
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of new cyclic analogues of the antimicrobial lipopeptide tridecaptin A1†
Ross D.
Ballantine
 a,
Yong-Xin
Li
b,
Pei-Yuan
Qian
b and
Stephen A.
Cochrane
*a
a,
Yong-Xin
Li
b,
Pei-Yuan
Qian
b and
Stephen A.
Cochrane
*a
aSchool of Chemistry and Chemical Engineering, David Keir Building, Stranmillis Road, Queen's University Belfast, Belfast BT9 5AG, UK. E-mail: s.cochrane@qub.ac.uk
bDepartment of Ocean Science and Division of Life Science, Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong, China
First published on 4th September 2018
Abstract
Non-ribosomal peptides (NRPs) are a rich source of antibiotic candidates. However, it was recently discovered that resistance to NRPs can be mediated by D-stereoselective peptidases. The tridecaptins, a class of NRPs that selectively target Gram-negative bacteria, are degraded by the D-peptidase TriF. Through analysis of a solution NMR structure of tridecaptin A1, we have rationally synthesized new cyclic tridecaptin analogues that retain strong antimicrobial activity and are resistant to TriF.
Antimicrobial resistance is a major global concern. It has been estimated that if the current rise in infections resulting from multidrug resistant bacteria is not subdued, by 2050 they will cause more deaths than cancer.1 Due to the inherent ability of bacteria to develop resistance mechanisms, new antimicrobial compounds and targets will always be needed.2 In recent years, there has been a worrying lack of new antibiotics that target Gram-negative bacteria.3 These pathogens have an extra layer of protection in the form of the outer-membrane, which precludes the entry of many large-scaffold antibiotics.4 Several of the Gram-negative-targeting antibiotics currently undergoing clinical trials are derivatives of known classes (e.g. β-lactams/β-lactamase inhibitors) and could therefore be more susceptible to resistance development.5 Non-ribosomal peptides (NRPs) are becoming increasingly important in the fight against MDR bacteria, with many new classes being discovered in recent years.6D-Amino acid-containing NRPs (DNRPs) are often resistant to peptidases as the vast majority of peptidases only cleave L-peptide bonds. However, recent studies have identified the widespread distribution of D-stereoselective peptidases. For example, the D-peptidase BogQ can degrade the DNRPs bacitracin, rampoplanin and daptomycin, all of which are clinically used antibiotics. Therefore, D-peptidases could pose a major threat to the longevity of NRP antibiotics.7
The tridecaptins are a class of linear DNRPs isolated from Bacillus and Paenibacillus species.8 Tridecaptin A1 (TriA1) is the archetypical member of this class and shows strong activity against Gram-negative bacteria, including multidrug resistant (MDR) strains of Escherichia coli, Klebsiella pneumoniae and Acinetobacter baumannii (Fig. 1A).9 TriA1 exerts its bactericidal effect by binding to lipid II on the inner-membrane and disrupting the proton-motive force.10 The interaction between TriA1 and lipid II imbedded in dodecylphosphocholine (DPC) micelles was recently studied by NMR.10 In the absence of lipid II, TriA1 adopts a tight hairpin-like amphiphilic structure, however a more open looped structure is adopted upon lipid II binding (Fig. 1B). Analysis of this structure suggests that the loop is stabilized by a π-stacking interaction between D-Trp5 and L-Phe9. A previously reported alanine scan of TriA1 corroborates the importance of these residues, as substitution of either D-Trp5 or L-Phe9 significantly decreases antimicrobial activity.11
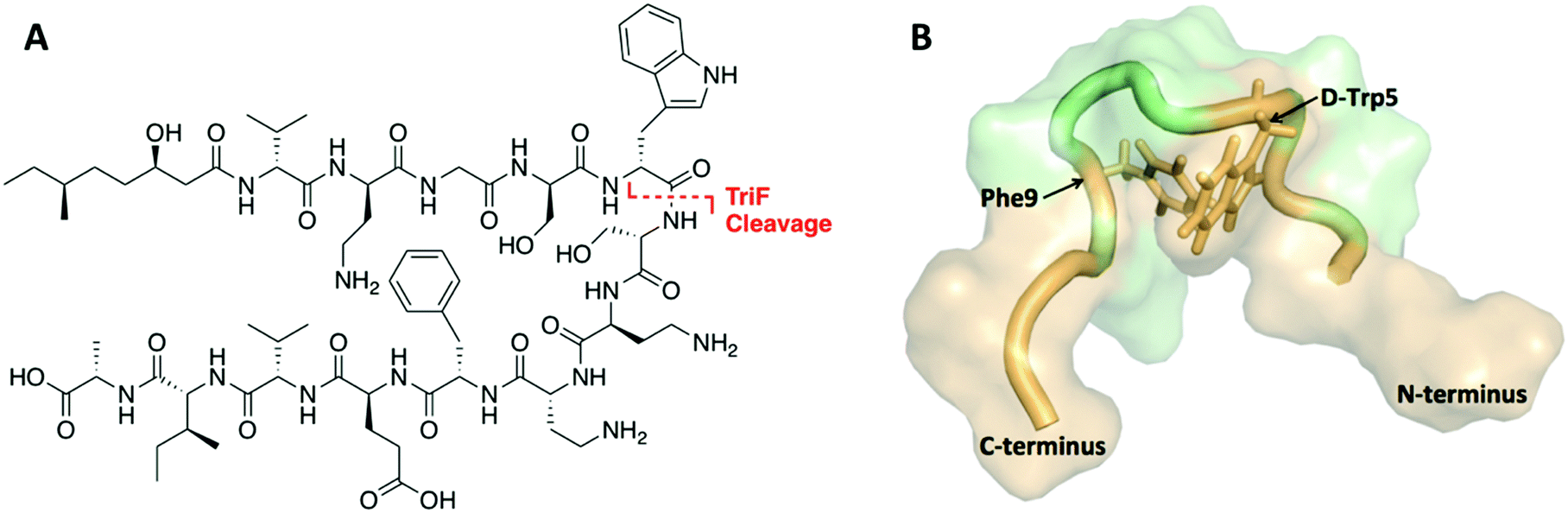 | ||
| Fig. 1 (A) Structure of TriA1 with TriF cleavage site shown in red. (B) NMR solution structure of TriA1 bound to lipid II (not shown) in DPC micelles (PDB ID: 2N5W). A π-stacking interaction that may stabilize the looped structure is shown. | ||
The tridecaptins are attractive antibiotic candidates, owing to their selective activity against Gram-negative bacteria and ease of preparation by solid-phase peptide synthesis (SPPS). However, it was recently discovered that the tridecaptins are hydrolytically cleaved by the D-peptidase TriF at the amide bond between D-Trp5 and Ser6, rendering them inactive.7 Although this is likely a self-protection mechanism, similar resistance mechanisms could develop in more pathogenic bacteria, rendering these DNRPs inactive. Macrocyclization is often an effective strategy to improve the stability of peptides towards peptidases.12 However, N to C cyclization, which is one of the most commonly used methods to cyclize peptides, is not appropriate for the tridecaptins. Firstly, they are N-acylated, making N to C cyclization more difficult. Secondly and most importantly, the lipid II-bound conformation of TriA1 places the N- and C-termini far apart as it wraps around lipid II on the cell membrane (Fig. 1B). Given that D-Trp5 and L-Phe9 are in close proximity through a π-stacking interaction, and that D-Trp5 is cleaved by TriF, we rationalized that replacement of this π-stacking interaction with a covalent linkage could impart resistance to TriF, as well as providing a new scaffold of macrocyclic peptides that specifically target Gram-negative bacteria. The generation of cyclic TriA1 (cTriA1) analogues cyclized between these positions would also corroborate the importance of the looped secondary structure in the mechanism of action of this peptide. Herein, we discuss our synthesis of novel cyclic TriA1 analogues that retain strong antimicrobial activity and are resistant to the D-peptidase TriF.
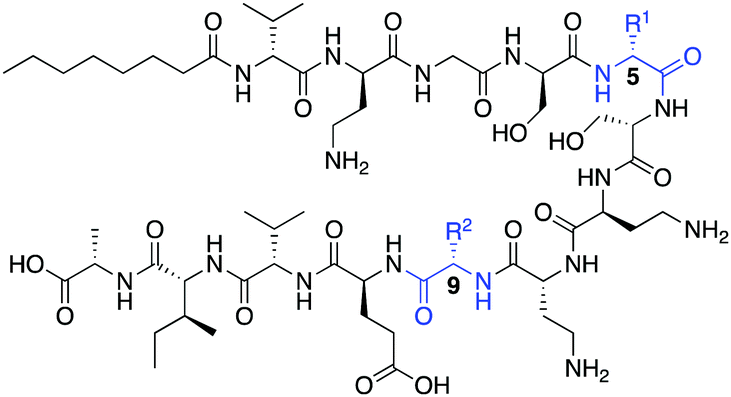
Oct-TriA1 (1) was first synthesized by Fmoc-SPPS and tested against a model Gram-negative (Escherichia coli) and Gram-positive (Staphylococcus aureus) indicator strain (Table 1). Replacement of the chiral lipid tail on TriA1 with octanoic acid has no effect on antimicrobial activity, therefore N-terminal octanoylation was employed for all analogues.9 Consistent with previous reports, Oct-TriA1 (1) showed potent activity against E. coli and weak activity against S. aureus. Our initial efforts to prepare cyclic TriA1 (cTriA1) analogues focused on replacing the D-Trp5-Phe9 π-stacking interaction with an alkene bridge using on-resin ring-closing metathesis (RCM). Derksen and Vederas previously used this strategy to replace a disulfide in the antimicrobial peptide leucocin A.13 Oct-TriA1-(5-D-Agl, 9-Agl) (2) was synthesized by SPPS, however, the full-length peptide proved refractory to on-resin RCM. We rationalized that the peptide might be aggregating, therefore we also attempted the cyclization at the nonapeptide stage of synthesis. However, this also failed. Further attempts involving increased catalyst loading, alternate solvents, increased reaction temperature or chaotropic salts13 were also unsuccessful. We postulated that extension of the alkene chains as S-allylcysteine (Sac) could facilitate cyclization, however exposure of Oct-TriA1-(5-D-Sac, 9-Sac) (3) to RCM conditions failed to yield any cyclic product. Neither alkene-containing peptide showed antimicrobial activity <50 μg mL−1, suggesting that hydrophobic interactions are not sufficient to stabilize the active conformation of TriA1. Previous studies have shown that not all peptides undergo RCM, with yields often highly dependent on peptide sequence.14 This situation often arises in peptides containing a large number of hydrophobic amino acids.15 Therefore, we believe that the significant hydrophobicity of the C-terminal region of Oct-TriA1 causes aggregation on-resin that hinders the RCM reaction. This limitation could be overcome in solution using an aqueous/organic solvent mix, as Oct-TriA1 is conformationally flexible in these solvent systems.11 However, as the vast excess of chaotropic salts required makes purification difficult,16 we directed our efforts towards an alternative cyclization method (Scheme 1).
|
|
||||
|---|---|---|---|---|
| Peptide | Amino acid | MICa | ||
| 5 | 9 | E. coli | S. aureus | |
| a MIC = minimum inhibitory concentration. Determined by microbroth dilutions assays and experiments run in duplicate. Values are shown to two significant figures and reported in μg mL−1. b Strain NCTC 12241. c Strain NCTC 10788. | ||||
| 1 | D-Trp | Phe | 0.39 | 25 |
| 2 | D-Agl | Agl | >50 | >50 |
| 3 | D-Sac | Sac | >50 | >50 |
| 4 | D-Cys | Cys | >50 | >50 |
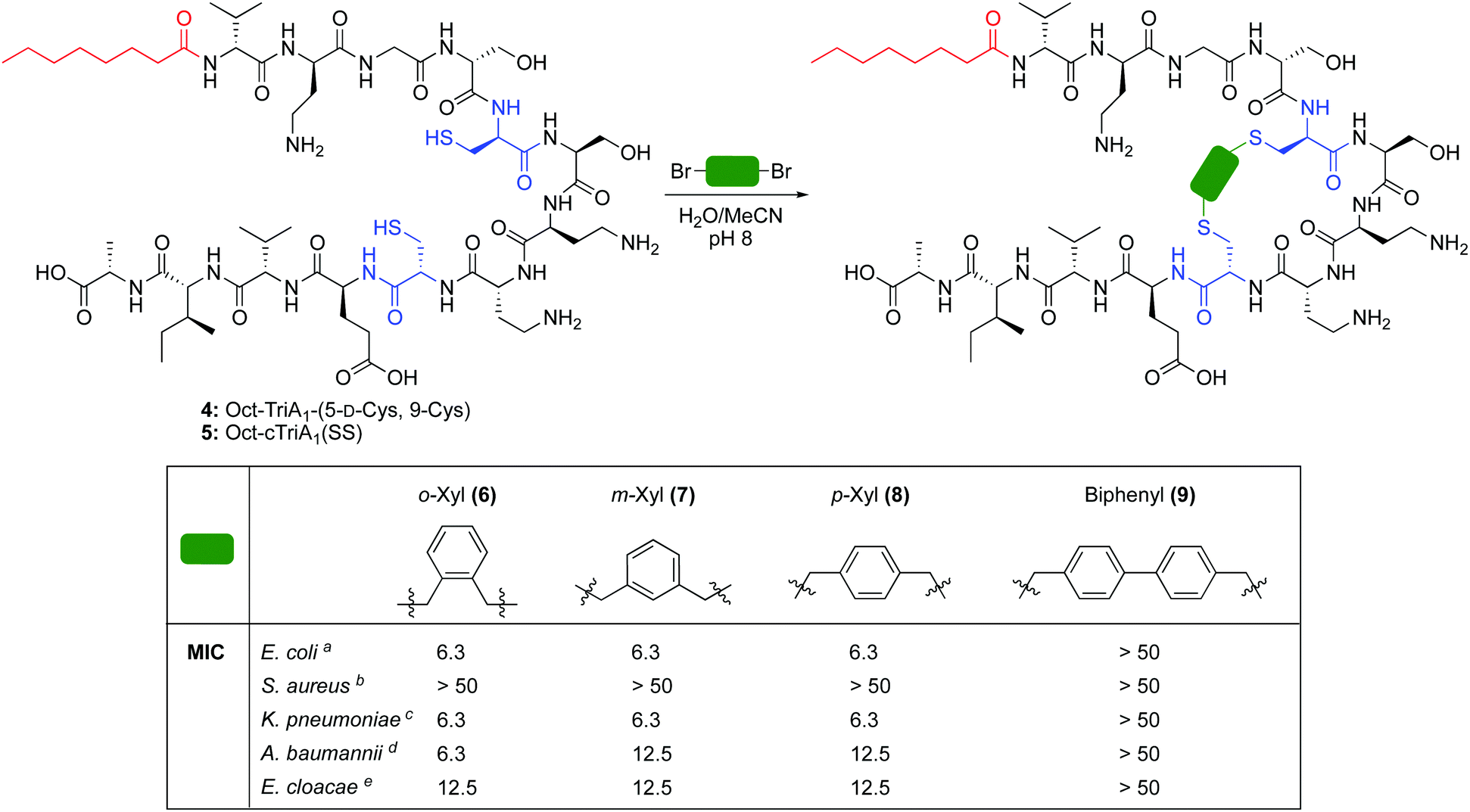 | ||
| Scheme 1 Synthesis of novel cyclic analogues of TriA1. MIC = minimum inhibitory concentration. Determined by microbroth dilutions assays and experiments run in duplicate. Values are shown to two significant figures and reported in μg mL−1. a Strain NCTC 12241. b Strain NCTC 10788. c Strain NCTC 9633. d Strain NCTC 13304. e Strain NCTC 5920. | ||
The cross-linking of cysteine residues is another method used to staple peptides,17 which has been used to prepare cell-penetrant inducers of autophagy18 and p53 inhibitors.19 In both studies the macrocycle ring size affected activity. Therefore, we directed our efforts towards the generation of cTriA1 analogues with varying ring-sizes. Oct-TriA1-(5-D-Cys, 9-Cys) (4) was first synthesized by SPPS. Next, disulfide 5, the smallest possible macrocycle, was synthesized from dithiol 4 but showed no antimicrobial activity. Dithiol 4 was then cross-linked with different benzylic cross-linkers to yield Oct-cTriA1 analogues 6–9, which contain increasingly larger macrocyclic rings. Peptides 6–8, wherein cysteines have been crosslinked with o-, m- and p-Xyl linkers respectively, retained strong activity against E. coli (6.3 μg mL−1), although none were active against S. aureus. Further ring expansion using a biphenyl linker yielded peptide 9, however this peptide showed no activity at the highest concentrations tested (50 μg mL−1). Encouraged by these results, we tested the activity of peptides 6–9 against more clinically significant Gram-negative bacteria, including Klebsiella pneumoniae, and the critical tier pathogens Acinetobacter baumannii and Enterobacter cloacae.20 Gratifyingly, peptides 6–8 showed strong activity against all strains. Activity against A. baumannii NCTC 13304, which is a carbapenem resistant strain,21 is particularly promising as this is currently the WHO's No. 1 priority pathogen.20 We next attempted to synthesize cTriA1 analogues containing saturated hydrocarbon cross-links. Performing these reactions under the same conditions did not yield any cyclic product, with just linear starting material recovered. Increasing reaction temperatures thermally, or by microwave irradiation, lead to the formation of several side-products, including linear peptides in which one of the cysteine residues had been converted to dehydroalanine. This likely occurs through bisalkylation of a single Cys, followed by elimination of the resulting sulfonium. We therefore moved forward with TriA1 analogues 1–9 to study their susceptibility to degradation by the D-peptidase TriF.
The D-stereoselective peptidase TriF is a membrane associated protein found in Paenibacillus polymyxa CICC 10580.7 TriFpep, the soluble periplasmic peptidase domain of TriF, which lacks the signal peptide and four hydrophobic transmembrane helices, was expressed in E. coli BL21 cells as a C-terminal His6-tag protein construct.7 TriA1 analogues 1–9 were incubated with TriFpep at 37 °C for 12 h and the reaction mixtures analysed by UPLC-MS. Both Oct-TriA1 (1) and Oct-TriA1(5-D-Sac, 9-Sac) (3) were degraded by TriFpep (Fig. 2 and Fig. S1, ESI†), whereas Cys analogue 4 and cTriA1 analogues 5–8 were completely resistant (Fig. 2). We were unable to test the stability of Oct-cTriA1(biphenyl) (9) as it is insoluble in the TriF assay mixture. Although 4 and 5 are resistant to TriF, they also have negligible antimicrobial activity. In contrast, peptides 6–8, which have been crosslinked with Xyl linkers, retain strong antimicrobial activity and are resistant to TriF. These analogues therefore represent a new scaffold of macrocyclic peptides with selective activity against Gram-negative bacteria. Furthermore, this work highlights the importance of the π-stacking interaction to TriA1's mechanism of action. Improvements in the antimicrobial activity of the cTriA1 analogues should be possible through further structure–activity relationship studies.
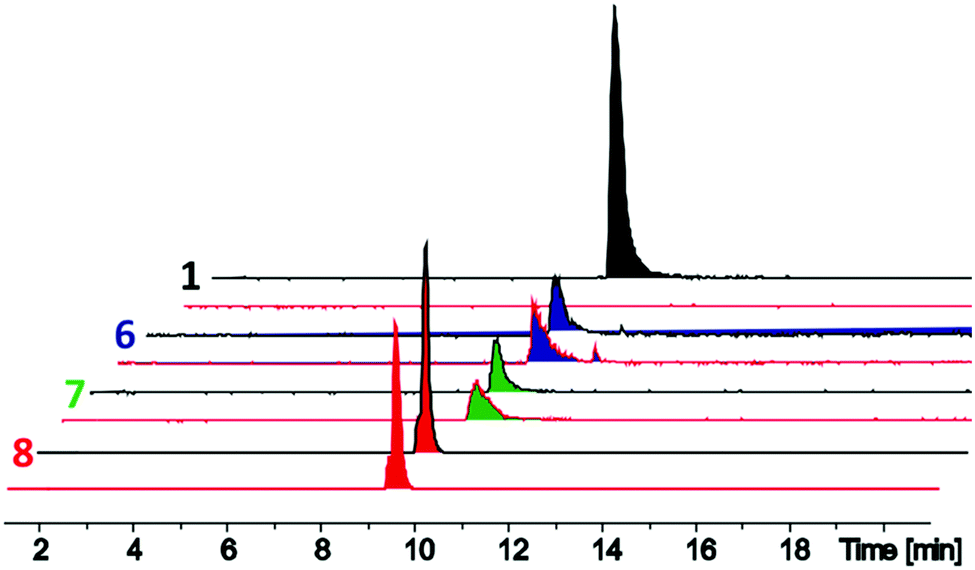 | ||
| Fig. 2 Liquid chromatography-mass spectrometry traces of in vitro assays of TriFpep against Oct-TriA1 (1, black peaks) and o-Xyl- (6, blue peaks), m-Xyl- (7, green peaks) and p-Xyl- (8, red peaks) crosslinked peptides. Standards (black line) without TriFpep and experiments with TriFpep (red line) are shown. Whereas Oct-TriA1 is cleaved by TriFpep, no corresponding cleavage was observed in TriFpep treated 6–8. | ||
In conclusion, we have employed a rational design approach to generate new cyclic analogues of tridecaptin A1 that are resistant to the D-peptidase TriF. Analysis of an NMR solution structure of TriA1 identified a possible cyclization point between positions 5 and 9, which are in close proximity due to a π-stacking interaction. Substitution of D-Trp5 and L-Phe-9 with D-Cys and L-Cys respectively, followed by cross-linking with benzylic di-bromo linkers yielded cyclic TriA1 analogues that retain selective activity against Gram-negative bacteria and are resistant to TriF. To the best of our knowledge, the replacement of π-stacking interactions with a covalent linkage to impart peptidase stability is a novel strategy. These structures constitute a new class of Gram-negative-targeting macrocyclic peptides and could be the basis for new antibiotic candidates.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. O’Neill, Review on Antimicrobial Resistance, 2014 Search PubMed.
- L. J. V. Piddock, Nat. Rev. Microbiol., 2017, 15, 639–640 CrossRef PubMed.
- World Health Organization, Antibacterial Agents in Clinical Development – An analysis of the antibacterial clinical development pipeline, including tuberculosis, Sept 2017.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef PubMed.
- The PEW Charitable Trusts, Antibiotics Currently in Global Clinical Development, Feb 2018.
- (a) B. M. Hover, S. H. Kim, M. Katz, Z. Charlop-Powers, J. G. Owen, M. A. Ternei, J. Maniko, A. B. Estrela, H. Molina, S. Park, D. S. Perlin and S. F. Brady, Nat. Microbiol., 2018, 3, 415–422 CrossRef PubMed; (b) P. 't Hart, T. M. Wood, K. H. M. E. Tehrani, R. M. van Harten, M. Śleszyńska, I. Rentero Rebollo, A. P. A. Hendrickx, R. J. L. Willems, E. Breukink and N. I. Martin, Chem. Sci., 2017, 8, 7991–7997 RSC; (c) A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brötz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511–516 CrossRef PubMed; (d) J. Chu, X. Vila-Farres, D. Inoyama, M. Ternei, L. J. Cohen, E. A. Gordon, B. V. Reddy, Z. Charlop-Powers, H. A. Zebroski, R. Gallardo-Macias, M. Jaskowski, S. Satish, S. Park, D. S. Perlin, J. S. Freundlich and S. F. Brady, Nat. Chem. Biol., 2016, 12, 1004–1006 CrossRef PubMed; (e) S. Cociancich, A. Pesic, D. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noëll, I. Pieretti, M. Hügelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Süssmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef PubMed.
- Y. X. Li, Z. Zhong, P. Hou, W. P. Zhang and P. Y. Qian, Nat. Chem. Biol., 2018, 14, 381–387 CrossRef PubMed.
- (a) S. A. Cochrane, C. T. Lohans, M. J. van Belkum, M. A. Bels and J. C. Vederas, Org. Biomol. Chem., 2015, 13, 6073–6081 RSC; (b) C. T. Lohans, M. J. van Belkum, S. A. Cochrane, Z. Huang, C. S. Sit, L. M. McMullen and J. C. Vederas, ChemBioChem, 2014, 15, 243–249 CrossRef PubMed; (c) T. Kato, R. Sakazaki, H. Hinoo and J. Shoji, J. Antibiot., 1979, 32, 305–312 CrossRef PubMed; (d) T. Kato, H. Hinoo and J. Shoji, J. Antibiot., 1978, 31, 652–661 CrossRef PubMed.
- S. A. Cochrane, C. T. Lohans, J. R. Brandelli, G. Mulvey, G. D. Armstrong and J. C. Vederas, J. Med. Chem., 2014, 57, 1127–1131 CrossRef PubMed.
- S. A. Cochrane, B. Findlay, A. Bakhtiary, J. Z. Acedo, E. M. Rodriguez-Lopez, P. Mercier and J. C. Vederas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11561–11566 CrossRef PubMed.
- S. A. Cochrane, B. Findlay, J. C. Vederas and E. S. Ratemi, ChemBioChem, 2014, 15, 1295–1299 CrossRef PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef PubMed.
- D. J. Derksen, J. L. Stymiest and J. C. Vederas, J. Am. Chem. Soc., 2006, 128, 14252–14253 CrossRef PubMed.
- B. J. van Lierop, A. N. Whelan, S. Andrikopoulos, R. Mulder, W. R. Jackson and A. J. Robinson, Int. J. Pept. Res. Ther., 2010, 16, 133–144 CrossRef.
- B. J. van Lierop, C. Bornschein, W. R. Jackson and A. J. Robinson, Aust. J. Chem., 2011, 64, 806–811 CrossRef.
- S. A. Cochrane, Z. Huang and J. C. Vederas, Org. Biomol. Chem., 2013, 11, 630–639 RSC.
- L. Peraro, T. R. Siegert and J. A. Kritzer, Methods Enzymol., 2016, 580, 303–332 Search PubMed.
- L. Peraro, Z. Zou, K. M. Makwana, A. E. Cummings, H. L. Ball, H. Yu, Y. S. Lin, B. Levine and J. A. Kritzer, J. Am. Chem. Soc., 2017, 139, 7792–7802 CrossRef PubMed.
- G. Gerona-Navarro, Y. Rodríguez, S. Mujtaba, A. Frasca, J. Patel, L. Zeng, A. N. Plotnikov, R. Osman and M. M. Zhou, J. Am. Chem. Soc., 2011, 133, 2040–2043 CrossRef PubMed.
- World Health Organization, Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics, Feb 2017.
- M. Afzal-Shah, N. Woodford and D. M. Livermore, Antimicrob. Agents Chemother., 2001, 45, 583–588 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc05790g |
| This journal is © The Royal Society of Chemistry 2018 |