
Reconstitution of full-length P450BM3 with an artificial metal complex by utilising the transpeptidase Sortase A†
Keita
Omura
 a,
Yuichiro
Aiba
a,
Hiroki
Onoda
a,
Joshua Kyle
Stanfield
a,
Shinya
Ariyasu
a,
Hiroshi
Sugimoto
bc,
Yoshitsugu
Shiro
d,
Osami
Shoji
*ab and
Yoshihito
Watanabe
*e
a,
Yuichiro
Aiba
a,
Hiroki
Onoda
a,
Joshua Kyle
Stanfield
a,
Shinya
Ariyasu
a,
Hiroshi
Sugimoto
bc,
Yoshitsugu
Shiro
d,
Osami
Shoji
*ab and
Yoshihito
Watanabe
*e
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-0802, Japan. E-mail: shoji.osami@a.mbox.nagoya-u.ac.jp
bCore Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency, 5 Sanban-cho, Chiyoda-ku, Tokyo, 102-0075, Japan
cRIKEN SPring-8 Center, 1-1-1 Kouto, Sayo-cho, Hyogo, 679-5148, Japan
dDepartment of Life Science, Graduate School of Life Science, University of Hyogo, 3-2-1 Kouto, Kamighori, Akoh, Hyogo, 678-1297, Japan
eResearch Center for Materials Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-0802, Japan
First published on 22nd May 2018
Abstract
Haem substitution is an effective approach to tweak the function of haemoproteins. Herein, we report a facile haem substitution method for self-sufficient cytochrome P450BM3 (CYP102A1) from Bacillus megaterium utilising the transpeptidase Sortase A from Staphylococcus aureus. We successfully constructed Mn-substituted BM3 and investigated its catalytic activity.
Cytochrome P450s (P450s) are haem-dependent monooxygenases, which catalyse the oxidation of organic substrates and are intimately involved in the biosynthesis of hormones and the metabolism of drugs.1 As P450s are capable of regio- and stereoselectively activating inert C–H bonds under mild conditions, they are garnering much attention in the growing field of green chemistry as environmentally friendly biocatalysts.2–5 In reactions involving P450s, redox-partners are indispensable for the reductive activation of dioxygen molecules to form the active oxygen species ‘oxoferryl porphyrin π-cation radical’ known as ‘compound I’.1 For the generation of compound I, two electrons must be supplied from redox partners of which several different types exist.1 For example, prokaryotic class I P450 systems utilise an NAD(P)H-dependent FAD-containing reductase and a [2Fe–2S] ferredoxin (e.g., P450cam). On the other hand, eukaryotic class II P450 systems are composed of a membrane-bound P450 and a membrane-bound NAD(P)H-dependent diflavin [FAD/FMN] reductase (e.g., human liver P450s). Although the majority of P450s are expressed independently of their redox partners, P450BM3 (CYP102A1, BM3) from Bacillus megaterium is fused to its redox partner, and this unique structural feature facilitates efficient interdomain electron transfer (Fig. 1). The C-terminal FAD domain accepts electrons from NADPH and the FMN domain relays them to the haem in the active site of BM3.
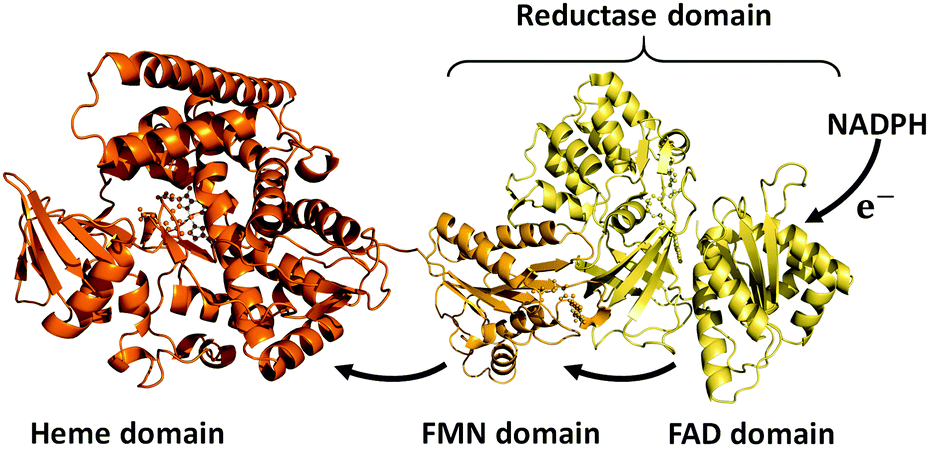 | ||
| Fig. 1 Cartoon of the structure of self-sufficient P450BM3. PDB: 1BVY (haem domain and FMN domain), 4DQK (FAD domain). | ||
Owing to the efficient electron transfer, BM3 exhibits a very high catalytic activity among P450s, hence BM3 is regarded as a prime candidate for the development of an outstanding biocatalyst. Its applications have been studied extensively, advanced by the aid of various types of mutagenesis,2–4,6–8 and more recently also by the use of decoy molecules.9–13
The haem in haemoproteins plays a pivotal role in governing their biological function and catalytic activity, and haem substitution is thus seen as a potent means to manipulate haemoprotein behaviour. Several methodologies for haem substitution have been developed to create novel metalloproteins in the past decades.14–17 Conventionally, acid/organic solvent is used to remove the haem from target haemoproteins.14 However, the harsh conditions inherent to these methods are not compatible with every haemoprotein, leaving some P450s irreversibly denatured, thus prohibiting reconstitution with artificial metal complexes. To accomplish the haem substitution of hitherto unreconstructed P450s, several mild methods have been developed that do not rely on harsh denaturing conditions.16,17 Nevertheless, although the haem domain of BM3 can be reconstituted with synthetic metal complexes employing these methods,16–19 there is no report on the preparation of haem-substituted BM3 in its full length form. This is likely due to the irreversible denaturation of full-length BM3, as well as the loss of other cofactors (FAD and FMN). To prepare reconstituted full-length BM3, incorporating synthetic metal complexes in its haem domain, we focused on a transpeptidase, Sortase A (SrtA),20 that can connect two protein domains. A simple ligation of the haem-substituted haem domain to the reductase domain by SrtA was envisioned to allow the construction of reconstituted full-length BM3. As a proof of concept, we were able to show that, using the method described herein, reconstituted full-length BM3 retained its high-catalytic activity and that the domain–domain ligation using SrtA presents a promising approach for haem-substitution. Subsequently, we found that Mn-substituted full-length BM3 can catalyse the monooxygenase reaction of fatty acids. Propane hydroxylation was also catalysed by Mn-substituted full-length BM3 with the assistance of decoy molecules.
SrtA from Staphylococcus aureus recognises a short amino acid sequences, known as a ‘sorting motif’ (LPXTG: Leu-Pro-Xaa-Thr-Gly, where Xaa can be any amino acid), and sequence-specifically forms a new peptide bond (LPXTGG…)21–23 with proteins containing an oligoglycine (two or more glycines) at their N-terminus, as shown in Fig. 2a. The peptide bond between Thr and Gly is cleaved by a nucleophilic cysteine of SrtA, yielding a thioacyl-intermediate. This intermediate is subject to nucleophilic attack by the amino group of oligoglycine at the N-terminus of another protein, to form a new peptide bond between Thr and incoming Gly, culminating in protein–protein ligation.22–24
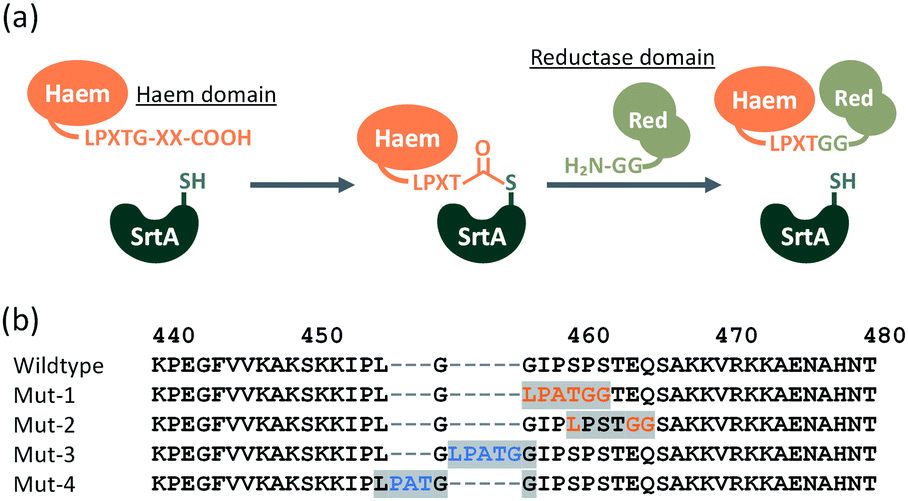 | ||
| Fig. 2 (a) Protein–protein ligation by SrtA. X represents any amino acid residue. (b) Amino acid sequence of linker region (Lys440–Thr480) connecting the N-terminal haem domain and C-terminal reductase domain. Point mutations are shown in red, insertions are shown in blue. The peptide sequence LPXTGG resulting from SrtA-mediated ligation are highlighted in grey. | ||
Considering the resulting amino acid sequence following ligation by SrtA, a series of BM3 mutants (Mut-1 to 4) containing LPXTGG sequence between the haem and reductase domain were prepared to determine the optimum joint sequence for ligation with SrtA. Mut-1 and -2 were prepared by replacement of amino acids in the linker region, whereas Mut-3 and -4 were prepared via the insertion of additional amino acids into the linker region (Fig. 2b). As the linker sequence is believed to affect smooth electron transfer and therefore the catalytic activity of BM3, we looked into whether these four mutants exhibited any decrease in catalytic activity.24,25 The catalytic activity of these mutants was evaluated by a p-nitrophenoxycarboxylic acid (pNCA) assay, using 10-(4-nitrophenoxy)capric acid (10-pNCA), which was developed to evaluate the hydroxylation activity of BM3 for fatty acid26 (Fig. 3). 10-pNCA is a capric acid derivative possessing a p-nitrophenol group at its ω-terminus, which, upon hydroxylation by BM3, is released as a p-nitrophenolate anion. This p-nitrophenolate anion has absorption at 410 nm, permitting the catalytic activity of BM3 mutants to be monitored by UV-Vis spectroscopy. As shown in Fig. 3, alteration of the linker sequence greatly affected catalytic activity of BM3, with Mut-3 exhibiting the best reaction efficiency amongst the four mutants, resembling that of WT. We thus selected the linker of Mut-3 to connect the two domains of BM3.
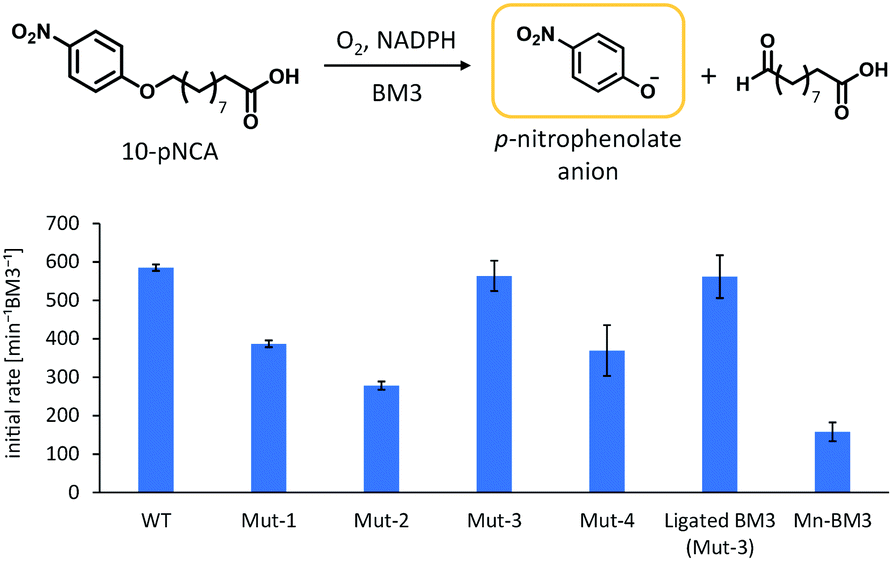 | ||
| Fig. 3 Initial rate of p-nitrophenolate formation. Reaction conditions: BM3 (0.1 μM), 10-pNCA (50 μM), NADPH (200 μM), at r.t. in Tris–HCl buffer (pH 8.2). Initial rates were calculated from the increment of the absorbance at 410 nm based on the extinction coefficient ε410 = 13.2 mM−1 cm−1 (pH 8.2). Error bar was given as the standard deviation from at least three measurements. | ||
Accordingly, we designed two expression vectors, one for the haem domain, and another for the reductase domain. The constructs were chosen so that, following ligation by SrtA, the resulting sequence was the same as Mut-3. The haem domain with the sorting motif at the C-terminus (1-446-LPATG) and the reductase domain composed of FMN and FAD domains with two glycine residues at the N-terminus (GG-447-1049) were expressed and purified. For efficient purification, Strep-tag II and TEV protease recognition sites were inserted at the N-terminus of the haem domain. The ligation reaction was performed by simply mixing the two domains in the presence of SrtA, and formation of full-length BM3 was confirmed via SDS-PAGE. Following a 15 h reaction, the SDS-PAGE showed a clear band corresponding to full-length BM3. After treatment with SrtA, ligated full-length BM3 was isolated from the reaction mixture by using a Strep-Tactin column followed by gel-filtration column chromatography (see ESI,† Fig. S1).
The catalytic activity of ligated BM3 for fatty acid hydroxylation was examined by a pNCA assay and we found that the catalytic activity was the same as that of Mut-3 (Fig. 3), indicating that full-length BM3 maintained its high catalytic activity and was reconstituted correctly.
Given that full-length P450BM3 can be prepared by simply connecting the haem and reductase domain with SrtA, we attempted to prepare full-length P450BM3 incorporating Mn-protoporphyrin IX (Mn-PPIX). Apo-form of BM3 was prepared by expressing BM3 using E. coli. under iron-limiting conditions followed by reconstitution with Mn-PPIX, as reported previously.17 The UV-vis spectrum of Mn-substituted haem domain was comparable to previously reported spectra of Mn-substituted P450s16,17,27 (see ESI,† Fig. S3). ICP-OES analysis showed that the concentration of manganese correlated well with protein concentration, with a ratio of approximately one-to-one, indicating that the haem domain and Mn-PPIX formed a stoichiometric protein–Mn-PPIX complex (see ESI,† Fig. S4). It should be noted that no significant amount of iron could be detected by ICP-OES analysis. X-ray crystallographic analysis of reconstituted haem domain also confirmed the substitution of haem by Mn-PPIX (PDB code: 5ZIS). The anomalous difference Fourier map calculated from the X-ray diffraction data collected at a wavelength of 1.75 Å displayed a significant peak at the centre of protoporphyrin IX. As shown in Fig. 4a, the anomalous scattering showed a manganese atom located at the centre of protoporphyrin IX. The sulphur atom of Cys400 can be observed coordinating to manganese (Fig. 4b), signifying correct folding of the Mn-substituted haem domain. The overall structure of the Mn-substituted haem domain was almost identical to that of haem-bound haem domain (Fig. 4c).
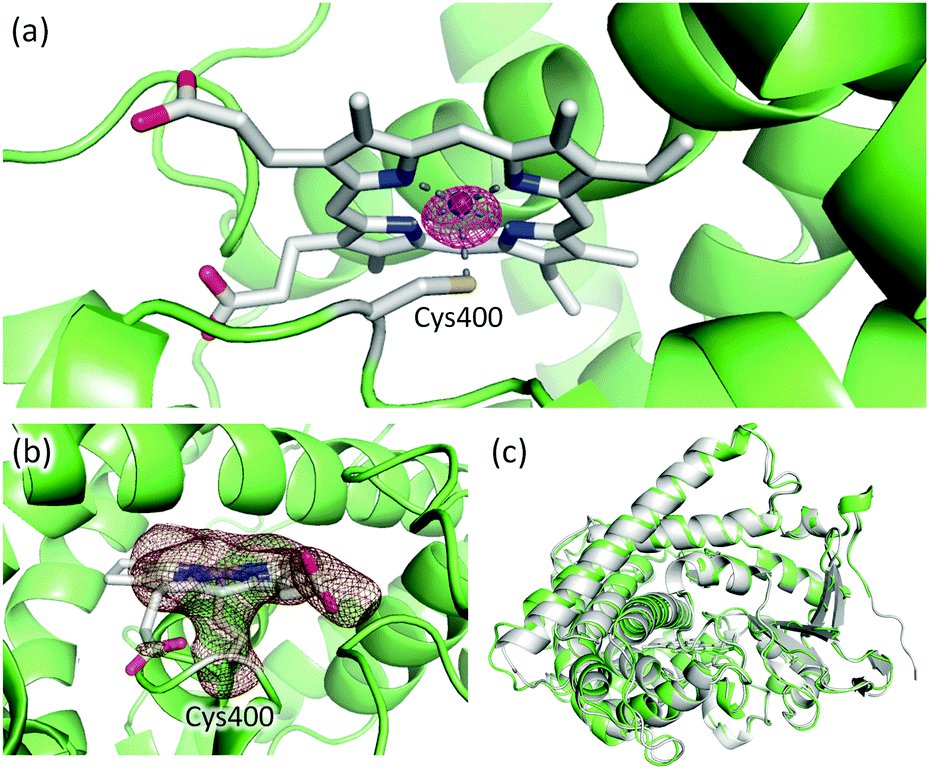 | ||
| Fig. 4 (a) Mn-PPIX and Cys400 in the Mn-substituted haem domain. The anomalous difference Fourier map is shown as a purple mesh (contoured at 5σ). The diffraction data for the map calculation was collected at a wavelength of 1.75 Å. (b) Omit map of Mn-PPIX and Cys400 is shown as a brown mesh (3σ). (c) Structural alignment of Mn-substituted (green) and haem-bound (grey, PDB: 1BU7) haem domain. | ||
The Mn-substituted haem domain was ligated to the reductase domain by SrtA, affording Mn-substituted full-length BM3 (Mn-BM3). The catalytic activity was evaluated by pNCA assay (Fig. 3). Formation of the p-nitrophenolate anion was observed via time course analysis of UV-Vis spectra on the reaction with Mn-BM3. Decreasing absorption at 340 nm was also observed, which was assigned to the consumption of NADPH as the reaction proceeded (see ESI,† Fig. S6). These results indicate that Mn-BM3 catalyses the hydroxylation of fatty acids by using NADPH as an electron source for the activation of molecular oxygen in the same manner as other P450s. Still, the catalytic efficiency was lower than that of the haem-bound wild-type BM3 expressed in its full-length form. The fact that Mn-BM3 can catalyse the hydroxylation of fatty acids encouraged us to examine propane hydroxylation with the assistance of a substrate analogue (decoy molecule) that can activate P450BM3.10 In the presence of decoy molecules, such as perfluorinated pelargonic acid (PFC9), P450BM3 can hydroxylate propane to give 2-propanol as the main product (Scheme 1). In the case of Mn-substituted P450BM3, propane was also hydroxylated, but the ratio of 1-propanol to 2-propanol was larger than that of WT-P450BM3 with iron-haem, presumably reflecting differences in the Mn-base active species (Table 1). Although the active oxygen species of manganese has not yet been identified, MnV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O represents the most likely candidate, as it can promote two electron oxidations, such as hydroxylation.
O represents the most likely candidate, as it can promote two electron oxidations, such as hydroxylation.
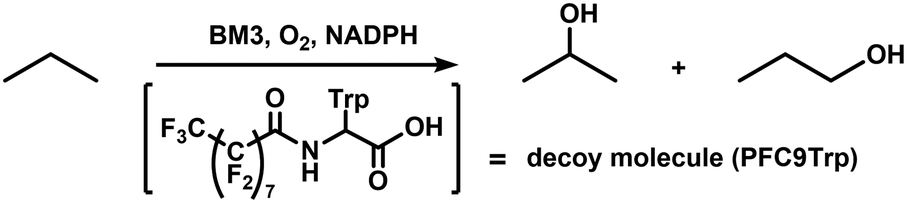 | ||
| Scheme 1 Propane oxidation using BM3 with decoy molecule (PFC9Trp). | ||
| TOF [min−1] | Regioselectivity | ||
|---|---|---|---|
| 1-Propanol [%] | 2-Propanol [%] | ||
| The reaction was performed in 1 mL of O2/propane buffer containing 0.5 μM BM3, 100 μM PFC9Trp, and 5 mM NADPH at r.t. for 10 min. | |||
| Wild-type BM3 (haem-binding) | 176 ± 11 | 4.8 | 95.2 |
| Mn-BM3 | 13 ± 1 | 17.5 | 82.5 |
Although the electron transfer from redox partners to Mn-PPIX is difficult, due to the relatively negative redox potential of Mn-PPIX,27 we speculated that the structural features of self-sufficient P450s enabled us to realise this challenging electron transfer.
In conclusion, we have successfully developed a simple method for the haem substitution of full-length P450 BM3 (BM3), exploiting the transpeptidase Sortase A (SrtA). We utilised SrtA for the ligation of reconstituted haem domain to its reductase domain to generate reconstituted full-length BM3, thus circumventing the challenging reconstitution of full-length BM3. Full-length BM3 reconstituted by SrtA maintained its original high catalytic activity, proving that this method can be used for the facile reconstitution of BM3. Subsequently, we successfully prepared Mn-substituted full-length BM3 (Mn-BM3). Surprisingly, Mn-BM3 retained catalytic activity for fatty acid hydroxylation even though, to the best of our knowledge, the catalytic activity of haem-substituted P450 with its redox partners using molecular oxygen has never been reported before. These results suggest that this method for haem substitution permits the modification of BM3 reactivity and selectivity by altering the central metal and/or tuning the redox potential of prosthetic groups. Ligation with a redox partner also enables P450s to activate molecular oxygen without the requirement for peroxides such as mCPBA, hydrogen peroxide, or iodosylbenzene, and hence represents an effective approach for the construction of environmentally friendly biocatalysts. The procedure described herein is in principle applicable to other self-sufficient P450s or other kinds of haemoproteins consisting of several domains. Moreover, as haem determines the reactivity of haemoenzymes, this method can expand the reaction scope of BM3 by substituting haem with diverse artificial metal complexes. Thus, we believe our method brings new perspective to the construction of novel redox-active artificial metalloenzymes.
This work was supported by Grants-in-Aid for Scientific Research (B) to Y. W. (17H03087) from the Ministry of Education, Culture, Sports, Science, and Technology (Japan) and JST CREST Grant Number JPMJCR15P3, Japan. This work was also supported by JSPS KAKENHI Grant Number JP15H05806 in Precisely Designed Catalysts with Customized Scaffolding to O. S.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. R. Ortiz de Montellano, Cytochrome P450: structure, mechanism, and biochemistry, Plenum Press, New York, 3rd edn, 2005 Search PubMed.
- R. Fasan, ACS Catal., 2012, 2, 647–666 CrossRef.
- S. T. Jung, R. Lauchli and F. H. Arnold, Curr. Opin. Biotechnol., 2011, 22, 809–817 CrossRef PubMed.
- O. Shoji and Y. Watanabe, Chem. Lett., 2017, 46, 278–288 CrossRef.
- H. M. Girvan and A. W. Munro, Curr. Opin. Chem. Biol., 2016, 31, 136–145 CrossRef PubMed.
- C. J. C. Whitehouse, S. G. Bell and L.-L. Wong, Chem. Soc. Rev., 2012, 41, 1218–1260 RSC.
- B. C. Chenna, B. A. Shinkre, J. R. King, A. L. Lucius, S. V. L. Narayana and S. E. Velu, Bioorg. Med. Chem. Lett., 2008, 18, 380–385 CrossRef PubMed.
- M. A. Noble, C. S. Miles, S. K. Chapman, D. A. Lysek, A. C. Mackay, G. A. Reid, R. P. Hanzlik and A. W. Munro, Biochem. J., 1999, 339, 371–379 CrossRef PubMed.
- N. Kawakami, O. Shoji and Y. Watanabe, Angew. Chem., Int. Ed., 2011, 50, 5315–5318 CrossRef PubMed.
- Z. Cong, O. Shoji, C. Kasai, N. Kawakami, H. Sugimoto, Y. Shiro and Y. Watanabe, ACS Catal., 2015, 5, 150–156 CrossRef.
- K. Suzuki, J. K. Stanfield, O. Shoji, S. Yanagisawa, H. Sugimoto, Y. Shiro and Y. Watanabe, Catal. Sci. Technol., 2017, 7, 3332–3338 Search PubMed.
- O. Shoji, S. Yanagisawa, J. K. Stanfield, K. Suzuki, Z. Cong, H. Sugimoto, Y. Shiro and Y. Watanabe, Angew. Chem., Int. Ed., 2017, 56, 10324–10329 CrossRef PubMed.
- S. Dezvarei, H. Onoda, O. Shoji, Y. Watanabe and S. G. Bell, J. Inorg. Biochem., 2018, 183, 137–145 CrossRef PubMed.
- F. W. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef.
- L. Fruk, C.-H. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem., Int. Ed., 2009, 48, 1550–1574 CrossRef PubMed.
- V. S. Lelyveld, E. Brustad, F. H. Arnold and A. Jasanoff, J. Am. Chem. Soc., 2011, 133, 649–651 CrossRef PubMed.
- N. Kawakami, O. Shoji and Y. Watanabe, ChemBioChem, 2012, 13, 2045–2047 CrossRef PubMed.
- E. W. Reynolds, M. W. McHenry, F. Cannac, J. G. Gober, C. D. Snow and E. M. Brustad, J. Am. Chem. Soc., 2016, 138, 12451–12458 CrossRef PubMed.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef PubMed.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef PubMed.
- S. K. Mazmanian, H. Ton-That and O. Schneewind, Mol. Microbiol., 2001, 40, 1049–1057 CrossRef PubMed.
- S. D. Zink and D. L. Burns, Infect. Immun., 2005, 73, 5222–5228 CrossRef PubMed.
- S. Govindaraj and T. L. Poulos, Biochemistry, 1995, 34, 11221–11226 CrossRef PubMed.
- S. Govindaraj and T. L. Poulos, Protein Sci., 1996, 5, 1389–1393 CrossRef PubMed.
- U. Schwaneberg, C. Schmidt-Dannert, J. Schmitt and R. D. Schmid, Anal. Biochem., 1999, 269, 359–366 CrossRef PubMed.
- M. H. Gelb, W. A. Toscano and S. G. Sligar, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 5758–5762 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc02760a |
| This journal is © The Royal Society of Chemistry 2018 |