
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentifying reactive organo-selenium precursors in the synthesis of CdSe nanoplatelets†
Andreas
Riedinger
 ab,
Aniket S.
Mule
a,
Philippe N.
Knüsel
a,
Florian D.
Ott
a,
Aurelio A.
Rossinelli
a and
David J.
Norris
*a
ab,
Aniket S.
Mule
a,
Philippe N.
Knüsel
a,
Florian D.
Ott
a,
Aurelio A.
Rossinelli
a and
David J.
Norris
*a
aOptical Materials Engineering Laboratory, Department of Mechanical and Process Engineering, ETH Zurich, 8092 Zurich, Switzerland. E-mail: dnorris@ethz.ch
bMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
First published on 2nd October 2018
Abstract
In the synthesis of CdSe nanoplatelets, the selenium-to-selenide reduction pathway is unknown. We study solvent-free growth of CdSe nanoplatelets and identify bis(acyl) selenides as key reactive intermediates. Based on our findings, we prepare a series of bis(acyl) selenides that provide useful precursors with tailored reactivity for liquid-phase syntheses of nanoplatelets.
CdSe nanoplatelets (NPLs) are quasi-two-dimensional semiconductor nanocrystals with precise atomic-scale thickness.1–3 Due to their spectrally pure fluorescence,4–6 large absorption cross-sections,7 and enhanced energy-transfer rates,8 they have been heavily studied for applications.8–12 To obtain these materials, the standard synthesis involves heating long-chain cadmium carboxylates (e.g. cadmium myristate or cadmium oleate) with elemental selenium in a non-coordinating solvent (1-octadecene, ODE). At a certain temperature, cadmium acetate powder is introduced, triggering zincblende NPL formation.1,13,14 We have recently exploited a simple solvent-free route to unravel the growth mechanism that leads to the highly anisotropic shape of NPLs.15 However, the critical selenium reduction pathway in this process remains unclear. Understanding this chemistry can lead to further control of NPL growth and improved production of high-quality materials.
Our solvent-free route to CdSe NPLs involves heating a solid blend of cadmium propionate and elemental selenium. CdSe NPLs grow in the melt from cadmium propionate coordination polymers.15 We stress that the utility of this approach is not to obtain high-quality NPLs for applications but rather to study the underlying chemistry under simplified conditions. Here, we take advantage of this solvent-free reaction and investigate the reactive organo-chalcogenide intermediates that form by performing in situ thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and electron ionization mass spectrometry (EI-MS). Based on our results, we then synthesize a series of bis(acyl) selenide molecules at the multi-gram scale. The reactivity of these selenide precursors towards cadmium carboxylates can be tuned by molecular design. Thus, useful molecular precursors are obtained for the growth of CdSe NPLs.
When solid mixtures of cadmium carboxylate [e.g. Cd(propionate)2] and Se powder are heated to 200 °C under N2, CdSe NPLs are obtained along with CdO (Fig. 1). The same reaction with S powder yields analogous results for CdS NPLs. The presence of CdO is providing an important clue about the reaction mechanism. Our goal is to use this and other clues to identify the chalcogen reduction pathway in this reaction. Because Cd is already in its highest oxidation state, Se and S must be reduced by the only other reactant, the propionate (or its decomposition products). To determine the intermediates involved, we collected TGA and DSC data in situ while measuring EI-MS of volatile species (Fig. 2). We analyzed reactions with Cd(propionate)2 alone and mixed with equimolar amounts of S or Se. In all three cases, weight loss below 180 °C was negligible (Fig. 2a). Above this temperature, differences between Cd(propionate)2 alone or with Se or S were observed in DSC (Fig. 2b). In addition to the melting of Cd(propionate)2 at 180 °C, the samples with chalcogen showed melting of elemental Se (220 °C) and S (115 °C), respectively. These findings can be combined with the simultaneously recorded EI-MS data. In general, EI-MS of transition-metal propionates shows two distinct decomposition products: propionyl radicals, observed as propionyl cations with a molecular ion peak of M+ = 57, and 3-pentanone with M+ = 86.17,18 For Cd(propionate)2 alone, both species were observed below 250 °C. With Se or S, these were absent (see Fig. 2c and Fig. S1 in the ESI†).
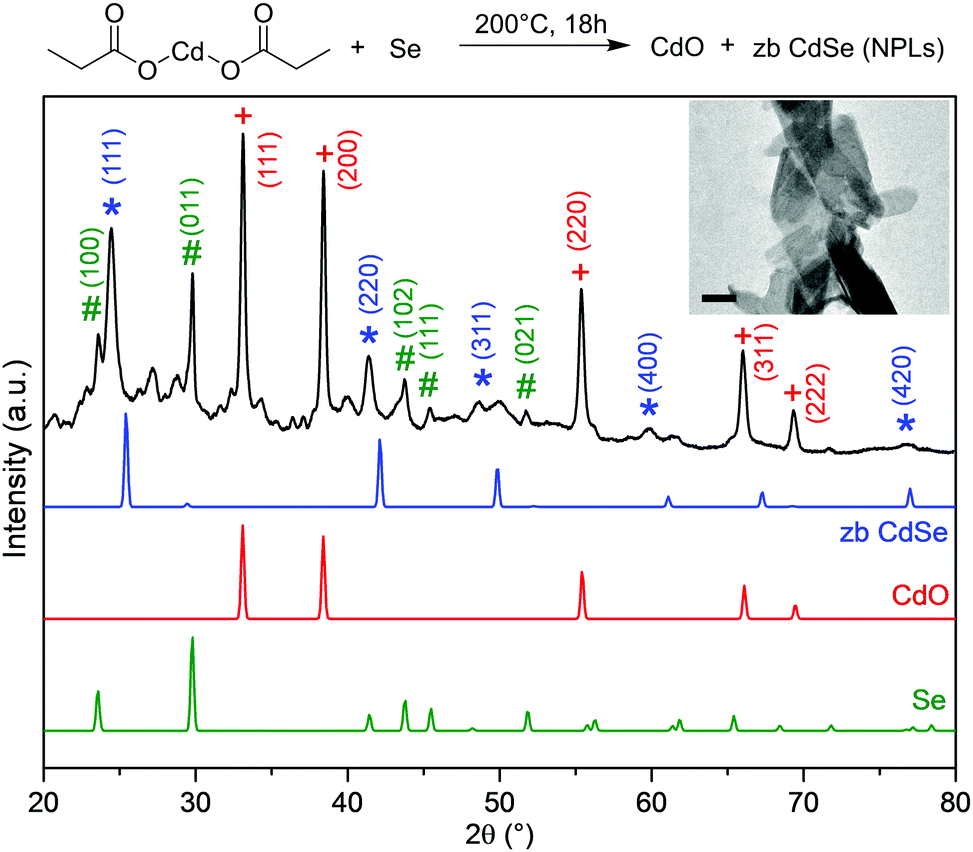 | ||
| Fig. 1 Heating cadmium propionate and Se powder to 200 °C for 18 h yields zincblende (zb) CdSe NPLs (inset: transmission electron micrograph, 100 nm scale bar). The powder X-ray diffraction pattern of the product is plotted (black). The reflections for CdSe NPLs are shifted to lower angles compared to bulk zb CdSe (blue, PDF: 01-088-2346 19-191).16 Other observed reflections are consistent with CdO (red, PDF: 01-073-2245 5-640) and Se (green, PDF: 01-083-2437 6-362). Several additional weak reflections are present presumably due to unreacted Cd(propionate)2 or Se in various modifications. Such phases can be present in the crude reaction product of the simple solvent-free reaction examined here. | ||
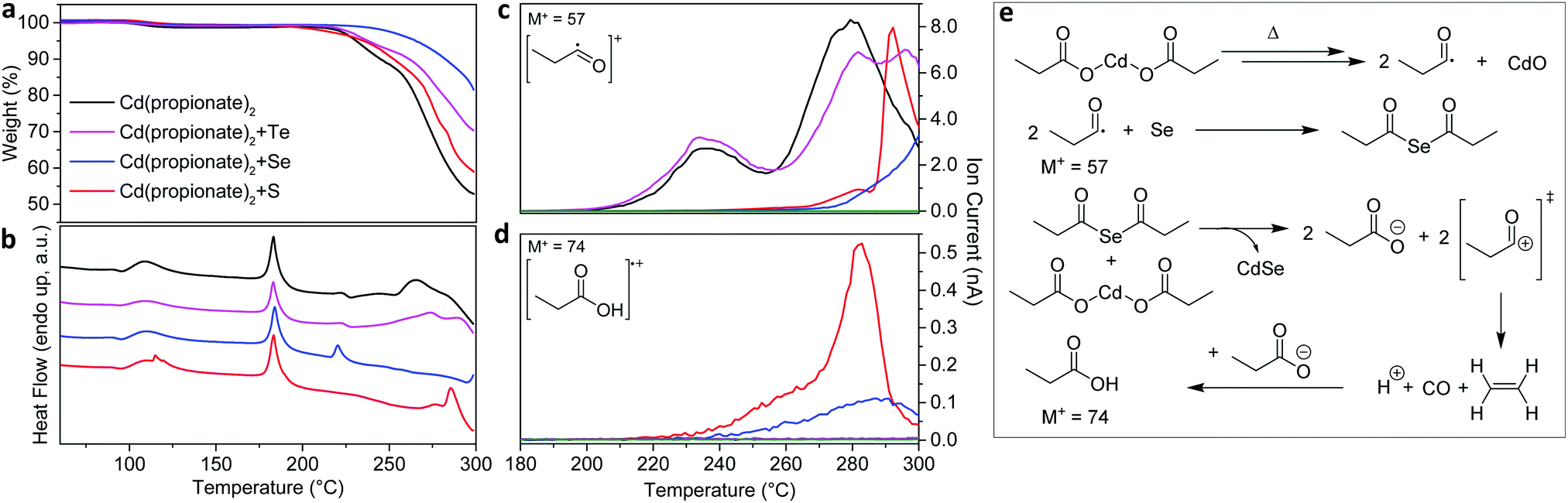 | ||
| Fig. 2 In situ analysis of the reactive chalcogenide species formed in melts of Cd(propionate)2. (a) Thermogravimetric analysis (TGA) and (b) differential scanning calorimetry (DSC) data for Cd(propionate)2 alone or with one equivalent of S, Se, or Te. Cd(propionate)2 with Se or S showed melting of Se (220 °C) or S (115 °C). The melting of Cd(propionate)2 occurred in all samples (180 °C). (c and d) During TGA/DSC measurements, volatile species were analyzed by electron-ionization mass spectrometry (EI-MS). Below 250 °C, propionyl radicals (M+, mass-to-charge ratio of 57) were detected only for Cd(propionate)2 alone or with Te (c). Release of propionic acid (M+ = 74) was observed only with Se or S (d). (e) The reaction mechanism consistent with TGA/DSC/EI-MS data. Decomposition of Cd(propionate)2 yields propionyl radicals, which are captured by Se (or S). The resulting bis(propionyl) selenide (sulfide) reacts with Cd(propionate)2 to produce CdSe (CdS) NPLs, releasing propionate anions and propionyl cations. The latter decompose to ethene, carbon monoxide, and protons. Free propionate can capture these protons. Indeed, propionic acid was detected only in samples containing Se (or S). See also Fig. S1 in the ESI.† | ||
These data lead to our proposed reaction mechanism for CdSe (or CdS) NPLs (Fig. 2e). The absence of propionyl radicals in the presence of Se (S) strongly suggests the formation of bis(propionyl) selenide (sulfide). These molecules can then react with Cd(propionate)2 to yield CdSe (CdS), releasing propionate anions and propionyl cations. The latter can decompose to carbon monoxide, ethene, and protons (Fig. 2e). These protons can then be captured by free propionate to yield propionic acid. Indeed, traces of this molecule were detected, but only with Se and S (M+ = 74, Fig. 2d). Thus, bis(propionyl) selenide (or sulfide) appears to be the reactive chalcogenide intermediate in NPL growth. Note that CdO, which was present in Fig. 1, is an expected by-product of the in situ formation of bis(propionyl) selenide (or sulfide) (Fig. 2e).
Fig. 2 also presents results for Te. However, the equimolar mixture of Cd(propionate)2 and Te behaves similarly to Cd(propionate)2 alone. This is consistent with the observation that when blends of Te powder and Cd(propionate)2 are heated to 200 °C, CdTe NPLs do not form. This may be due to the higher melting point of Te (450 °C). Unfortunately, the reaction temperature cannot simply be increased; Cd(propionate)2 starts to decompose above 200 °C (Fig. 2c). Thus, this synthetic approach cannot yield CdTe, because reactive Te compounds do not form. This is reflected in the mass trace of Cd(propionate)2 with Te, which closely resembles the trace of Cd(propionate)2 alone at useful reaction temperatures.
However, for Se and S, our work suggests that bis(acyl) selenides and sulfides are involved. More generally, they could be exploited as precursors in nanocrystal syntheses, where elemental Se or S is often combined with metal carboxylates. Recently, substituted thio and selenourea precursors with tailored reactivity were used to improve PbS and PbSe quantum dots.19,20 Libraries of bis(acyl) selenide or sulfide precursors could similarly allow for tailored reactivity. In contrast to substituted thio or selenourea, bis(acyl) chalcogenides yield only carbonyl byproducts after reaction with cadmium carboxylates. It is well known that the by-products of nanocrystal syntheses with thio or selenoureas (amines and most likely carbamides) can significantly influence growth.21,22 Hence, bis(acyl) selenides or sulfides should be better for syntheses where amines and carbamides have a negative impact. This is expected for CdSe and CdS NPL syntheses, which rely on phase separation of cadmium carboxylate coordination polymers.15 Amines and carbamides can break down these polymers23 and should be avoided.
To investigate the utility of bis(acyl) selenides as precursors, we adapted a known synthesis24,25 (Scheme 1) to obtain multiple grams of bis(acyl) selenides from a one-pot reaction. First, lithium aluminum hydride is reacted at −10 °C with elemental Se in tetrahydrofuran (THF) to form LiAlHSeH, a greyish solid. Without further purification, addition of acyl chlorides to this selenating agent yielded bis(acyl) selenides. We synthesized three different examples: bis(stearoyl) selenide (1), bis(benzoyl) selenide (2), and bis(4-chloro-benzoyl) selenide (3). After purification by recrystallization, we characterized the products by 1H, 13C, and 77Se nuclear magnetic resonance spectroscopy (NMR, see ESI†).
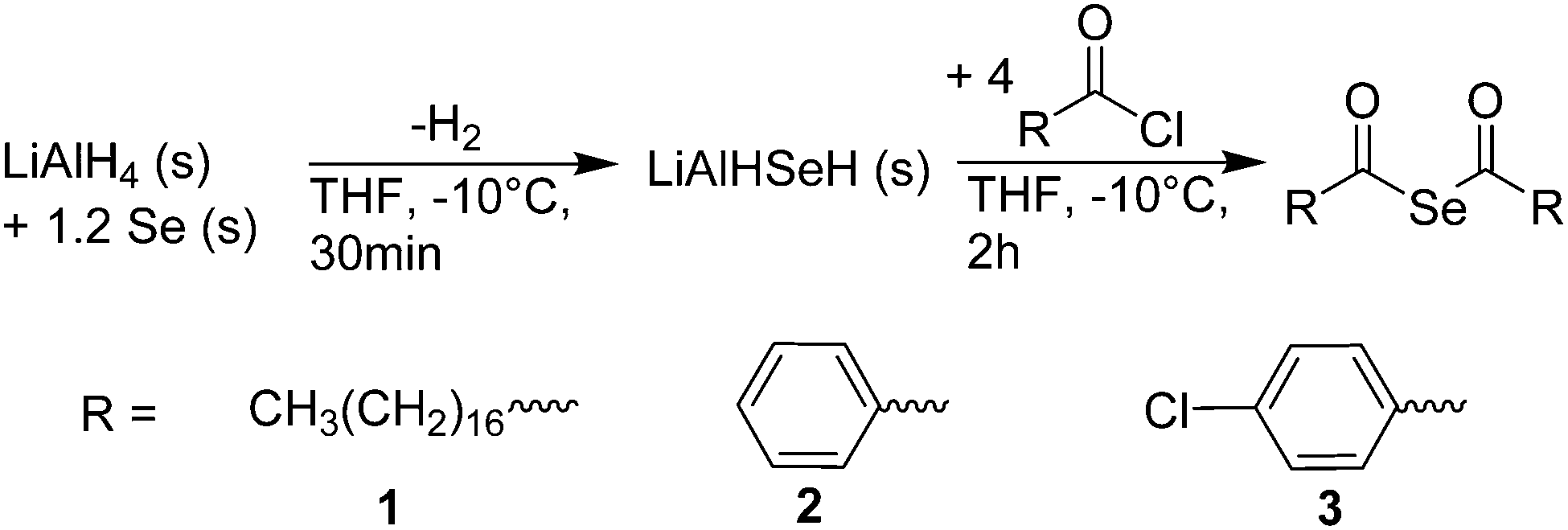 | ||
| Scheme 1 One-pot reaction for multi-gram synthesis of bis(acyl) selenides. | ||
As the reaction between bis(acyl) selenide and Cd(carboxylate)2 to yield CdSe is expected to be of the Lewis acid/base type,26,27 the Lewis basicity of the selenide precursor determines its reactivity. In aliphatic compound 1, only inductive electron pushing depolarizes the C–Se bond. In aromatic compounds 2 and 3, mesomeric effects cause further depolarization of this bond. Moreover, in compound 3, the lone-pair electrons of Cl are conjugated with the aromatic system resulting in a lower Lewis basicity compared to 2. Thus, the reactivity towards Cd(carboxylate)2 should decrease from 1 to 2 to 3. We confirmed this trend with a liquid-phase synthesis of quasi-spherical CdSe quantum dots (QDs). 1, 2, or 3 was combined with Cd(oleate)2 in ODE at 180 °C, as described in the ESI.† The resulting QD size (Fig. S2, ESI†) increased from 1 to 2 to 3, as expected from the predicted reactivity.20
We then tested our bis(acyl) selenide precursors in the liquid-phase reaction of CdSe NPLs. The goal was not to improve the quality of CdSe NPLs obtained, but to test if the reactivity of the selenide precursor influenced the nucleation and growth. Following classical nucleation theory, control over the nucleation process can be gained through changes in surface energy (γ), temperature (T), and supersaturation (S). In NPL syntheses, γ is given by the carboxylate surface passivation and largely fixed. Current NPL syntheses primarily control nucleation via T (i.e. changing the temperature at which the cadmium acetate is added). Here, we demonstrate that the precursor reactivity (via changes in S) also allows control over the nucleation process in NPL syntheses.
For this, Cd(myristate)2 in ODE was degassed at room temperature and heated under N2 to 240 °C. Acetic anhydride (1 eq.) was injected, and the mixture was allowed to stand for 1 min at 240 °C. Acetic acid, which forms in situ, reacts with Cd(myristate)2 to yield mixed coordination polymers Cd(myristate)2−x(acetate)x, which phase separate from ODE.15 Then, precursor 1, 2, or 3 was injected and the reaction was allowed to continue at 240 °C for 9 min, followed by quick cooling to room temperature. Fig. 3a plots the resulting absorption spectra of the unpurified reaction products. Fig. 3b and c show the corresponding transmission electron microscopy (TEM) images for 1 and 2, respectively. We intentionally avoided any size-selective purification to obtain a full picture of the reaction products.
 | ||
| Fig. 3 (a) Absorption spectra of CdSe NPLs synthesized by combining 1, 2, or 3 with Cd(myristate)2−x(acetate)x in ODE at 240 °C (aliquots after 10 min, measured without further purification). Precursor 1 yields 3 monolayer (ML) NPLs [heavy-hole optical transition (hh) at ∼460 nm], 2 yields 4 ML NPLs (hh at ∼510 nm), and 3 yields mostly quantum dots. (b and c) Transmission electron micrographs of the unpurified reaction product for NPLs synthesized with (b) 1 and (c) 2. Scale bars are 100 nm. We intentionally avoided any size-selective purification before analysis to see all products. XRD patterns for the products are shown in the ESI.† | ||
The product obtained correlates with the precursor reactivity. The most reactive precursor 1 yielded mostly thin 3-monolayer (ML) thick NPLs [heavy-hole optical transition (hh) at ∼460 nm].16 The less-reactive precursor 2 gave primarily 4 ML CdSe NPLs (hh at ∼510 nm).16 Precursor 3 was apparently not sufficiently reactive, yielding polydisperse CdSe QDs.
The spectra at early time points further clarify these observations (Fig. S3, ESI†). In the synthesis with 1, just traces of 4 ML NPLs are present after 2 min, while with 2 and 3, they are much more evident. This is seen from their absorption features at ∼510 nm. With 3 however, larger quantities of QDs are also observed early. As we have recently shown,15 thin NPLs grow faster than thicker NPLs but are less stable. Indeed, the thermodynamically most stable shape for CdSe nanocrystals are quasi-spherical QDs. Hence, in all three syntheses, 3 ML NPLs are expected to appear early but disappear due to Ostwald ripening as the reaction proceeds.28 The released [CdSe] monomers can redeposit either on other 3 ML NPLs (Ostwald ripening within a thickness population) or on thermodynamically more stable 4 ML NPL seeds or QDs. In the synthesis with 1, in which very little 4 ML NPLs or QDs are available, mainly lateral Ostwald ripening within a thickness population is observed.28 However, in the synthesis with 2, 4 ML NPL seeds are present in sufficient quantities that the monomers mainly deposit on them, leading after 9 min predominantly to 4 ML NPLs. In the synthesis with 3, even though 4 ML NPL seeds are present, QDs are the most abundant species. Over time, the material transferred from NPLs to QDs only increases, driven by the high initial QD concentration. These results demonstrate effects on nucleation by tailoring the precursor reactivity via their molecular design.
In summary, we have identified bis(acyl) selenides (and sulfides) as reactive organo-selenium precursors for nanocrystal growth. We uncovered such species as intermediates during a detailed analysis of the solvent-free synthesis of CdSe (CdS) NPLs. While we focused here on the selenides, we note that some of the bis(acyl) sulfides are commercially available. In addition to the utility of these chalcogenide precursors in liquid-phase synthesis of semiconductor nanocrystals, such precursors can provide tailored reactivity for studying the fundamentals of nucleation and growth of these materials.
This work was supported by ETH Research Grant ETH-38 14-1 and by the Swiss National Science Foundation under Grant No. 200021-165559. We are grateful to H. Schönberg and H. Grützmacher for technical assistance and discussion of the TGA/DSC/EI-MS data. We thank V. Lin, K. McNeill, O. Waser, F. Starsich, and S. Pratsinis for help with the 77Se NMR and XRD measurements and K. Boldt for stimulating discussions. We utilized facilities at the Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zurich.
Conflicts of interest
The authors declare no competing financial interests.Notes and references
- S. Ithurria and B. Dubertret, J. Am. Chem. Soc., 2008, 130, 16504–16505 CrossRef CAS PubMed.
- J. Joo, J. S. Son, S. G. Kwon, J. H. Yu and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 5632–5633 CrossRef CAS PubMed.
- Y.-H. Liu, F. Wang, Y. Wang, P. C. Gibbons and W. E. Buhro, J. Am. Chem. Soc., 2011, 133, 17005–17013 CrossRef CAS PubMed.
- A. W. Achtstein, A. Schliwa, A. Prudnikau, M. Hardzei, M. V. Artemyev, C. Thomsen and U. Woggon, Nano Lett., 2012, 12, 3151–3157 CrossRef CAS PubMed.
- S. Ithurria, M. D. Tessier, B. Mahler, R. Lobo, B. Dubertret and A. Efros, Nat. Mater., 2011, 10, 936–941 CrossRef CAS PubMed.
- M. Olutas, B. Guzelturk, Y. Kelestemur, A. Yeltik, S. Delikanli and H. V. Demir, ACS Nano, 2015, 9, 5041–5050 CrossRef CAS PubMed.
- A. Yeltik, S. Delikanli, M. Olutas, Y. Kelestemur, B. Guzelturk and H. V. Demir, J. Phys. Chem. C, 2015, 119, 26768–26775 CrossRef CAS.
- C. E. Rowland, I. Fedin, H. Zhang, S. K. Gray, A. O. Govorov, D. V. Talapin and R. D. Schaller, Nat. Mater., 2015, 14, 484–489 CrossRef CAS PubMed.
- J. Q. Grim, S. Christodoulou, F. Di Stasio, R. Krahne, R. Cingolani, L. Manna and I. Moreels, Nat. Nanotechnol., 2014, 9, 891–895 CrossRef CAS PubMed.
- C. She, I. Fedin, D. S. Dolzhnikov, A. Demortière, R. D. Schaller, M. Pelton and D. V. Talapin, Nano Lett., 2014, 14, 2772–2777 CrossRef CAS PubMed.
- E. Lhuillier, S. Pedetti, S. Ithurria, H. Heuclin, B. Nadal, A. Robin, G. Patriarche, N. Lequeux and B. Dubertret, ACS Nano, 2014, 8, 3813–3820 CrossRef CAS PubMed.
- Z. Chen, B. Nadal, B. Mahler, H. Aubin and B. Dubertret, Adv. Funct. Mater., 2014, 24, 295–302 CrossRef CAS.
- S. Ithurria, G. Bousquet and B. Dubertret, J. Am. Chem. Soc., 2011, 133, 3070–3077 CrossRef CAS PubMed.
- B. Mahler, B. Nadal, C. Bouet, G. Patriarche and B. Dubertret, J. Am. Chem. Soc., 2012, 134, 18591–18598 CrossRef CAS PubMed.
- A. Riedinger, F. D. Ott, A. Mule, S. Mazzotti, P. N. Knüsel, S. J. Kress, F. Prins, S. C. Erwin and D. J. Norris, Nat. Mater., 2017, 16, 743–748 CAS.
- D. Chen, Y. Gao, Y. Chen, Y. Ren and X. Peng, Nano Lett., 2015, 15, 4477–4482 CrossRef CAS PubMed.
- M. Nasui, R. B. Mos, T. Petrisor Jr, M. S. Gabor, R. A. Varga, L. Ciontea and T. Petrisor, J. Anal. Appl. Pyrolysis, 2011, 92, 439–444 CrossRef CAS.
- J. P. Kercher, E. A. Fogleman, H. Koizumi, B. Sztáray and T. Baer, J. Phys. Chem. A, 2005, 109, 939–946 CrossRef CAS PubMed.
- M. P. Campos, M. P. Hendricks, A. N. Beecher, W. Walravens, R. A. Swain, G. T. Cleveland, Z. Hens, M. Y. Sfeir and J. S. Owen, J. Am. Chem. Soc., 2017, 139, 2296–2305 CrossRef CAS PubMed.
- M. P. Hendricks, M. P. Campos, G. T. Cleveland, I. Jen-La Plante and J. S. Owen, Science, 2015, 348, 1226–1230 CrossRef CAS PubMed.
- T. Qi, H.-Q. Yang, D. M. Whitfield, K. Yu and C.-W. Hu, J. Phys. Chem. A, 2016, 120, 918–931 CrossRef CAS PubMed.
- Z. H. Sun, H. Oyanagi, H. Nakamura, Y. Jiang, L. Zhang, M. Uehara, K. Yamashita, A. Fukano and H. Maeda, J. Phys. Chem. C, 2010, 114, 10126–10131 CrossRef CAS.
- R. Garcia-Rodriguez and H. Liu, Chem. Commun., 2013, 49, 7857–7859 RSC.
- H. Ishihara, M. Koketsu, Y. Fukuta and F. Nada, J. Am. Chem. Soc., 2001, 123, 8408–8409 CrossRef CAS PubMed.
- M. Koketsu, F. Nada, S. Hiramatsu and H. Ishihara, J. Chem. Soc., Perkin Trans. 1, 2002, 737–740 RSC.
- J. S. Owen, E. M. Chan, H. T. Liu and A. P. Alivisatos, J. Am. Chem. Soc., 2010, 132, 18206–18213 CrossRef CAS PubMed.
- H. T. Liu, J. S. Owen and A. P. Alivisatos, J. Am. Chem. Soc., 2007, 129, 305–312 CrossRef CAS PubMed.
- F. D. Ott, A. Riedinger, D. R. Ochsenbein, P. N. Knüsel, S. C. Erwin, M. Mazzotti and D. J. Norris, Nano Lett., 2017, 17, 6870–6877 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc06326e |
| This journal is © The Royal Society of Chemistry 2018 |