
Arylation of benzyl amines with aromatic nitriles†
Yingjie
Lei
,
Ju
Yang
,
Rupeng
Qi
,
Shan
Wang
,
Rui
Wang
 * and
Zhaoqing
Xu
*
* and
Zhaoqing
Xu
*
Key Laboratory of Preclinical Study for New Drugs of Gansu Province, The Institute of Pharmacology, School of Basic Medical Sciences, Lanzhou University, 199 West Donggang Road, Lanzhou 730000, China. E-mail: wangrui@lzu.edu.cn; zqxu@lzu.edu.cn
First published on 25th September 2018
Abstract
In the past years, the activations of aromatic nitriles for radical arylations under photoirradiation have been developed. We here report the first example of radical arylations using aromatic nitriles without the assistance of photoirradiation. Importantly, with this method, the direct arylation of C(sp3)–H in benzyl amines provided a practical method for the synthesis of diarylmethylamines without the use of precious transition metal catalysts.
Diarylmethylamines are important structural motifs in medicinal chemistry and often act as central components of pharmaceuticals, bioactive natural products, and agrochemicals.1 Importantly, recent statistical studies have indicated that the prominence of heteroaromatic rings in marketed oral drugs is increasing (Fig. 1).1e In this context, the synthesis of diarylmethylamines has attracted more and more attention. In the past decades, the transition-metal-catalyzed direct α-C(sp3)–H arylations of benzyl amines with aryl halides have been developed, which provided convenient methods to prepare diarylmethylamines.2 The reactions avoid the commonly employed nucleophilic addition of aryl-metal reagents (aryl lithium or Grignard reagent) to imines. In the reactions, precious transition-metal catalysts (mostly Pd) were required (Scheme 1, a).
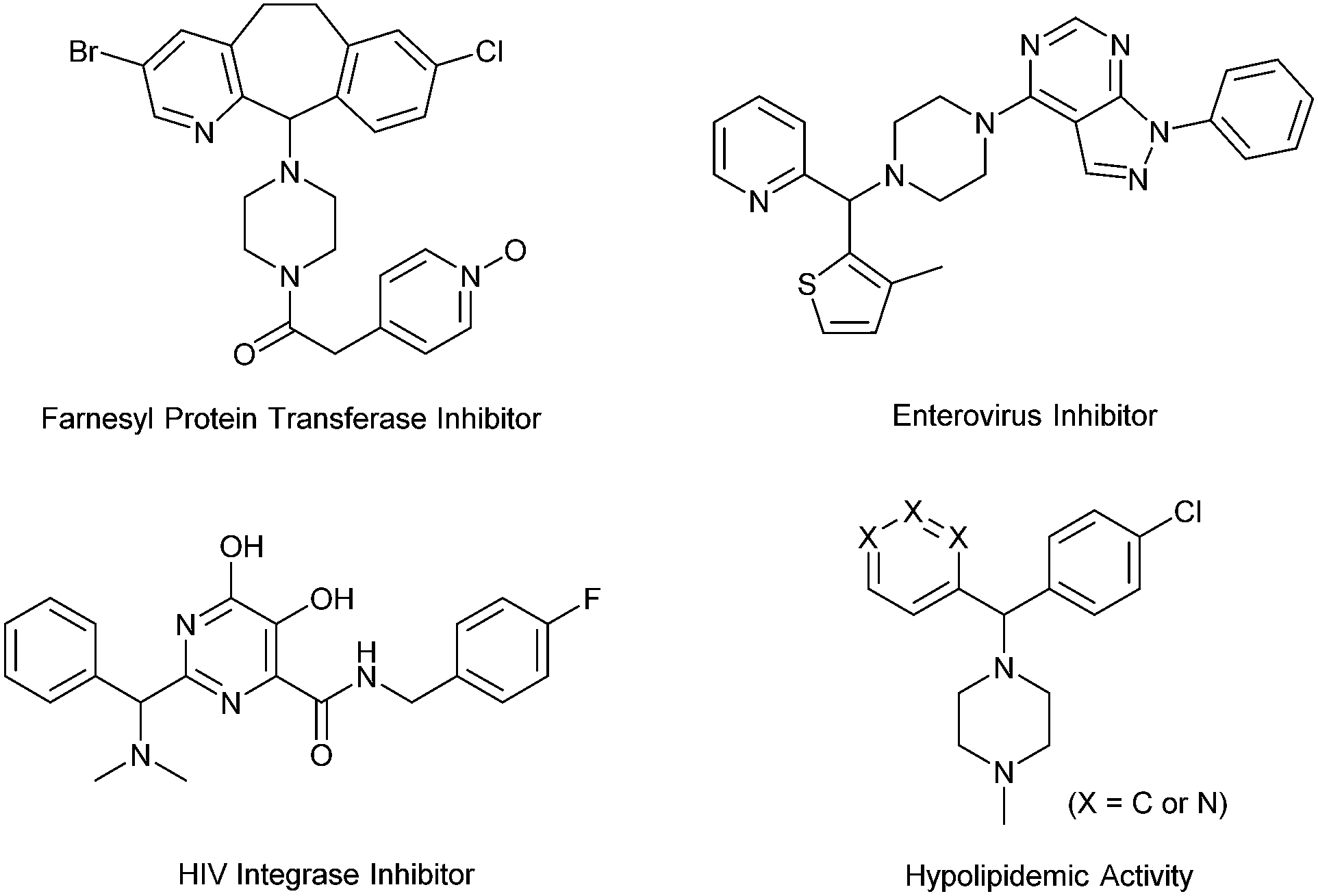 | ||
| Fig. 1 Pharmacologically active compounds containing diaryl (heteroaryl) methylamines. | ||
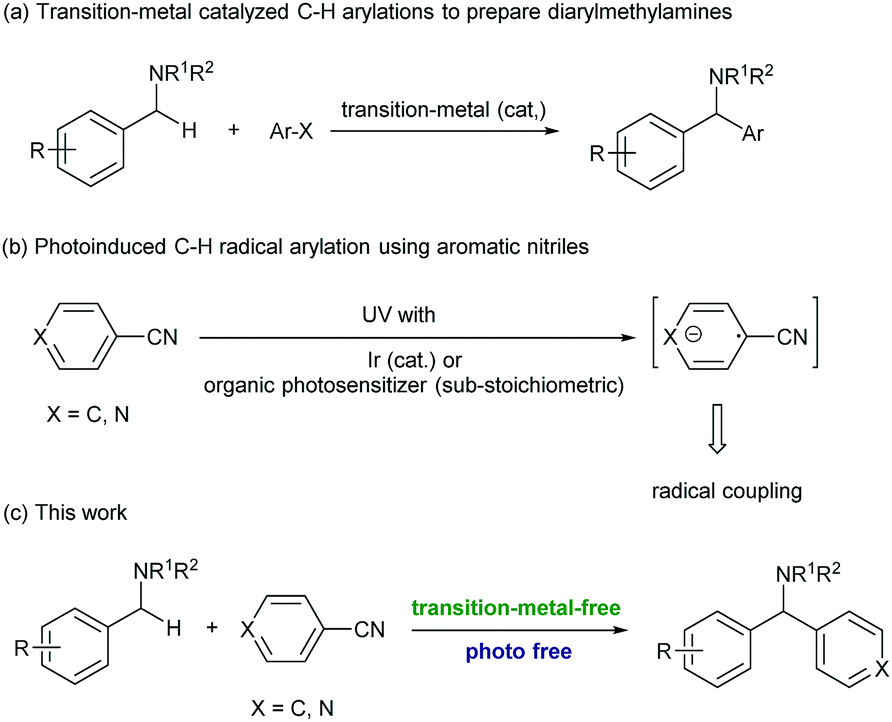 | ||
| Scheme 1 Research background introductions. | ||
In the past years, the application of aromatic nitriles for radical C(sp3)–H arylation has been developed.3 The reactions were usually proceeded under photoirradiation, in which the aromatic nitrile received an electron from the photoreductant and formed an aryl radical anion. This intermediate was then subjected to the subsequent radical coupling reactions (Scheme 1, b). For example, the MacMillan group recently realized the first example of photoinduced radical C–H arylation using aromatic nitriles as the arylation reagents.4 In these reactions, the sp3 C–H bonds at the allylic, benzylic, and α-positions of alkyl amines were successfully coupled with a variety of (hetero)aromatic nitriles. With this strategy, the reactions needed to be conducted under UV light irradiation or proceeded under visible light with the assistance of a precious Ir photocatalyst.5 In 2013, Inoue and co-workers reported a novel photoinduced transition-metal free arylation reaction with aromatic nitriles. In this reaction, a sub-stoichiometric amount of benzophenone (0.5 equiv.) and 100 W medium pressure mercury lamp irradiation were essentially required to mediate the electron transfer.6 So far, the strategy to activate aromatic nitriles and apply to radical arylation without the assistance of photoirradiation has been undeveloped, and is still highly desirable.
In the past years, organic molecule based electron donors have received much attention due to their tunable reduction ability and environmentally benign reaction conditions.7 Among them, one class of compounds called super-electron-donors (SEDs) possess reduction potentials as high as −1.50 eV (versus SCE),8 which were able to initiate the dissociation of a large variety of chemical bonds.9 In the reactions, organic SEDs are capable of spontaneous one- or two-electron transfer from a donor to an organic substrate, while the bond dissociation can take place either simultaneously or in a stepwise process under mild and homogeneous conditions. Different ranges of redox potentials and choices of reactivities and selectivities can be achieved by simple modulation of their structure and the reaction parameters. These unique properties are now attracting more and more attention in various original applications like substitutions, addition, cyclization, polymerization initiators, and greenhouse gas reduction.10 For example, the Walsh group recently developed a novel transition-metal free radical coupling reaction of 2-azaallyl species with vinyl, aryl and alkyl halides.11 The DFT computational studies and EPR experiment suggested that the 2-azaallyl anion served as an SED and mediated the SET (single electron transfer) with organic halides.
As a complementary method to the previously reported photoinduced activation of aromatic nitriles for radical arylations,4–6 we were interested in developing an alternative method without the assistance of photoirradiation. We envisioned that an SED, such as the 2-azaallyl anion, could possibly provide an electron to aromatic nitriles and promoted the generation of aryl radicals. Importantly, the direct arylation of 2-azaallyl based ketimines could provide a practical method for C(sp3)–H arylation for the synthesis of diarylmethylamines without the use of precious transition metal catalysts (Scheme 1, c).2 In this reaction, the ketimine served not only as the SED to activate the aromatic nitrile but also as the radical coupling partner after SET.
The investigation was initiated using ketimines 1a and 4-cyanopyridine 2a (2 equiv.) as model substrates, and the reaction was proceeded at room temperature for 4 h in 1 ml methyl tert-butyl ether (MTBE). It was delightful that the desired arylation product 3a was obtained in 40% yield using 3 equiv. of NaHMDS (NaN(SiMe3)2, Table 1, entry 1). Other basic additives were also tested in our reactions, such as LiOtBu, NaOtBu, KOtBu, K2CO3, Na2CO3, NaOAc, Et3N, TMEDA, DBU, DMAP, etc.; however, only LiHMDS and KHMDS gave the desired products but with lower yields (entries 2 and 3, see the ESI† for details). The solvent screening indicated that dimethoxyethane (DME) was the best choice (entries 4–8). Increasing the amount of NaHMDS to 4 equiv. did not improve the yield (entry 9). To our satisfaction, upon reducing the loading of NaHMDS to 2 equiv. and extending the time to 12 h, a complete conversion of 1a was observed with 95% yield (88% isolated yield, entries 10–12).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yield (%) |
| a Reaction conditions: unless otherwise noted, reactions were conducted using 0.2 mmol of 2a, 2 equiv. of 1, 2 equiv. of 1a, 3 equiv. of base, and 1 ml solvent. NaHMDS 2 M in THF was used. b 4 equiv. of NaHMDS used. c 2 equiv. of NaHMDS used. Yields were determined by 1H NMR using mesitylene as the internal standard; the isolated yield indicated in parentheses. | ||||
| 1 | NaHMDS | MTBE | 4 | 40 |
| 2 | LiHMDS | MTBE | 4 | 28 |
| 3 | KHMDS | MTBE | 4 | 18 |
| 4 | NaHMDS | THF | 4 | 56 |
| 5 | NaHMDS | DME | 4 | 72 |
| 6 | NaHMDS | Toluene | 4 | 21 |
| 7 | NaHMDS | Dioxane | 4 | 44 |
| 8 | NaHMDS | Et2O | 4 | 22 |
| 9b | NaHMDS | DME | 4 | 68 |
| 10c | NaHMDS | DME | 4 | 38 |
| 11 | NaHMDS | DME | 12 | 95 (88) |
| 12 | NaHMDS | DME | 24 | 69 |
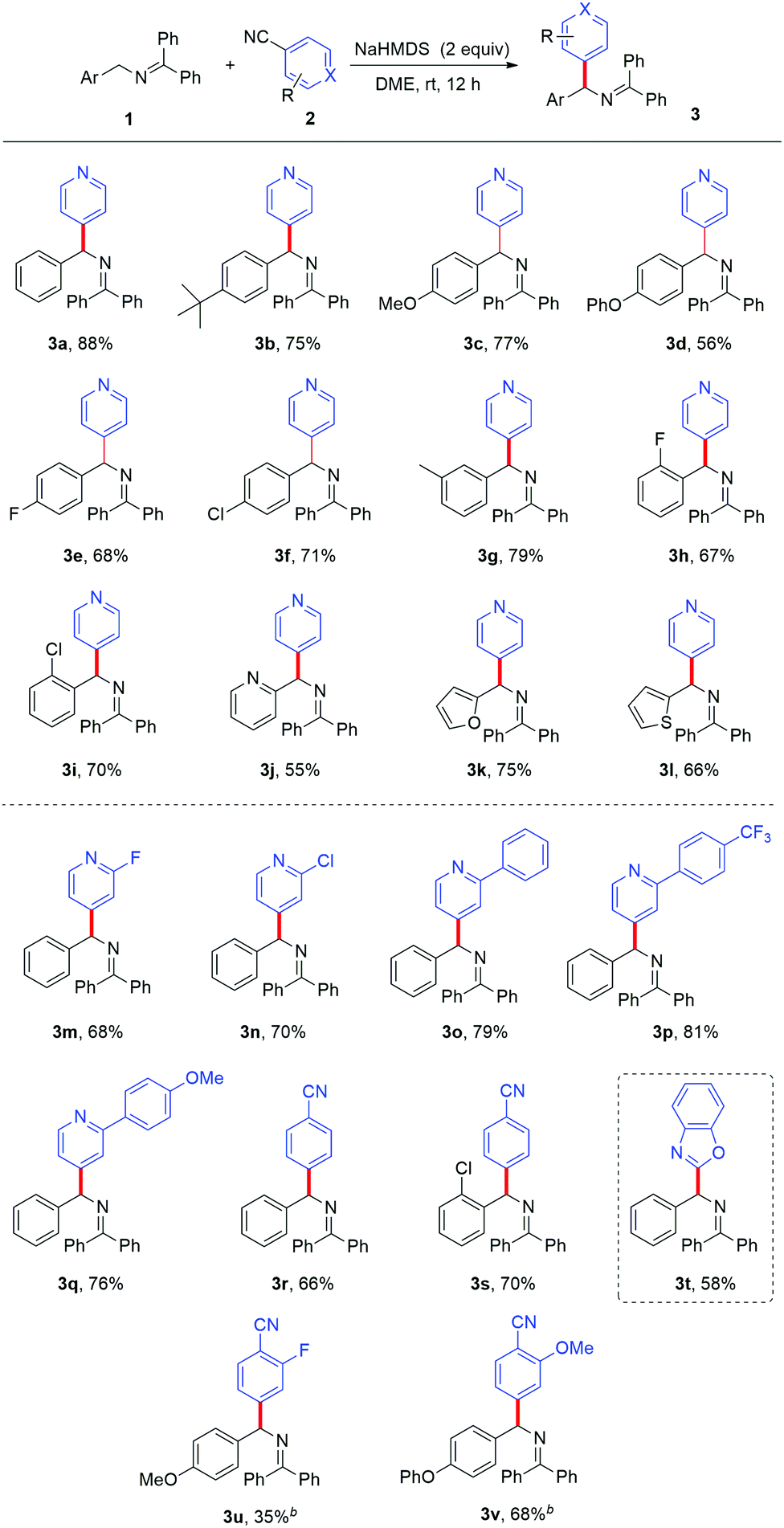
With the optimized reaction conditions in hand, we first studied the substrate scope with respect to the ketimine with different substituents. The substrates with -tert-Bu, -OMe, -OPh, -F, and -Cl at the para-position of the aryl ring all gave good results (3b–3f). The aromatic ketimines with -3-CH3, -2-F, and -2-Cl afforded compatible yields (3g–3i). It was worth noting that heteroaryl substituted substrates, such as 2-pyridyl, 2-furyl, and 2-thiophenyl ketimines, also underwent the reaction smoothly and afforded the corresponding products in good yields (3j–3l). Our attention then turned to the variation of the aromatic nitriles. Cyano-substituted pyridines with halide or phenyl substituents at the 2-position of pyridine were tolerated and afforded the corresponding heteroarylated products with good results (3m–3q). Unfortunately, 2-cyanopyridine and 3-cyanopyridine did not give the desired products, which might be attributed to the competing side-reaction under basic conditions.12 The cyanopyridine substrates were consumed in the system with complex by-products formed. The site specificity of the coupling process can be further exploited through the use of 1,2-dicyanobenzene, efficiently forming diarylmethylamines 3r and 3s in 66% and 70% yields respectively. The substrates with the –F (3u) and –OMe (3v) groups were also well tolerated. A simple chloride can function equivalently to –CN as a suitable leaving group; this enables heteroarylation of ketimines with chloro-substituted benzoxazole (3t) (Table 2).
To test the scalability of this protocol, we performed the gram-scale synthesis of a heterocyclic product (3k). Ketimine 1k was prepared from the corresponding furyl amine and benzophenone imine precursors and directly used for the next step without further purification. The radical arylation step was performed with 4-cyanopyridine with a standard process and afforded the desired product (3k) in 61% overall yield (1.03 g, Scheme 2, eqn (1)). Hydrolysis of 3s was conducted to afford 4-(amino(2-chlorophenyl)methyl)benzonitrile (4) in 80% yield.
 | ||
| Scheme 2 Synthetic applications. | ||
In order to gain some information on the reaction mechanism, radical inhibition experiments were carried out. When 2 equiv. of radical scavenger TEMPO (2,2,6,6-tetromethyl-1-piperidinyloxy) was added under the standard conditions, the reaction was completely suppressed (Scheme 3, eqn (1)). Addition of butylated hydroxytoluene (BHT) led to a dramatic decrease of the yield (eqn (2)). These results suggested that a radical pathway might be involved in the current reaction.
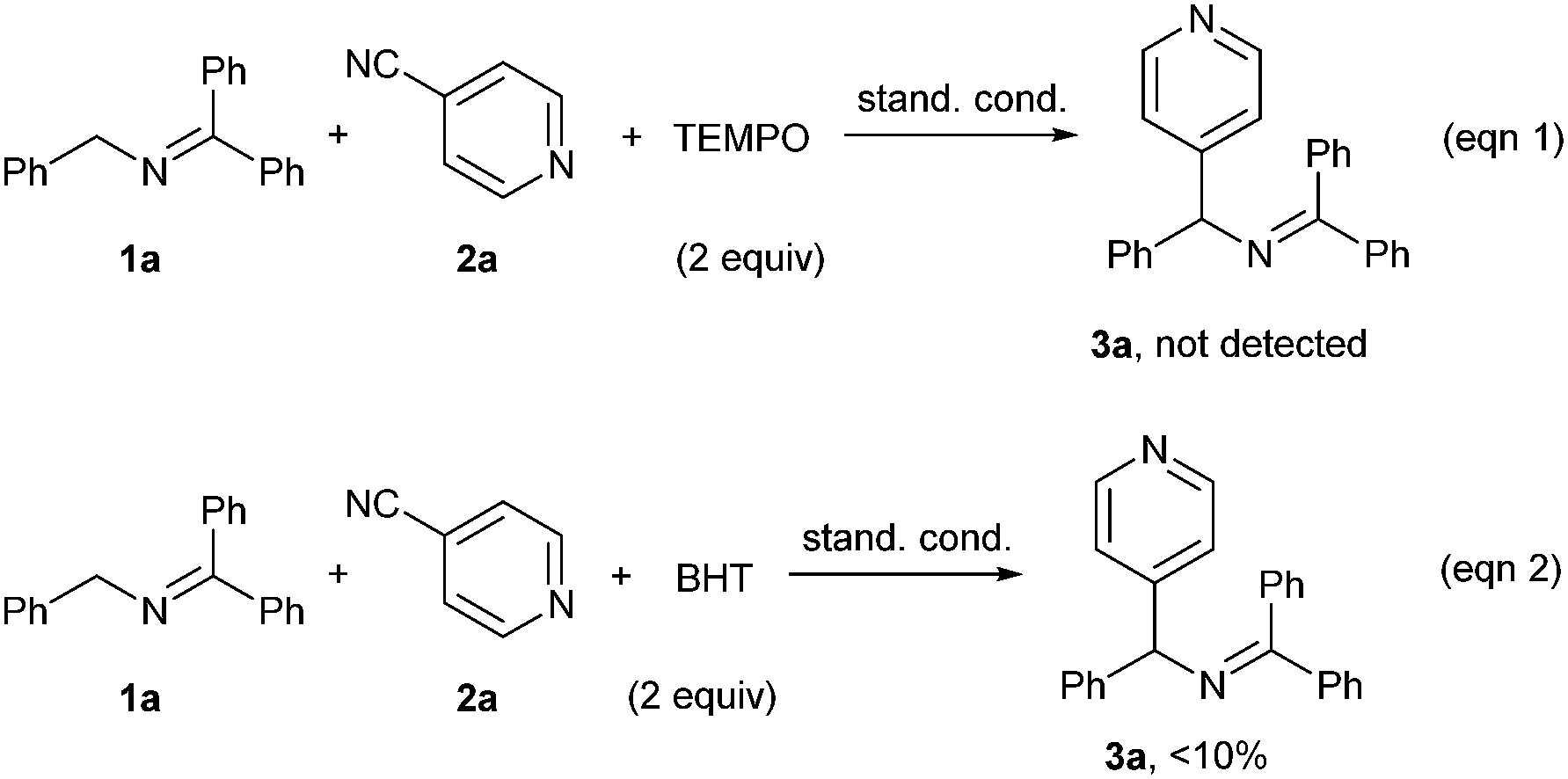 | ||
| Scheme 3 Mechanistic studies. | ||
Although the precise reaction mechanism remains to be clarified, guided by these supporting pieces of evidence and known literature reports,11 a plausible mechanism for the reaction is depicted in Scheme 4. At the beginning, the azaallyl anion A was formed after deprotonation of 2-azaallyl by NaHMDS, which could serve as an SED to donate an electron to nitriles and formed the corresponding arene radical anion C. In the meantime, 2-azaallyl radical B was simultaneously formed during this SET process. The subsequent coupling of the relatively long lived persistent 2-azaallyl radical B with the short lived transient aromatic radical provided the desired arylation product owing to the persistent radical effect.13
 | ||
| Scheme 4 Proposed mechanism. | ||
In summary, we reported that organic SEDs (2-azaallyl anions) drove SET, which was able to activate aromatic nitriles for radical arylations without the assistance of photoirradiation. The reaction required only commercially available base and proceeded under mild conditions. The azaallyl based ketimine acts not only as an electron donor reagent but also a benzyl amine substrate, which facilitated the C(sp3)–H arylation of benzyl amines for the synthesis of diarylmethylamines under transition metal free conditions.
We are grateful for the grants from the NSFC (No. 21432003) and Fundamental Research Funds for the Central Universities (lzujbky-2017-127 and lzujbky-2018-k09).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Some selected examples: (a) M. Liu, M. S. Bryant, J. Chen, S. Lee, B. Yaremko, Z. Li, J. Dell, P. Lipari, M. Malkowski, N. Prioli, R. R. Rossman, W. A. Korfmacher, A. A. Nomeir, C. C. Lin, A. K. Mallams, R. J. Doll, J. J. Catino, V. M. Girijavallabhan, P. Kirschmeier and W. R. Bishop, Cancer Chemother. Pharmacol., 1998, 43, 50 CrossRef PubMed; (b) J.-H. Chern, K.-S. Shia, T.-A. Hsu, C.-L. Tai, C.-C. Lee, Y.-C. Lee, C.-S. Chang, S.-N. Tseng and S.-R. Shih, Bioorg. Med. Chem. Lett., 2004, 14, 2519 CrossRef CAS PubMed; (c) V. Summa, A. Petrocchi, V. G. Matassa, C. Gardelli, E. Muraglia, M. Rowley, O. G. Paz, R. Laufer, E. Monteagudo and P. Pace, J. Med. Chem., 2006, 49, 6646 CrossRef CAS PubMed; (d) H. Anagnostopulos, R. R. Bartlett, U. Elben and P. Stoll, Eur. J. Med. Chem., 1989, 24, 227 CrossRef CAS; (e) T. J. Ritchie, S. J. F. Macdonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2011, 16, 164 CrossRef CAS PubMed.
- Some selected examples: (a) T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 4689 CrossRef CAS PubMed; (b) T. Niwa, T. Suehiro, H. Yorimitsu and K. Oshima, Tetrahedron, 2009, 65, 5125 CrossRef CAS; (c) G. I. McGrew, J. Temaismithi, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2010, 49, 5541 CrossRef CAS PubMed; (d) G. I. McGrew, C. Stanciu, J. Zhang, P. J. Carroll, S. D. Dreher and P. J. Walsh, Angew. Chem., Int. Ed., 2012, 51, 11510 CrossRef CAS PubMed; (e) M. Li, S. Berritt and P. J. Walsh, Org. Lett., 2014, 16, 4312 CrossRef CAS PubMed; (f) B.-S. Kim, J. Jimenez, F. Gao and P. J. Walsh, Org. Lett., 2015, 17, 5788 CrossRef CAS PubMed; (g) J. A. FernandezSalas, E. Marelli and S. P. Nolan, Chem. Sci., 2015, 6, 4973 RSC; (h) M. Li, B. Yücel, J. Adrio, A. Bellomo and P. J. Walsh, Chem. Sci., 2014, 5, 2383 RSC.
- (a) B. Lipp, A. M. Nauth and T. Opatz, J. Org. Chem., 2016, 81, 6875 CrossRef CAS PubMed; (b) A. M. Nauth, A. Lipp, B. Lipp and T. Opatz, Eur. J. Org. Chem., 2017, 2099 CrossRef CAS; (c) B. Lipp, A. Lipp, H. Detert and T. Opatz, Org. Lett., 2017, 19, 2054 CrossRef CAS PubMed.
- (a) M. Andrew, K. P. Christopher and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef PubMed; (b) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS PubMed; (c) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74 CrossRef CAS PubMed.
- (a) J. A. Vega, J. M. Alonso, G. Mendez, M. Ciordia, F. Delgado and A. A. Trabanco, Org. Lett., 2017, 19, 938 CrossRef CAS PubMed; (b) C. Yan, L. Li, Y. Liu and Q. Wang, Org. Lett., 2016, 18, 4686 CrossRef CAS PubMed.
- T. Hoshikawa and M. Inoue, Chem. Sci., 2013, 4, 3118 RSC.
- Some selected examples: (a) S. S. Hanson, N. A. Richard and C. A. Dyker, Chem. – Eur. J., 2015, 21, 8052 CrossRef CAS PubMed; (b) S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236 CrossRef CAS PubMed; (c) E. Doni, S. O'Sullivan and J. A. Murphy, Angew. Chem., Int. Ed., 2013, 52, 2239 CrossRef CAS PubMed; (d) E. Doni, B. Mondal, S. O'Sullivan, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2013, 135, 10934 CrossRef CAS PubMed; (e) E. Cahard, F. Schoenebeck, J. Garnier, S. P. Y. Cutulic, S. Zhou and J. A. Murphy, Angew. Chem., Int. Ed., 2012, 51, 3673 CrossRef CAS PubMed.
- H. S. Farwaha, G. Bucher and J. A. Murphy, Org. Biomol. Chem., 2013, 11, 8073 RSC.
- (a) Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001 Search PubMed; (b) Electron Transfer Reactions in Organic Synthesis, ed. P. Vanelle, Research Signpost, Trivandrum, 2002 Search PubMed; (c) R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71 CrossRef CAS PubMed; (d) A. Houmam, Chem. Rev., 2008, 108, 2180 CrossRef CAS PubMed.
- (a) M. Rueping, P. Nikolaienko, Y. Lebedev and A. Adams, Green Chem., 2017, 19, 2571 RSC; (b) Y. Su, Y. Li, R. Ganguly and R. Kinjo, Chem. Sci., 2017, 8, 7419 RSC; (c) F. Cumine, S. Zhou, T. Tuttle and J. A. Murphy, Org. Biomol. Chem., 2017, 15, 3324 RSC; (d) Y. Liu, J. Xu, J. Zhang, X. Xu and Z. Jin, Org. Lett., 2017, 19, 5709 CrossRef CAS PubMed; (e) D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886 CrossRef CAS PubMed; (f) J. Broggi, M. Rollet, J. L. Clément, G. Canard, T. Terme, D. Gigmes and P. Vanelle, Angew. Chem., Int. Ed., 2016, 55, 5994 CrossRef CAS PubMed; (g) J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402 CrossRef CAS PubMed; (h) R. Rayala, A. Giuglio-Tonolo, J. Broggi, T. Terme, P. Vanelle, P. Theard, M. Médebielle and S. F. Wnuk, Tetrahedron, 2016, 72, 1969 CrossRef CAS PubMed.
- (a) M. Li, O. Gutierrez, S. Berritt, A. Pascual-Escudero, A. Yeşilçimen, X. Yang, J. Adrio, G. Huang, E. Nakamaru-Ogiso, M. C. Kozlowski and P. J. Walsh, Nat. Chem., 2017, 9, 997 CrossRef CAS PubMed; (b) M. Li, S. Berritt, L. Matuszewski, G. Deng, A. Pascual-Escudero, G. B. Panetti, M. Poznik, X. Yang, J. J. Chruma and P. J. Walsh, J. Am. Chem. Soc., 2017, 139(45), 16327 CrossRef CAS PubMed.
- N. G. R. Hearns, R. Clérac, M. Jennings and K. E. Preuss, Dalton Trans., 2009, 3193 RSC.
- (a) H. Fisher, J. Am. Chem. Soc., 1986, 108, 3925 CrossRef; (b) H. Fisher, Chem. Rev., 2001, 101, 3581 CrossRef; (c) A. Studer, Chem. – Eur. J., 2001, 7, 1159 CrossRef CAS PubMed; (d) K.-S. Focsaneanu and J. Scaiano, Helv. Chim. Acta, 2006, 89, 2473 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc06408c |
| This journal is © The Royal Society of Chemistry 2018 |