Blue LED | KOAc | DMSO | Trace |
| 8 | Blue LED | KF | DMSO | Trace |
| 9 | Blue LED | DMPU | DMSO | NR |
| 10 | Blue LED | TMEDA | DMSO | NR |
| 11 | Blue LED | Et3N | DMSO | NR |
| 12 | Blue LED | Cs2CO3 | MeOH | NR |
| 13 | Blue LED | Cs2CO3 | DCE | NR |
| 14 | Blue LED | Cs2CO3 | DMF | Trace |
| 15 | Blue LED | Cs2CO3 | MeCN | Trace |
| 16 | Green LED | Cs2CO3 | DMSO | 67 |
| 17 | Yellow LED | Cs2CO3 | DMSO | 69 |
| 18 | — | Cs2CO3 | DMSO | NR |
| 19 | Blue LED | — | DMSO | NR |
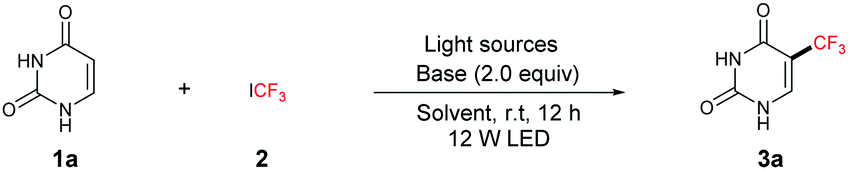
As illustrated in Table 2, we found that this new photochemical protocol allows the direct trifluoromethylation of a range of uracils. Initially, the substituent effect on nitrogen was examined. 1-Methyluracil and 5-bocuracil give the trifluoromethylated product in 77% and 71% yields, respectively. While 1,3-dimethyluracil afforded a lower yield (45%) (Table 2, 3b–d). Functional group substitution at the non-heteroatom position, such as alkoxycarbonyl and methyl, is also amenable to this trifluoromethylation protocol, leading to the corresponding products in 64–80% yields (Table 2, 3e–f). 2,4-Dimethyl-6-hydroxypyrimidine was also a suitable substrate, and provided the corresponding product in 77% yield (Table 2, 3g). 1-(Tetrahydrofuran-2-yl)-5-trifluoromethyluracil, an important intermediate used in medicinal chemistry, can be accessed in 67% yield (Table 2, 3h). Trifluridine, an antiviral drug, can also be readily prepared from abundant 2′-deoxyuridine via the current photo-induced process without protection (Table 2, 3i), thus providing a facile route for applications in drug discovery and development. Remarkably, when the reaction was performed on a 10 gram scale (3a), a comparable yield still could be obtained, thus demonstrating the synthetic utility of this protocol.
| a Reaction conditions: 1 (0.4 mmol, 1.0 equiv.), 2 (1.2 mmol, 3.0 equiv.), Cs2CO3 (0.8 mmol, 2.0 equiv.) in anhydrous DMSO (3.0 mL), r.t., 12 W blue LED, for 24 h. b Yield of the isolated product. c The reaction was performed for 12 h. d (100 mmol, 1.0 equiv.), CF3I (300 mmol, 3.0 equiv.), Cs2CO3 (2.0 equiv.), DMSO (400 mL), r.t., 24 W blue LED, for 48 h. e 1.0 mL DMSO was used. |
|---|

|
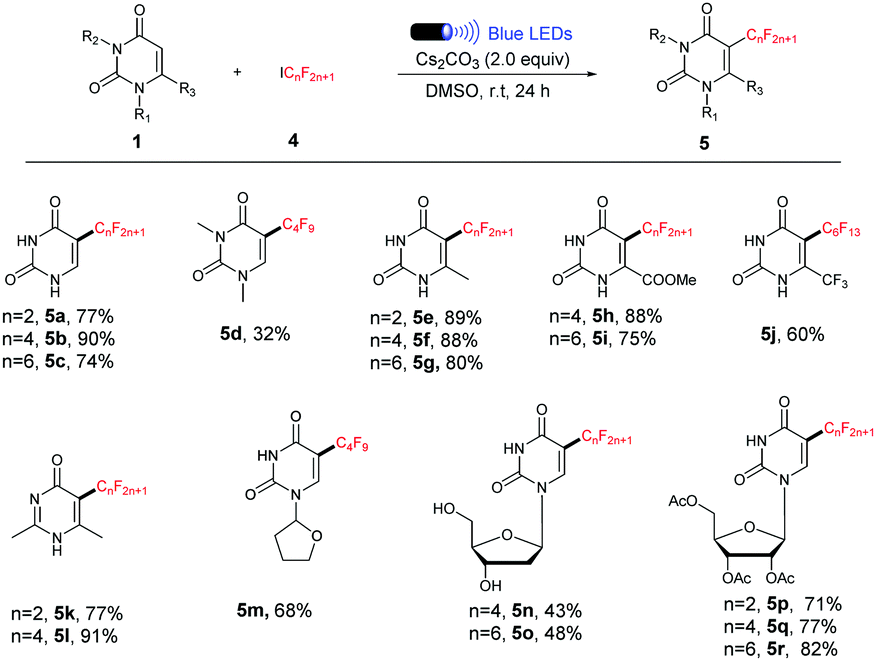

We next investigated the scope of the reaction with respect to the perfluoroalkyl reagents (Table 3). Other commercially available perfluoroalkyl iodides such as C2F5I, C4F9I and C6F13I underwent the current process smoothly, and provided the corresponding products in moderate to excellent yields. However, 1,3-dimethyluracil showed less reactivity and only 32% yield of 5d was obtained, suggesting that the existence of the unprotected N–H bond is critical to the reaction efficiency (Table 3, 5d).

Cytosines are also important heterocyclic motifs found in numerous bioactive molecules;15 however, methods for introducing fluoroalkyl groups to these structures are still limited.16 Therefore, the reaction of cytosines with fluoroalkyl iodides was also examined (Table 4). No reaction occurred when cytosine was tested, but good to excellent yields can be obtained when N4-benzoylcytosine and N4-acetylcytosine were treated (Table 4, 7b–f). Further experiments demonstrated that the successful negative ionization of the N–H bond on these heterocycles is the key step for the reaction efficiency (for details, see the ESI†).
The utility of the current reaction can also be demonstrated by the rapid fluoroalkylation of 4-pyridones.17 As shown in Table 5, 54% yield of 2,6-dimethyl-3,5-bis(trifluoromethyl)pyridin-4(1H)-one (Table 5, 9a) along with 11% yield of the mono-substituted product was generated under the optimized conditions. 82% yield of the di-substituted product was obtained when C4F9I was used (Table 5, 9b). Quinolones were also suitable substrates in the reaction system, and moderate to good yields could still be obtained (Table 5, 9c–9f). When 1-methyl-4-quinolone was treated with CF3I, the reaction become complex, and only a trace amount of the desired product was detected (Table 5, 9g).
To gain insight into the mechanism of this transformation, a series of experiments were carried out. Initially, radical inhibition experiments were conducted. No desired product was observed when a radical scavenger TEMPO (100 mol%) was added under the standard reaction conditions, suggesting a radical pathway is involved in this transformation. Radical clock experiments were performed subsequently. When cyclopropylstyrene (10) was treated with CF3I in the presence or absence of 1a under the standard reaction conditions, a ring-opened product 11 was formed. Finally, the optical absorption spectra of the reactants were investigated, which indicated that an EDA complex might be generated between the negatively ionized uracil and CF3I (for details, see the ESI†).
On the basis of these preliminary results, a plausible mechanism was proposed, as shown in Fig. 1, for this transformation. Firstly, the negatively ionized uracil (A) was generated in the presence of Cs2CO3. A fluoroalkyl radical can be produced under the irradiation of blue LEDs via the EDA complex. Two pathways are feasible for the propagation step: (1) the newly formed radical then reacts with A to produce intermediate B, which can activate the fluoroalkyl iodides via a SET process and meanwhile generate the desired products (3/5); or (2) the Rf radical can add to uracils (1) and generate the carbon radical intermediate D, which abstracts an iodine atom from RfI to afford E and regenerate the Rf radical. The desired products (3/5) were obtained via the fast rearomatization of E through HI elimination.
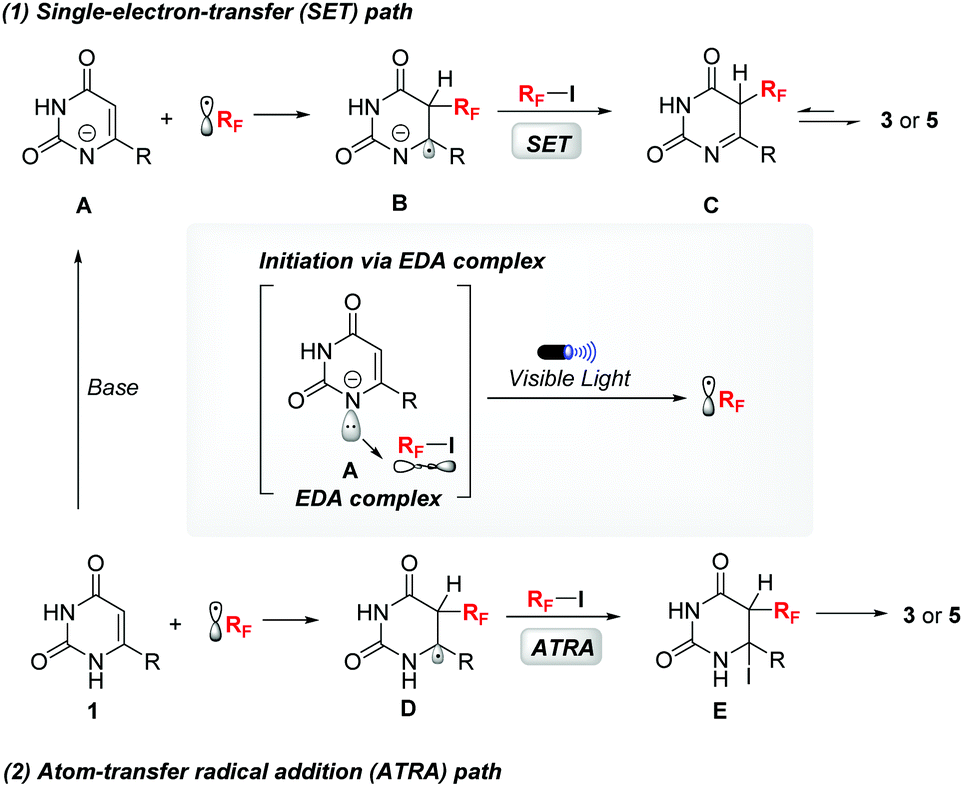 | ||
| Fig. 1 Proposed reaction mechanism. | ||
In conclusion, we have developed a useful and operationally green method for the synthesis of trifluoromethylated/perfluoroalkylated uracils and cytosines. This new protocol was also successfully applied to 4-pyridones. This reaction is distinguished by its mild reaction conditions, wide substrate scope, and especially good applicability. The application of this methodology in life sciences is underway in our lab.
The Young Elite Scientists Sponsorship Program by Cast of the China Association for Science and Technology (No. 2015-41), the National Natural Science Foundation of China (No. 81760624 and 21702241), and Programs of Guizhou Province (No. 2017-1225 and 2018-1427) are warmly acknowledged for funding this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected books and reviews, see: (a) I. Ojima, Fluorine in Medicinal Chmeistry and Chemical Biology, Wiley-Blackwell, Oxford, UK, 2009 CrossRef; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (d) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (b) C. Alonso, E. M. Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (c) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (d) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed; (e) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed.
- For selected papers, see: (a) T. Shirai, M. Kanai and Y. Kuninobu, Org. Lett., 2018, 20, 1593 CrossRef CAS PubMed; (b) Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270 CrossRef CAS PubMed; (c) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2012, 134, 1298 CrossRef CAS PubMed; (d) M. G. Mormino, P. S. Fier and J. F. Hartwig, Org. Lett., 2014, 16, 1744 CrossRef CAS PubMed; (e) M. Nagase, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2016, 138, 6103 CrossRef CAS PubMed.
- For selected reviews, see: (a) P. Weigele and E. A. Raleigh, Chem. Rev., 2016, 126, 12655 CrossRef PubMed; (b) E. Romero, J. R. G. Castellanos, G. Gadda, M. W. Fraaije and A. Mattevi, Chem. Rev., 2018, 118, 1742 CrossRef CAS PubMed . For selected papers, see: ; (c) P. S. Pallan, E. M. Greene, P. A. Jicman, R. K. Pandey, M. Manoharan, E. Rozners and M. Egli, Nucleic Acids Res., 2011, 39, 3482 CrossRef CAS PubMed; (d) M. Košutić, L. Jud, C. Da Veiga, M. Frener, K. Fauster, C. Kreutz, E. Ennifar and R. Micura, J. Am. Chem. Soc., 2014, 136, 6656 CrossRef PubMed.
- N. P. Dolman, J. C. A. More, A. Alt, J. L. Knauss, O. T. Pentikainen, C. R. Glasser, D. Bleakman, M. L. Mayer, G. L. Collingridge and D. E. Jane, J. Med. Chem., 2007, 50, 1558 CrossRef CAS PubMed.
- (a) H.-J. Lenz, S. Stintzing and F. Loupakis, Cancer Treat. Rev., 2015, 41, 777 CrossRef CAS PubMed; (b) E. De Clercq, Antiviral Chem. Chemother., 2013, 23, 93 CrossRef CAS PubMed.
- (a) T. Fuchikami and I. Ojima, Tetrahedron Lett., 1982, 23, 4099 CrossRef CAS; (b) T. Fuchikami, A. Yamanouchi and I. Ojima, Synthesis, 1984, 766 CrossRef CAS.
- (a) D. Uraguchi, K. Yamamoto, Y. Ohtsuka, K. Tokuhisa and T. Yamakawa, Appl. Catal., A, 2008, 342, 137 CrossRef CAS; (b) F. Sladojevich, E. McNeill, J. Boergel, S.-L. Zheng and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3712 CrossRef CAS PubMed; (c) A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868 CrossRef PubMed.
- For representative reviews on photoredox catalysis, see: (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532 CrossRef CAS PubMed; (c) J. W. Battey and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474 CrossRef PubMed; (d) M. N. Hopkinson, B. Sahoo, J. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (e) J. Xie, H. Jin, P. Xu and C. Zhu, Tetrahedron Lett., 2014, 55, 36 CrossRef CAS; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (g) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284 CrossRef CAS PubMed . For recent examples on photoredox catalysis, see: ; (h) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 6174 CrossRef PubMed; (i) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (j) J. A. Terrett, J. D. Cuthbertson, V. W. Shuvtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed; (k) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed; (l) Ł. Woźniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678 CrossRef PubMed.
- (a) M. Nappi, G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 4921 CrossRef CAS PubMed; (b) V. M. Fernández-Alvarez, M. Nappi, P. Melchiorre and F. Maseras, Org. Lett., 2015, 17, 2676 CrossRef PubMed; (c) S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adán and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485 CrossRef CAS PubMed.
- For catalyst free and visible-light mediated reactions, see: (a) B. Janhsen and A. Studer, J. Org. Chem., 2017, 82, 11703 CrossRef CAS PubMed; (b) C. Qu, Z. Wu, W. Li, H. Du and C. Zhu, Adv. Synth. Catal., 2017, 359, 1672 CrossRef CAS; (c) Y. Wang, J. Wang, G.-X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442 CrossRef CAS PubMed; (d) X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638 CrossRef CAS PubMed; (e) L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809 CrossRef CAS PubMed; (f) Q. Guo, M. Wang, H. Liu, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 4747 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- (a) N. J. W. Straathof, S. E. Cramer, V. Hessel and T. Noël, Angew. Chem., Int. Ed., 2016, 55, 15549 CrossRef CAS PubMed; (b) J. L. Monteiro, P. F. Carneiro, P. Elsner, D. M. Roberge, P. G. M. Wuts, K. C. Kurjan, B. Gutmann and C. O. Kappe, Chem. – Eur. J., 2017, 23, 176 CrossRef CAS PubMed.
- C.-Y. He, J.-W. Gu and X. Zhang, Tetrahedron Lett., 2017, 58, 3939 CrossRef CAS.
- For selected reviews, see: (a) S. U. Siriwardena, K. Chen and A. S. Bhagwat, Chem. Rev., 2016, 116, 12688 CrossRef CAS PubMed; (b) M. J. Booth, E.-A. Raiber and S. Balasubramanian, Chem. Rev., 2015, 115, 2240 CrossRef CAS PubMed; (c) A. C. Drohat and C. T. Coey, Chem. Rev., 2016, 116, 12711 CrossRef CAS PubMed.
- D. Musumeci, C. Irace, R. Santamaria and D. Montesarchio, MedChemComm, 2013, 4, 1405 RSC.
- For the direct fluoroalkylation of 4-pyridones, see: (a) Y.-Y. Yu, A. R. Ranade and G. I. Georg, Adv. Synth. Catal., 2014, 356, 3510 CrossRef CAS; (b) Z. Fang, Y. Ning, P. Mi, P. Liao and X. Bi, Org. Lett., 2014, 16, 1522 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07759b |
| ‡ Y. Huang and Y.-Y. Lei contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |