
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocompatible metal-assisted C–C cross-coupling combined with biocatalytic chiral reductions in a concurrent tandem cascade†
Patricia
Schaaf
 a,
Thomas
Bayer
b,
Moumita
Koley
c,
Michael
Schnürch
a,
Uwe T.
Bornscheuer
b,
Florian
Rudroff
*a and
Marko D.
Mihovilovic
*a
a,
Thomas
Bayer
b,
Moumita
Koley
c,
Michael
Schnürch
a,
Uwe T.
Bornscheuer
b,
Florian
Rudroff
*a and
Marko D.
Mihovilovic
*a
aInstitute of Applied Synthetic Chemistry, 163-OC, TU Wien, 1060 Vienna, Austria. E-mail: florian.rudroff@tuwien.ac.at; marko.mihovilovic@tuwien.ac.at
bInstitute of Biochemistry, Dept. of Biotechnology & Enzyme Catalysis, Greifswald University, Felix-Hausdorff-Str. 4, 17487 Greifswald, Germany
cIndian Institute of Science, Bangalore-560012, India
First published on 1st November 2018
Abstract
In this study, we present a concurrent chemo/biocatalytic one pot reaction cascade by combining a metal (Pd/Cu) assisted Liebeskind–Srogl (L–S) coupling with an enantioselective enzymatic reduction for the production of chiral amines and alcohols. The latter transformation was realized by applying enantiocomplementary alcohol dehydrogenases from Lactobacillus kefir (R-selective) and Rhodococcus ruber (S-selective). Compatibility issues were solved by investigating first the L–S-coupling protocol in water at room temperature. Subsequently, we investigated two different biphasic systems and applied a biomimicking approach to separate enzyme-deactivating components. By using a lipophilic membrane in a smart reactor design, we were able to perform concurrent catalytic cascades with overall concentrations up to 100 mM substrate and to produce 1-phenylethylamine and several chiral alcohols in high yields (up to 81% over 2 steps) and enantiomeric purity ((+) and (−)-enantiomers both with 99% ee).
Cascade-type reactions have gained substantial attention in the fields of chemistry and biocatalysis in the past decade.1 Tremendous time and efforts have been devoted to the combination of various kinds of reactions and different types of catalysis to establish novel, sustainable, and environmentally benign methods.2–4 Especially interesting are combinations of metal assisted and biocatalytic reactions since their chemo-orthogonality offers high potential for accessing new, highly efficient, and stereoselectively controlled synthetic strategies.1,2,5–7 So far, only a few successful chemo- (especially metal assisted) and enzyme catalyzed concurrent reactions have been published.8–16 A major constraint of such combinations is the different conditions of chemical and enzymatic reactions leading prospectively to the mutual inactivation of the catalytic species. Whereas chemo-catalyzed reactions operate within a broad range of feasible reaction conditions such as temperature, solvent, and pH, enzyme mediated transformations are often restricted to aqueous reaction media, ambient temperature, a narrow pH range, and low substrate concentrations.17 Hence, concurrent tandem-type cascades, in which all reagents are present in the reaction mixture from the beginning, are even more challenging. In this particular case, the bond forming step or a functional group interconversion is a particular prerequisite to enable the subsequent step of the cascade. In addition, all involved catalytic species have to display exclusive chemo selectivity to avoid side reactions, which potentially reduce the overall productivity.2
Different concepts have been applied to overcome such limitations for the combination of metal assisted and biocatalytic tandem cascades. The most prominent concept is based on the spatial separation of both catalyst types and is inspired by nature. Biological compartmentalization allows living organisms to maintain different reaction conditions (e.g., pH) and operate numerous tandem reactions in a ‘one-pot’ system. In the past, chemists and engineers have imitated this concept with biphasic systems,5,18 supramolecular hosts,13 artificial metalloenzymes,14 and spatial separation by flow chemistry19 to realize and optimize multi-catalytic reactions.2 Lately, Ríos-Lombardía et al. demonstrated a ruthenium-catalysed isomerisation of allylic alcohols, coupled with an asymmetric bioamination in a one-pot process.15 Recently, the Gröger group developed a two-step one-pot transformation of styrenes into the corresponding chiral alcohols by combination of the Wacker oxidation with subsequent reduction by an alcohol dehydrogenase (ADH) from Lactobacillus kefir.18 However, to the best of our knowledge, only one chemoenzymatic tandem process in a concurrent fashion has been published, which does not require an intermediate work-up step of the chemocatalytic reaction mixture (e.g., dilution, adaption of the pH value, or separation of inhibiting components) prior to performing the biocatalytic transformation.20
In the following, we envisaged the simultaneous operation of a chemical C–C coupling reaction, which generates a new functional group that, subsequently, undergoes a biocatalytic functional group interconversion leading to enantiomerically pure products (Fig. 1). This work focused on different compartmentalization concepts to overcome the necessity of intermediate manipulation steps and tackled the compatibility issue of chemo- and enzyme catalysed reactions.
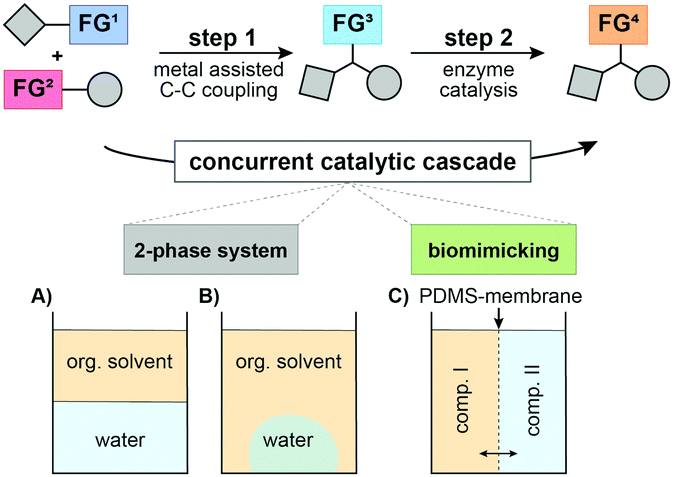 | ||
| Fig. 1 Concurrent chemo/biocatalytic cascade. Different engineering strategies were applied: (A) biphasic system (heptane/buffer); (B) heptane/superabsorber (sodium polyacrylate); and (C) polydimethylsiloxane (PDMS) membrane. | ||
In 2000, Liebeskind and Srogl (L–S) published a copper mediated, palladium catalyzed C–C cross-coupling protocol of thioesters (TEs) with boronic acids for the formation of ketones under aerobic conditions (Scheme 1).21–23 The method was later successfully extended to aryl coupling reactions based on the orthogonal behavior of the S-alkyl leaving group.24 This coupling reaction operates at neutral pH (base free conditions), in organic solvents (e.g., THF) and with a phosphite additive, activating the catalyst to work at room temperature.25 Recently, we reported the first example of a L–S reaction in water at 50 °C that led to bi(hetero)aryl products; while yields remained modest for this reaction, it opened up the principal applicability of the methods to operate in protic solvents.26
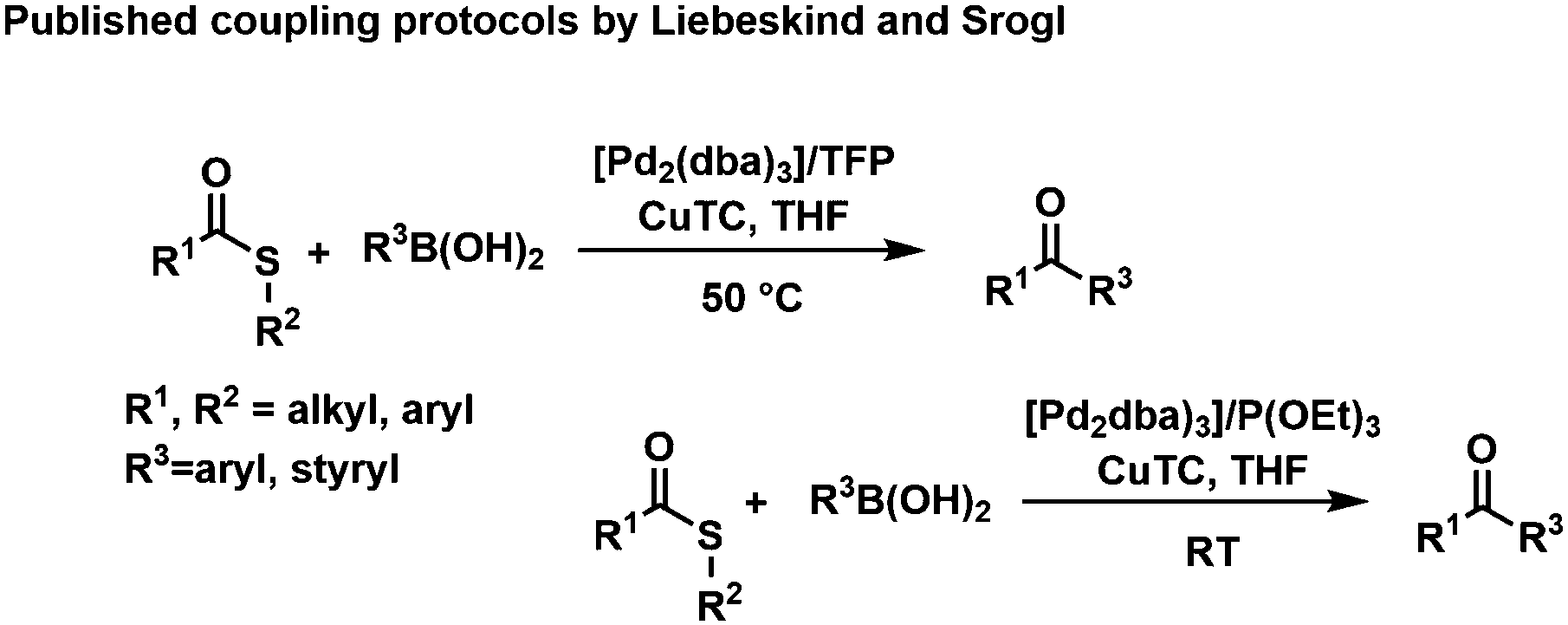 | ||
| Scheme 1 “Classical” L–S coupling (top) and optimized reaction conditions (bottom). | ||
Based on this finding we envisioned to exploit such aqueous conditions for the formation of ketones at room temperature, with the newly formed carbonyl group serving as a prospective substrate for redox biocatalysts (ADHs or ω-transaminases/ω-TAs) to produce chiral primary alcohols or amines, respectively (Scheme 2). According to this concept, intermediate formation of the new ketone function via L–S coupling of thioesters with boronic acids was expected to enable a true concomitant tandem transformation in combination with suitable biocatalysts, as the actual substrate function for the biotransformation was generated in situ. To realize such a concurrent chemoenzymatic tandem process, we had to overcome the general compatibility issues of the individual cascade steps: differences in the required solvent (water vs. THF) and enzyme inhibition/inactivation by both substrates and copper-(I)-thiophene-2-carboxylate (CuTC), an additive typically required in stoichiometric amounts in L–S reactions. The inhibition of biocatalysts by heavy metals and especially copper salts is well-known1,18 and represents the most important issue needed to be solved.
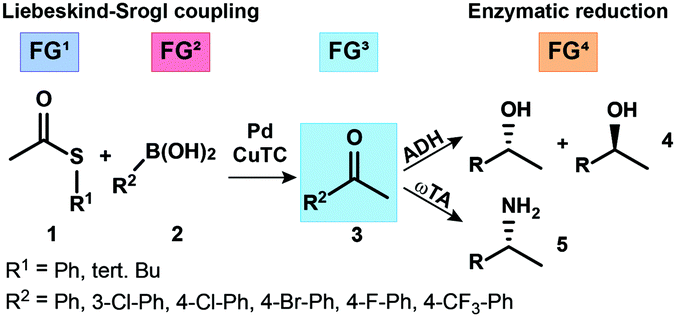 | ||
| Scheme 2 Liebeskind–Srogl coupling reaction and the subsequent biocatalytic reduction or transamination of the newly formed prochiral ketone. | ||
First, we investigated different L–S coupling procedures to gain deeper insights into the limitations towards a more biocompatible protocol. We tested two TEs (phenyl thioester (1a) and t-butyl-thioester (1b)), varied their concentrations, tested phenylboronic acid (2a) as a coupling partner, and screened three different temperatures (Table 1). Initially, we applied standard conditions (Pd(0), THF, and 50 °C; Table 1, entry #1) and obtained the desired ketone 3a in 69% GC-yield. As the first compartmentalization strategy, we changed the solvent to heptane to establish a two-phase system for the target chemoenzymatic cascade (Fig. 1A).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | TEa (R) | TE [mM] | Solv. system | T [°C] | Catalytic system | Conv. 3a 24 h [%] |
| a TE = thioester; Ph = phenyl; t-Bu = t-butyl. b Phenyl boronic acid (1.1 equiv.), CuTC (1.2 equiv.), Pd(PPh3)4 (5 mol%). c Phenyl boronic acid (1.7 equiv.), CuTC (1.6 equiv.), Pd2(dba)3 (2.5 mol%), P(OEt)3 (20 mol%); RT = room temperature; i-PrOH = isopropanol. | ||||||
| #1 | Ph (1a) | 250 | THF | 50 | Pd(PPh3)4b | 69 |
| #2 | Ph (1a) | 250 | Heptane | 50 | Pd(PPh3)4b | 99 |
| #3 | Ph (1a) | 250 | Heptane | 37 | Pd(PPh3)4b | 30 |
| #4 | Ph (1a) | 100 | Heptane | 37 | Pd(PPh3)4b | 24 |
| #5 | Ph (1a) | 100 | THF | RT | Pd2(dba)3c P(OEt)3 | 99 |
| #6 | Ph (1a) | 100 | Heptane | RT | Pd2(dba)3c P(OEt)3 | 99 |
| #7 | Ph (1a) | 100 | H2O/i-PrOH(90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) :10) |
RT | Pd2(dba)3c P(OEt)3 | TE hydrolysis |
| #8 | t-Bu (1b) | 100 | H2O/i-PrOH(90:10) |
RT | Pd2(dba)3c P(OEt)3 | 78 |
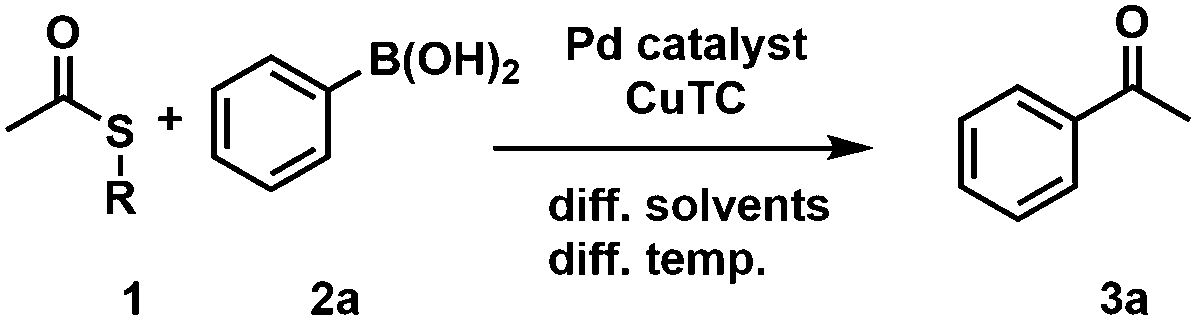
The conversion increased to 99% (Table 1, entry #2) and prompted us to decrease the temperature to be biocompatible (37 °C) (Table 1, #3) and to additionally reduce the TE concentration from 250 mM to 100 mM (Table 1 #4). Unfortunately, the yield dropped significantly in both cases from 99% to 30% and 24%, respectively (Table 1, entries #3 and #4). This indicates that the temperature had a more pronounced effect than the overall concentration of the reaction. Next, we investigated a different protocol, with Pd2dba3 as a catalyst precursor (tris(dibenzylideneacetone)-dipalladium(0)) and triethyl phosphite in THF at room temperature (Table 1, entry #5), which gave the desired acetophenone 3a in perfect yields.25
 | ||
| Fig. 2 Reactor device for a chemoenzymatic reaction sequence in a one-pot fashion. | ||
|
|
|||||
|---|---|---|---|---|---|
| Product | Method | Enzyme | Mode of operation | Yield [%] | ee [%] |
| Method A: heptane/water: (a) 50 mM TE 1a, phenyl boronic acid 2a (1.1 equiv.), CuTC (1.2 equiv.), Pd(PPh3)4 (5 mol%), 50 °C, heptane; (b) centrifuged coupling reaction 10% (v/v), ADH (10 mg whole-cell lyophilisates), NADP+, glucose (15 mM), glucose dehydrogenase (GDH, (1 U mL−1)), 30 °C; method B: heptane/superabsorber: (a) 100 mM TE 1a, phenyl boronic acid 2a (1.7 equiv.), CuTC (1.6 equiv.), Pd2(dba)3 (2.5 mol%), P(OEt)3 (20 mol%); (b) ADH (40 mg CFE), NADP+, glucose (15 mM), glucose dehydrogenase (GDH, (1 U mL−1)), sodium polyacrylate (2 × 5 mg); (c) ω-TA (40 mg, ATA025 Codexis screening kit), isopropylamine (1 M); method C: (a) coupling chamber: 50 mM TE 1b, phenyl boronic acid 2a (1.7 equiv.), CuTC (1.6 equiv.), Pd2(dba)3 (2.5 mol%), P(OEt)3 (20 mol%), water/i-PrOH (10% v/v coupling chamber), (b) biocatalytic chamber: ADH (200 mg whole-cell lyophilisates), Tris–HCl buffer (320 mM, pH 8), i-propanol (30% v/v). | |||||
| 4a | A | LK-ADH | Sequential | 57 | >99 |
| 5a | A | ATA025 | Sequential | n.a. | n.a. |
| 4a | B | LK-ADH | Sequential | 81 | >99 |
| 5a | B | ATA025 | Sequential | 51 | >99 |
| 4a | C | LK-ADH | Concurrent | 65 | >99 |
| 5a | C | ATA025 | Concurrent | n.a. | n.a. |

Subsequently, we moved to heptane and were delighted to obtain 3a still in 99% yield (Table 1, entry #6). Furthermore, we tested a 90:10-mixture of water and isopropanol (i-PrOH) under otherwise identical conditions.
Unfortunately, we observed prominent hydrolysis of the phenyl thioester 1a. We changed to the more stable t-butyl-thioester 1b to avoid hydrolysis of the TE and were able to produce the desired ketone 3a in 78% yield (Table 1, entry #8). Thus, we were confident to have established a biocompatible L–S protocol for an application in a biphasic system or in the biomimetic approaches as described above. For our target chemoenzymatic cascade, we selected a solvent tolerant (S)-selective ADH from Rhodococcus ruber (ADH-a)27 and an (R)-selective, isopropylamine dependent ω-transaminase from Codexis (ATA025). As a model reaction, we focused on the formation of acetophenone 3a that undergoes a biochemical reduction towards the secondary alcohol 4a or the corresponding primary amine 5a. First, we tested a sequential approach (Fig. 1A), where the coupling reaction was performed in heptane at 50 °C. This mixture was centrifuged before the biocatalyst was added (Table 2, method A) and the procedure resulted in the formation of the desired alcohol 4a in 57% yield, whereas no conversion to the corresponding amine was observed. Additionally, we employed a second compartmentalization strategy: a biphasic system involving heptane as organic solvent and sodium polyacrylate/H2O for trapping the components of the biocatalytic reaction (Fig. 1B).
Remarkably, we could obtain both chiral products in good to very good yields with perfect enantiomeric purity (Table 2, method B). Furthermore, we aimed for the concurrent approach and applied the third concept of compartmentalization (Fig. 1C). Thereby, we designed a membrane reactor with two chambers separated by a polydimethylsiloxane (PDMS) membrane (Fig. 2).18,28 PDMS facilitates the transport of lipophilic compounds but restricts the diffusion of hydrophilic components between the two catalytic chambers. Since organic solvents such as heptane have a detrimental effect on the integrity of the PDMS membrane, we applied the L–S reaction under aqueous/i-PrOH conditions as depicted in Table 1, entry #8. The second chamber was charged with ADH-a, buffer, and i-PrOH (for nicotinamide adenine dinucleotide phosphate (NADP+) cofactor recycling). However, the reaction did not yield the desired product. This was due to the present boronic acid (1.7 equiv.) and the released thiophene-2-carboxylic acid (1.6 equiv.), which decreased the pH value in the enzymatic chamber to 5.1 after 24 h. This inactivated the ADH and decreased the yield significantly.
By changing the reaction parameters to 320 mM Tris–HCl buffer, maintaining pH 8.0, we were able to run the chemoenzymatic reaction in a concurrent fashion. We obtained the desired chiral alcohol 4a in 65% overall yield and perfect enantioselectivity (>99% ee). At the end of the reaction, alcohol 4a slowly diffused back into the coupling chamber. Under these optimized conditions, we also tested the ω-TA from Codexis but no conversion to the desired chiral amine 5a was observed.
Parameter investigations resulted in the strong enzyme deactivating effect by phenyl boronic acid (2a) or the corresponding i-propyl ester, which was lipophilic enough to penetrate the PDMS membrane. Furthermore, already 20 mol% of P(OEt)3 and the TE 1a resulted in complete enzyme inhibition or deactivation whereas CuTC was still tolerated by the ω-TA (for further details see the ESI,† Table S3).
Nevertheless, we could expand the substrate portfolio to different halogenated phenylboronic acids 2a–f (Table 3) and tested them with the (S)-selective ADH-a and an (R)-selective LK-ADH29 to obtain the antipodal chiral alcohols 4a–f. To demonstrate the synthetic applicability we performed an experiment on the preparative scale and isolated both enantiomeric alcohols 4a (S) and (R) in 47% and 51% over two steps respectively (see the ESI,† Table S4).
| # | R | ADH-a | LK-ADH | ||
|---|---|---|---|---|---|
| Yield [%] | ee [%] | Yield [%] | ee [%] | ||
| ADH-a from Rhodococcus ruber; LK-ADH from Lactobacillus kefir. Reaction conditions: 100 mM concentration of 1b, boronic acid (1.7 equiv.), CuTC (1.6 equiv.), Pd2(dba)3 (2.5 mol%), P(OEt)3 (20 mol%), ADHs in Tris–HCl buffer (320 mM, pH 8.0) and i-PrOH (30% (v/v) referring to the biocatalytic reaction chamber) as cosubstrate, 24 h reaction time. | |||||
| 4a | Ph | 65 | 99 (S) | 81 | 99 (R) |
| 4b | 4-Cl-Ph | 60 | 99 (S) | 57 | 99 (R) |
| 4c | 3-Cl-Ph | 47 | 99 (S) | 53 | 99 (R) |
| 4d | 4-Br-Ph | 99 | 99 (S) | 50 | 99 (R) |
| 4e | 4-F-Ph | 75 | 99 (S) | 64 | 99 (R) |
| 4f | 4-CF3-Ph | 61 | 99 (S) | 53 | 99 (R) |
In summary, we disclose two approaches for realizing a L–S coupling–enzymatic reduction/amination protocol. First, a sequential approach, which requires only a simple centrifugation step after the L–S reaction. Second, and more importantly, a simultaneous chemoenzymatic process, combining the metal (Pd/Cu) catalyzed L–S coupling reaction and the subsequent biocatalytic reduction of the newly created carbonyl group in one-pot generating optically pure alcohols by applying enantiodivergent ADHs, importantly, without any intermediate isolation. We understand the formation of a novel functional group within a C–C bond formation reaction and its subsequent interconversion, a particularly novel feature of this chemo-enzymatic cascade.
By employing a biomimetic compartmentalization approach, the desired chiral secondary alcohols were accessible in up to 99% yield and perfect enantioselectivity (>99% ee) over two steps. Even though the applied reactor is not in the standard laboratory inventory, the setup is very simple and readily available from commercial sources. Furthermore, the required membrane can be easily prepared, which overall allows the application of this method in any lab without much effort.
Financial support for this research program from TU Wien (internal grant ABC – Applied Biosynthetic Cell factories) and the DFG (grant no. Bo1862/6-1) is gratefully acknowledged. The authors thank Prof. Dr Werner Hummel, Bielefeld University, for the generous donation of the expression system for the ADH from Lactobacillus kefir.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 CrossRef PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC.
- C. A. Denard, J. F. Hartwig and H. M. Zhao, ACS Catal., 2013, 3, 2856–2864 CrossRef CAS.
- S. Schmidt, K. Castiglione and R. Kourist, Chem. – Eur. J., 2018, 24, 1755–1768 CrossRef CAS PubMed.
- Y. J. Wang and H. M. Zhao, Catalysts, 2016, 6 Search PubMed.
- S. Borchert, E. Burda, J. Schatz, W. Hummel and H. Gröger, J. Mol. Catal. B: Enzym., 2012, 84, 89–93 CrossRef CAS.
- M. Heidlindemann, G. Rulli, A. Berkessel, W. Hummel and H. Gröger, ACS Catal., 2014, 4, 1099–1103 CrossRef CAS.
- M. Heidlindemann, M. Hammel, U. Scheffler, R. Mahrwald, W. Hummel, A. Berkessel and H. Gröger, J. Org. Chem., 2015, 80, 3387–3396 CrossRef CAS PubMed.
- M. Fuchs, D. Koszelewski, K. Tauber, W. Kroutil and K. Faber, Chem. Commun., 2010, 46, 5500–5502 RSC.
- C. A. Denard, H. Huang, M. J. Bartlett, L. Lu, Y. C. Tan, H. M. Zhao and J. F. Hartwig, Angew. Chem., Int. Ed., 2014, 53, 465–469 CrossRef CAS PubMed.
- Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100–103 CrossRef CAS PubMed.
- V. Köhler, Y. M. Wilson, M. Durrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knorr, D. Haussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93–99 CrossRef PubMed.
- N. Rios-Lombardia, C. Vidal, M. Cocina, F. Moris, J. Garcia-Alvarez and J. Gonzalez-Sabin, Chem. Commun., 2015, 51, 10937–10940 RSC.
- P. Schaaf, V. Gojic, T. Bayer, F. Rudroff, M. Schnürch and M. D. Mihovilovic, ChemCatChem, 2018, 10, 920–924 CrossRef CAS.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- H. Sato, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2015, 54, 4488–4492 CrossRef CAS PubMed.
- M. J. Fink, M. Schön, F. Rudroff, M. Schnürch and M. D. Mihovilovic, ChemCatChem, 2013, 5, 724–727 CrossRef CAS.
- N. Rios-Lombardia, C. Vidal, E. Liardo, F. Moris, J. Garcia-Alvarez and J. Gonzalez-Sabin, Angew. Chem., Int. Ed., 2016, 55, 8691–8695 CrossRef CAS PubMed.
- L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS.
- J. M. Villalobos, J. Srogl and L. S. Liebeskind, J. Am. Chem. Soc., 2007, 129, 15734–15735 CrossRef CAS PubMed.
- H.-G. Cheng, H. Chen, Y. Liu and Q. Zhou, Asian J. Org. Chem., 2018, 7, 490–508 CrossRef CAS.
- L. Betancourt-Mendiola, I. Valois-Escamilla, T. Arbeloa, J. Bañuelos, I. López Arbeloa, J. O. Flores-Rizo, R. Hu, E. Lager, C. F. A. Gómez-Durán, J. L. Belmonte-Vázquez, M. R. Martínez-González, I. J. Arroyo, C. A. Osorio- Martínez, E. Alvarado-Martínez, A. Urías-Benavides, B. D. Gutiérrez- Ramos, B. Z. Tang and E. Peña-Cabrera, J. Org. Chem., 2015, 80, 5771–5782 CrossRef CAS PubMed.
- H. Yang, H. Li, R. Wittenberg, M. Egi, W. W. Huang and L. S. Liebeskind, J. Am. Chem. Soc., 2007, 129, 1132–1140 CrossRef CAS PubMed.
- M. Koley, L. Wimmer, M. Schnürch and M. D. Mihovilovic, J. Hetero. Chem., 2013, 50, 1368–1373 CrossRef CAS.
- M. Karabec, A. Lyskowski, K. C. Tauber, G. Steinkellner, W. Kroutil, G. Grogan and K. Gruber, Chem. Commun., 2010, 46, 6314–6316 RSC.
- M. B. Runge, M. T. Mwangi, A. L. Miller, II, M. Perring and N. B. Bowden, Angew. Chem., Int. Ed., 2008, 47, 935–939 CrossRef CAS PubMed.
- C. W. Bradshaw, W. Hummel and C. H. Wong, J. Org. Chem., 1992, 57, 1532–1536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc05304a |
| This journal is © The Royal Society of Chemistry 2018 |