
Effect of the metal–support interaction on the activity and selectivity of methanol oxidation over Au supported on mesoporous oxides†
Sunyoung
Oh‡
ab,
You Kyung
Kim‡
bc,
Chan Ho
Jung
b,
Won Hui
Doh
 b and
Jeong Young
Park
*abc
b and
Jeong Young
Park
*abc
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: jeongypark@kaist.ac.kr
bCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
cGraduate School of EEWS, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
First published on 19th June 2018
Abstract
To elucidate the factors affecting the catalytic properties of supported Au catalysts on the metal oxide support we investigated Au NPs deposited on crystallized mesoporous transition-metal oxides (m-oxides: Co3O4, NiO, and α-Fe2O3) prepared using the nanocasting method. The metal–oxide interaction in Au/mesoporous oxides resulted in higher catalytic activity for converting methanol to CO2 as a full oxidation product than pure m-oxides. Au/m-Fe2O3 exhibited high activity and low selectivity for methyl formate as a partial oxidative coupling product. We correlate the change in activity and selectivity with the interface between the Au and m-oxides.
Gold-based catalysts have attracted much attention since the discovery of the origin of the catalytic activity of supported Au nanoparticles (NPs) for low-temperature CO oxidation.1 Even though extensive experimental and theoretical studies have been performed, the intrinsic origin of the catalytic activity of Au is still controversial. For oxidation catalysis, O2 dissociation is a critical step because single-crystal Au surfaces have an extremely low oxygen dissociation probability (≪10−6) and weak interaction. Mechanistic studies have thus relied on more reactive oxygen sources, such as ozone or oxygen atoms.2–4 Yet, supported Au catalysts with diameters of 2 to 5 nm were quite active on suitable oxide supports, such as titania and ceria.5,6 Moreover, Au-based catalysts have been recently reported for a wide range of catalytic reactions, including low-temperature CO oxidation, H2 oxidation, and the selective oxidation of alcohols and hydrocarbons.7–11
Methanol (CH3OH) is one of the most important industrial chemicals and selective oxidation of alcohols is a key issue in important transformation processes for energy conversion and chemical synthesis of fine chemicals via green chemistry.9,12–14 Recently, novel unsupported transition metal materials have gained attention for addressing the intrinsic origin of the catalytic activity for selective oxidation of methanol. Studies on the size effect of Pt NPs have suggested that the production of valuable chemicals, such as formaldehyde and carbon dioxide, can be obtained via the oxidative reaction of methanol under ambient pressure at low temperature.15 As the size of the particles decreased, they showed higher selectivity toward partial oxidation of methanol to formaldehyde, although the catalytic activity was very low. In particular, unsupported nanoporous Au (np-Au) is catalytically more active for the selective oxidative coupling of methanol to methyl formate at temperatures below 80 °C.16 In oxidation reactions, the activation of molecular oxygen is essential and the support should be involved in creating dissociated oxygen at the perimeter sites of the metal particles. Since heterogeneous catalysts can be prepared by depositing NPs on oxide supports, the metal–oxide interface plays a crucial role in affecting the catalytic properties of oxidation reactions because of geometric changes and charge transfer between the metal and the oxide support.17–20 Despite the various studies, there are limitations for utilizing Au catalysts because of the low dissociation probability on single-crystal Au surfaces and the poor stability of supported Au catalysts. To overcome these problems and to address the intrinsic catalytic origins of supported Au catalysts, porous oxides have been used recently as suitable oxide supports because of their thermal stability and high surface area.21–23
To elucidate the factors affecting the catalytic properties of supported Au catalysts depending on the different metal oxide supports, we investigated Au NPs deposited on crystallized mesoporous transition-metal oxides (m-oxides: Co3O4, NiO, and α-Fe2O3) prepared using the nanocasting method. Methanol oxidation was carried out on all the Au supported on m-oxide catalysts, which serve as promising model catalysts to probe the catalytic origin of supported Au catalysts without the propensity for sintering the NPs. We found a comparable catalytic activity and selectivity of Au/m-oxide catalysts for methanol oxidation to carbon dioxide (i.e., the full oxidation product) and methyl formate (the partial oxidation and coupling product). From this study, we varied the different m-oxides to engineer the metal–oxide interface and evaluate its role in altering the catalytic activity and selectivity.
The hard-templating approach using high-quality large mesoporous silica with cubic Ia3d symmetry (KIT-6) was chosen to synthesize highly crystallized mesoporous oxides of Co3O4, NiO, and Fe2O3. Fig. 1 shows the procedure for preparing the mesoporous oxides and their supported Au catalysts. Fig. 2 shows representative transmission electron microscopy (TEM) images of the m-oxides as well as the supported Au catalysts. Such m-oxides exhibit ordered porosity and single crystallinity in larger domains (Fig. 2b–d). In addition, their structures have large pore diameters and high surface areas (see Table S1, ESI†). To demonstrate the role of the interface between the Au and the oxides during the methanol oxidation reaction, we deposited Au NPs on m-oxides using the urea reduction method (Fig. 2e–h). The mean diameter of the Au NPs for the prepared catalysts is quite narrow at 5 nm (Fig. S1, ESI†). Moreover, the structural information for the bare m-oxides and Au/m-oxides is shown in the X-ray diffraction patterns (Rigaku D/MAX-2500 at 40 kV, 300 mA) (see Fig. S2, ESI†), which indicates that the metal nitrate precursors were completely converted to crystallized metal oxide structures after the calcination process.24–26 The chemical bonding states of the Au/m-oxide catalysts were further examined using XPS analysis (Fig. S3, ESI†). The Au NPs were also deposited on the m-oxides.27 The Au compositions were 2.5%, 3.6%, 2%, and 2.7% for the Au/KIT-6, Au/m-Co3O4, Au/m-NiO, and Au/m-Fe2O3, respectively, measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES) (see Table S1, ESI†). The surface areas and pore size distributions of the prepared supported Au catalysts, obtained using the Brunauer–Emmett–Teller (BET) method with the adsorption of N2 at the temperature of liquid nitrogen, are quite high compared with other normal oxides (Fig. S4 and Table S1, ESI†).28,29
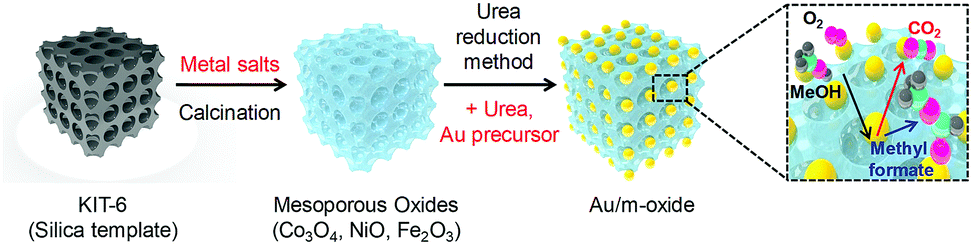 | ||
| Fig. 1 Schematic diagram of the hard-templating (nanocasting) approach for the preparation of mesoporous oxide supported Au nanoparticle catalysts. | ||
 | ||
| Fig. 2 TEM images of (a) the mesoporous silica template KIT-6, (b) m-Co3O4, (c) m-NiO, (d) m-Fe2O3, (e) Au/KIT-6, (f) Au/m-Co3O4, (g) Au/m-NiO, and (h) Au/m-Fe2O3. | ||
The oxidation of methanol (O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3OH = 1:1 v/v) was carried out on the pure mesoporous oxides and the Au-deposited mesoporous oxides at temperatures of 80–220 °C, following a reductive pre-treatment. The measured CO2 evolution is plotted in Fig. 3. The CO2 obtained was dependent upon the pure m-oxide (Fig. 3a). The mesoporous oxides have shown notable catalytic activity in the absence of noble metal particles,30–32 which indicates that the higher surface area of their structures can provide more active sites and that the porous structure promotes the adsorption and diffusion of reactant molecules compared with bulk metal oxides.33 Recent experiments on mesoporous structures, such as chromium oxide and cobalt oxide, have suggested that the high catalytic activity can be ascribed to the complete oxidation of toluene and propane.34,35 Also, CO oxidation results using different kinds of oxide supports are in agreement with our catalytic results, depending on the pure mesoporous oxides.36 This demonstrates that pure mesoporous oxides for methanol oxidation exhibit a redox capability consistent with previous results for CO oxidation.
:CH3OH = 1:1 v/v) was carried out on the pure mesoporous oxides and the Au-deposited mesoporous oxides at temperatures of 80–220 °C, following a reductive pre-treatment. The measured CO2 evolution is plotted in Fig. 3. The CO2 obtained was dependent upon the pure m-oxide (Fig. 3a). The mesoporous oxides have shown notable catalytic activity in the absence of noble metal particles,30–32 which indicates that the higher surface area of their structures can provide more active sites and that the porous structure promotes the adsorption and diffusion of reactant molecules compared with bulk metal oxides.33 Recent experiments on mesoporous structures, such as chromium oxide and cobalt oxide, have suggested that the high catalytic activity can be ascribed to the complete oxidation of toluene and propane.34,35 Also, CO oxidation results using different kinds of oxide supports are in agreement with our catalytic results, depending on the pure mesoporous oxides.36 This demonstrates that pure mesoporous oxides for methanol oxidation exhibit a redox capability consistent with previous results for CO oxidation.
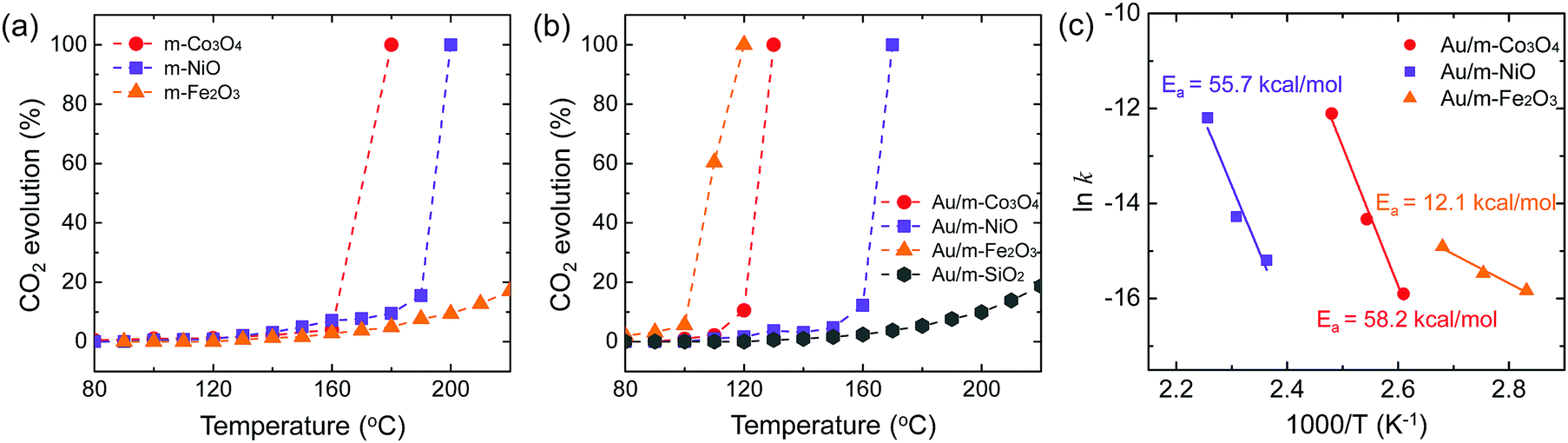 | ||
| Fig. 3 Catalytic activity of full-oxidation of methanol to CO2 as a function of temperature for (a) pure mesoporous oxides and (b) Au nanoparticles supported on mesoporous oxides. (c) Arrhenius plots of the rate of CO production on the Au/m-oxide catalysts. | ||
When Au NPs with an average diameter of 5 nm are incorporated into the mesoporous oxides, the observed catalytic activities for full methanol oxidation are considerably higher than for pure m-oxides (Fig. 3). Au NPs supported on porous metal oxides lead to an enhancement of the catalytic activity, and the metal–oxide interface provides active sites.10 As reported by Leppelt et al., this can be justified by the fact that catalytic active perimeter sites should exist at the interface between the gold and the support.37 Additionally, it is believed that the support is critical for catalytic activity because pure Au lacks high activity for molecular oxygen dissociation so that oxygen molecules can be activated by the presence of the Au metal that is in contact with the metal oxide supports.38 Recently, the interface between the Au support and metal oxide particles serves as an absorption site for CO reactants as well as allowing for efficient activation of molecular oxygen.39 Abad et al. reported that Au-deposited nanocrystalline ceria showed remarkable catalytic activity and selectivity for the oxidation of 3-octanol compared with Au-deposited carbon.8 The higher catalytic activity of the Au/m-oxides indicates that the mesoporous oxides increase the interface between the metal and the oxide compared with non-porous oxides. Among the Au nanoparticles supported on mesoporous oxide catalysts, Au/m-Fe2O3 shows the highest catalytic activity. It is suggested that a large amount of oxygen can adsorb onto the support as a reducible oxide, such as TiO2 and Fe3O4 (or at the metal–support interface), possibly on oxygen vacancies.40,41 Iron oxide can adsorb a large quantity of highly mobile oxygen, leading to a higher catalytic activity. Moreover, such support oxides can stabilize the Au nanoparticles and oxygen can transfer to the metal surface.42 The oxidation states of Au 4f for the Au/m-Fe2O3 catalysts before and after the methanol oxidation reaction are shown in Fig. S5 (ESI†). After the methanol oxidation reaction, Au3+ species are present, indicating that molecular oxygen can be activated by the Au supported on iron oxide as a reducible oxide. Despite the increasing number of studies, the mechanisms for oxygen adsorption and activation are highly controversial, but they are needed to explain the activity for oxidative reactions. To discover the contribution of just the Au nanoparticles to catalytic activity, Au supported on SiO2 (i.e., KIT-6, an inert support) was prepared. It shows the lowest CO2 evolution for methanol oxidation even up to 220 °C compared with Au supported on reducible oxides (Fig. 3b). In the case of inert support materials, it is a logical consequence that oxygen adsorption and dissociation should be possible on the Au surface.43 The activation energies for Au/m-Co3O4, Au/m-NiO, and Au/m-Fe2O3 obtained from Arrhenius plots for the rate of CO2 production are 58.2, 55.7, and 12.1 kcal mol−1, respectively (Fig. 3c). Au/m-oxide catalysts display a dependence on the mesoporous oxides for the catalytic activity for CO2 formation, implying that each oxide has a different interaction mechanism for the full methanol oxidation reaction.
In this experiment, the volume ratio of oxygen to methanol (1:1 v/v) allows the dominant coupling reaction and produces methyl formate as a partial oxidation product with comparable selectivity on the Au/m-oxide catalysts at 100 °C, as shown in Fig. 4. The selective oxidation of methanol resulted in 89.3% and 37.3% methyl formate production for the Au/m-Co3O4 and Au/m-Fe2O3 catalysts, respectively, while the Au/m-NiO catalyst only produced methyl formate. The apparent difference in selectivity dependent on the mesoporous oxide could be influenced by interactions between the Au NPs and the metal oxide supports. Liu et al. may provide a clarification that the initial adsorption of methanol on the Au sites induces the formation of a chemisorbed methoxy species, which facilitates the coupling reaction with a neighbouring methoxy species, thus generating methyl formate.44 Moreover, the oxide support of the supported Au catalysts can play a major role in supplying oxygen to the Au active center, leading to the oxidative coupling of methanol to produce methyl formate.45,46 The methoxy species is an intermediate of methanol oxidation that adsorbs on Ce4+ cations and is able to oxidize to the formate species when a gold metal is present with the CeO2 support; furthermore the Ce4+ cations are thus reduced to Ce3+.47,48 A suggested role of the Au is for Ce3+ formate generation from the ceria surface oxygen species, which further creates surface vacancies acting as oxygen pumps (the so-called “reverse spillover effect”). The reaction pathway for methyl formate production is still debated. Further investigation is needed to understand the detailed mechanism, but these results show that we can control the catalytic activity and selectivity for methanol oxidation by altering the oxide support.
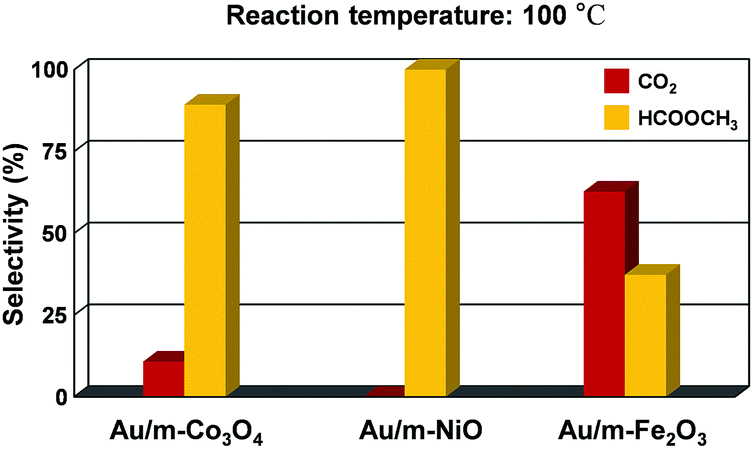 | ||
| Fig. 4 Selectivity showing the fraction of methanol converted to carbon dioxide and methyl formate at 100 °C for Au-supported mesoporous oxides (Co3O4, NiO, and Fe2O3). | ||
When using supported metal catalysts, a negative factor that may be important is the tendency for nanoparticles to sinter under reaction conditions. This leads to nanoparticle growth, and thus results in lower catalytic activity.49 A TEM image of Au/m-oxide after the methanol oxidation reaction reveals good thermal stability while retaining the original nanoparticle size and morphology (see Fig. S6, ESI†), which can be observed in the repetitive catalytic performance on Au/m-oxide (see Fig. S7, ESI†). This also demonstrates that the geometric properties and large surface area of the mesoporous oxide can prevent the sintering of Au NPs. Its thermal stability is also influenced by the strong interaction between the Au particles and the metal oxide support, which provides insight into the catalytic performance during the reaction.
In conclusion, we found the intrinsic methanol oxidation activity and selectivity of Au nanoparticles supported on crystallized mesoporous oxides (Co3O4, NiO, and α-Fe2O3). The catalytic activity for CO2 formation as a full oxidation product over Au/m-oxide catalysts is higher than that for pure mesoporous oxides, which can be attributed to the mesoporous oxides providing an increased interface between the Au and the oxide support compared with non-porous oxides. This approach reveals a promising route for controlling the activity as well as the selectivity by altering the oxide support, which governs the interface between the metal and the support.
This work was supported by the Institute for Basic Science [IBS-R004].
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef.
- X. Deng, B. K. Min, A. Guloy and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 9267–9270 CrossRef PubMed.
- B. Xu, X. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 48, 4206–4209 CrossRef PubMed.
- J. Gong, D. W. Flaherty, R. A. Ojifinni, J. M. White and C. B. Mullins, J. Phys. Chem. C, 2008, 112, 5501–5509 CrossRef.
- G. J. Hutchings, Catal. Today, 2005, 100, 55–61 CrossRef.
- J. Schwank, S. Galvagno and G. Parravano, J. Catal., 1980, 63, 415–424 CrossRef.
- K. Qadir, B. T. Quynh, H. Lee, S. Y. Moon, S. H. Kim and J. Y. Park, Chem. Commun., 2015, 51, 9620–9623 RSC.
- A. Abad, P. Concepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef PubMed.
- T. Ishida and M. Haruta, Angew. Chem., Int. Ed., 2007, 46, 7154–7156 CrossRef PubMed.
- B. Jorgensen, S. E. Christiansen, M. L. D. Thomsen and C. H. Christensen, J. Catal., 2007, 251, 332–337 CrossRef.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Top. Catal., 2004, 29, 95–102 CrossRef.
- R. A. Sheldon, I. W. C. E. Arends and A. Dijksman, Catal. Lett., 2000, 57, 157–166 Search PubMed.
- R. A. Sheldon, I. W. C. E. Arends, G.-J. T. Brink and A. R. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef PubMed.
- J. M. Tatibouët, Appl. Catal., A, 1997, 148, 213–252 CrossRef.
- H. Wang, Y. Wang, Z. Zhu, A. Sapi, K. An, G. Kennedy, W. D. Michalak and G. A. Somorjai, Nano Lett., 2013, 13, 2976–2979 CrossRef PubMed.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bädlani, Science, 2010, 327, 319–322 CrossRef PubMed.
- Y. Zhang, M. E. Grass, S. E. Habas, F. Tao, T. Zhang, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 12243–12253 CrossRef.
- G. A. Somorjai, H. Frei and J. Y. Park, J. Am. Chem. Soc., 2009, 131, 16589–16605 CrossRef PubMed.
- E. Gross and G. A. Somorjai, Top. Catal., 2013, 56, 1049–1058 CrossRef.
- H. Wang, K. An, A. Sapi, F. Liu and G. A. Somorjai, Catal. Lett., 2014, 144, 1930–1938 CrossRef.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef PubMed.
- K. An, S. Alayoglu, N. Musselwhite, K. Na and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 6830–6833 CrossRef PubMed.
- K. Zhu, B. Yue, W. Zhou and H. He, Chem. Commun., 2003, 98–99, 10.1039/b210065g.
- P. Dutta, M. S. Seehra, S. Thota and J. Kumar, J. Phys.: Condens. Matter, 2008, 20, 015218 CrossRef.
- Z. Lockman, X. Qi, A. Berenov, R. Nast, W. Goldacker and J. MacManus-Driscoll, Phys. C, 2001, 351, 34–37 CrossRef.
- B. K. Woo, H. J. Lee, J.-P. Ahn and Y. S. Park, Adv. Mater., 2003, 15, 1761–1764 CrossRef.
- Y. Chen, X. Gu, C.-G. Nie, Z.-Y. Jiang, Z. X. Xie and C.-J. Lin, Chem. Commun., 2005, 4181–4183, 10.1039/b504911c.
- S. H. Kim, C.-H. Jung, N. Sahu, D. Park, J. Y. Yun, H. Ha and J. Y. Park, Appl. Catal., A, 2013, 454, 53–58 CrossRef.
- C. H. Jung, J. Yun, K. Qadir, D. Park, J. Y. Yun and J. Y. Park, Res. Chem. Intermed., 2016, 42, 211–222 CrossRef.
- Y. Rao and D. M. Antonelli, J. Mater. Chem., 2009, 19, 1937–1944 RSC.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef PubMed.
- J. Jansson, A. E. C. Palmqvist, E. Fridell, M. Skoglundh, L. Österlund, P. Thormählen and V. Langer, J. Catal., 2002, 211, 387–397 CrossRef.
- Y. Xia, H. Dai, H. Jiang and L. Zhang, Catal. Commun., 2010, 11, 1171–1175 CrossRef.
- A. K. Sinha and K. Suzuki, Angew. Chem., Int. Ed., 2004, 44, 271–273 CrossRef PubMed.
- B. Solsona, T. E. Davies, T. Garcia, I. Vázquez, A. Dejoz and S. H. Taylor, Appl. Catal., B, 2008, 84, 176–184 CrossRef.
- Y. Yu, T. Takei, H. Ohashi, H. He, X. Zhang and M. Haruta, J. Catal., 2009, 267, 121–128 CrossRef.
- R. Leppelt, B. Schumacher, V. Plzak, M. Kinne and R. Behm, J. Catal., 2006, 244, 137–152 CrossRef.
- J. M. Gottfried, K. J. Schmidt, S. L. M. Schroeder and K. Christmann, Surf. Sci., 2003, 525, 184–196 CrossRef.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef PubMed.
- H. Liu, A. I. Kozlov, A. P. Kozlova, T. Shido, K. Asakura and Y. Iwasawa, J. Catal., 1999, 185, 252–264 CrossRef.
- H. Liu, A. I. Kozlov, A. P. Kozlova, T. Shido and Y. Iwasawa, Phys. Chem. Chem. Phys., 1999, 1, 2851–2860 RSC.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skala, A. Bruix, F. Illas, K. C. Prince, V. Matolin, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef PubMed.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef.
- X. Liu, R. J. Madix and C. M. Friend, Chem. Soc. Rev., 2008, 37, 2243–2261 RSC.
- S. Kegnæs, J. Mielby, U. V. Mentzel, C. H. Christensen and A. Riisager, Green Chem., 2010, 12, 1437–1441 RSC.
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2009, 11, 1366–1370 RSC.
- P. Bazin, S. Thomas, O. Marie and M. Daturi, Catal. Today, 2012, 182, 3–11 CrossRef.
- S. Rousseau, O. Marie, P. Bazin, M. Daturi, S. Verdier and V. Harle, J. Am. Chem. Soc., 2010, 132, 10832–10841 CrossRef PubMed.
- G. T. Whiting, S. A. Kondrat, C. Hammond, N. Dimitratos, Q. He, D. J. Morgan, N. F. Dummer, J. K. Bartley, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ACS Catal., 2015, 5, 637–644 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary figures, table, and references. See DOI: 10.1039/c8cc04295k |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |