
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Orthogonal, dual protein labelling by tandem cycloaddition of strained alkenes and alkynes to ortho-quinones and azides†
Jorick J.
Bruins
 a,
Daniel
Blanco-Ania
b,
Vincent
van der Doef
a,
Floris L.
van Delft
*a and
Bauke
Albada
*a
a,
Daniel
Blanco-Ania
b,
Vincent
van der Doef
a,
Floris L.
van Delft
*a and
Bauke
Albada
*a
aLaboratory of Organic Chemistry, Wageningen University & Research, Stippeneng 4, 6708 WE, Wageningen, the Netherlands. E-mail: floris.vandelft@wur.nl; bauke.albada@wur.nl
bInstitute for Molecules and Materials, Radboud University, Nijmegen, the Netherlands
First published on 7th June 2018
Abstract
Reaction of cyclopropanated trans-cyclooctene (cpTCO) with in situ generated ortho-quinone is an efficient tool for bioorthogonal protein conjugation. The (4+2)-cycloaddition of cpTCO with ortho-quinone is significantly faster than its cyclooctyne counterpart (BCN). Orthogonal, tandem cpTCO–quinone and BCN–azide cycloadditions afforded a homogeneous, dual labelled antibody–drug conjugate.
Bioorthogonal chemistry can be defined as the combined arsenal of reactions between two (or more) unique molecular functionalities that together rapidly form a covalent bond, but are inert to the remaining repertoire of chemical groups present in the surroundings.1 As a consequence of this high functional group compatibility, bioorthogonal reactions have inter alia been broadly applied for detection, immobilization and functionalization of biomolecules, such as carbohydrates, nucleic acid and proteins.2 For example, the Staudinger ligation3 paved the way for the selective imaging of cell-surface azidosugars, azide–alkyne click chemistry is a well-established tool in the field of DNA/RNA origami,4 and oxime ligation5 has proven a powerful technology for protein modification.6 Arguably the most powerful bioorthogonal reactions are those defined by metal-free, strain-promoted cycloadditions of alkynes or alkenes,7 of which stand out the strain-promoted azide–alkyne cycloaddition (SPAAC)8 and the inverse electron-demand Diels–Alder cycloaddition (SPIEDAC)9 of strained alkenes and tetrazines. Conveniently, it has been established that with judicious choice of functionalities, a next level of orthogonality can be achieved, involving mutually exclusive, bioorthogonal reactions proceeding in tandem or even concurrently.10 Besides inherent orthogonality in reactivity, or the application of targeting agents,11 the use of external stimuli to induce bioorthogonal reactivity with temporal and spatial control has also been described, mostly based on photolysis of tetrazole10a or cyclopropenone10b to nitrile imides or cyclooctyne derivatives, respectively. We,12 and others,13 recently described that enzymatic oxidation of aromatic rings can also be used for temporally controlled bioorthogonal chemistry. Specifically, we demonstrated that strain-promoted oxidation-controlled ortho-quinone cycloaddition (SPOCQ) with cyclooctyne,14 can be successfully applied for the site-specific conjugation of a cyclooctyne-functionalized fluorophore or a toxic payload to tyrosine-engineered proteins, including the monoclonal antibody trastuzumab,12 as well as for highly efficient and complete surface modification.15 Interestingly, with these developments, tyrosine is emerging as a very versatile amino acid for various peptide16 and protein17 derivatization strategies. With the present study, we have expanded the arsenal of SPOCQ chemistry by demonstrating that besides cyclooctynes strained alkenes also display high reactivity for ortho-quinones. Specifically, we show that cyclopropanated trans-cyclooctene (cpTCO)18 rapidly reacts with ortho-quinones (a.k.a. 1,2-quinones),19 enabling clean and efficient tyrosinase-mediated protein conjugation. In addition, the inertness of cpTCO for azides provides a next level of dual, orthogonal bioconjugation, which may be executed without additional purification steps in between reactions.
Before we embarked on the potential application of trans-cyclooctenes for SPOCQ modification of proteins, we tested if the strain-promoted click reaction between trans-cyclooctenes and ortho-quinone would take place. To this end, 4-tert-butyl-1,2-quinone 1 was mixed with cpTCO 2 (Scheme 1), and the disappearance of the absorbance that corresponds to the presence of ortho-quinone 1 at 395 nm was monitored.20 Much to our delight, the reaction of quinone 1 with cpTCO 2 not only occurs (Fig. 1 and Fig. S3–S5, ESI†), but was actually found to take place faster than that with bicyclo[6.1.0]non-4-yne (BCN)213. Specifically, the SPOCQ reaction of quinone 1 with cpTCO 2 proceeded with a rate-constant of 2900 ± 115 M−1 s−1, a near 3-fold increase versus the (corrected) rate constant of BCN 3 with quinone 1 (1112 ± 8 M−1 s−1, N = 3).12 In the same setup we found a rate constant of 10.4 ± 1.8 M−1 s−1 for the reaction of regular TCO with quinone 1. Apparently, the fused cyclopropane ring increases the rate approximately 300-fold, in analogy with enhancement of reaction rate with tetrazine.18 For comparison, cpTCO–SPOCQ is more than 103 times faster than reaction of quinone 1 with a cyclopropene (<1.7–1.9 M−1 s−1)22 and strain-promoted cycloadditions involving azides (0.01–1 M−1 s−1),7 however slightly slower than SPIEDAC reaction of regular TCO with tetrazines (2000 to >5000 M−1 s−1),13 and much slower than the fastest known cpTCO–tetrazine ligations (reaching above 106 M−1 s−1).23 Nevertheless, in contrast to azide and tetrazine, an ortho-quinone is readily generated from a canonical amino acid (tyrosine), and therefore has the inherent advantage of (inducible) bioconjugation chemistry to native proteins.
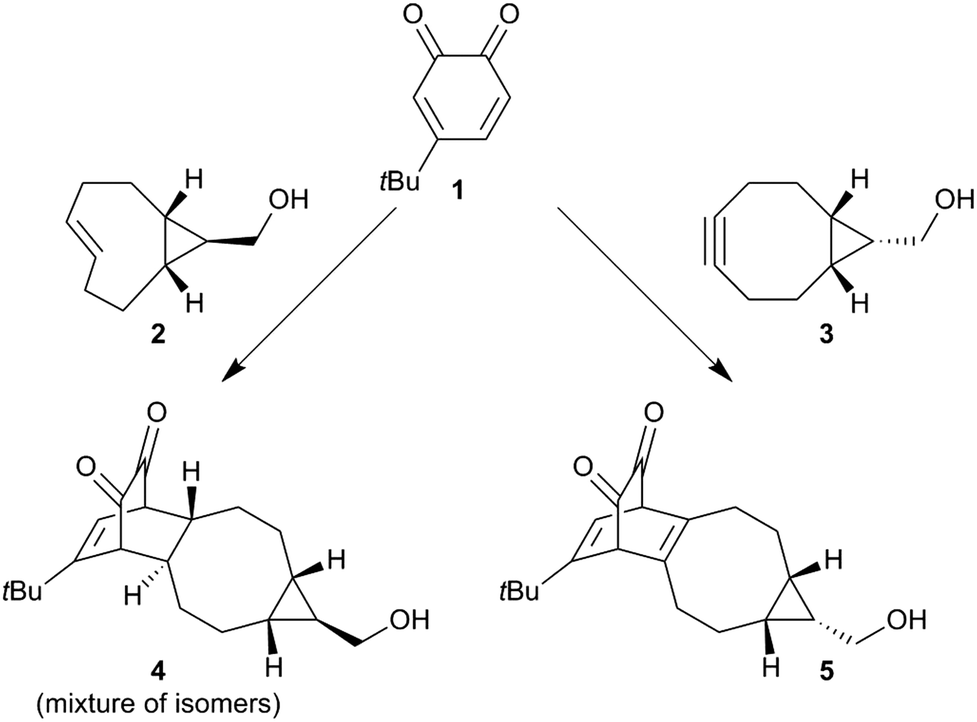 | ||
| Scheme 1 Reaction between quinone 1 and either exo-cpTCO 2 or endo-BCN 3 to give alkene-SPOCQ product 4 (this work) or alkyne-SPOCQ product 5.14 | ||
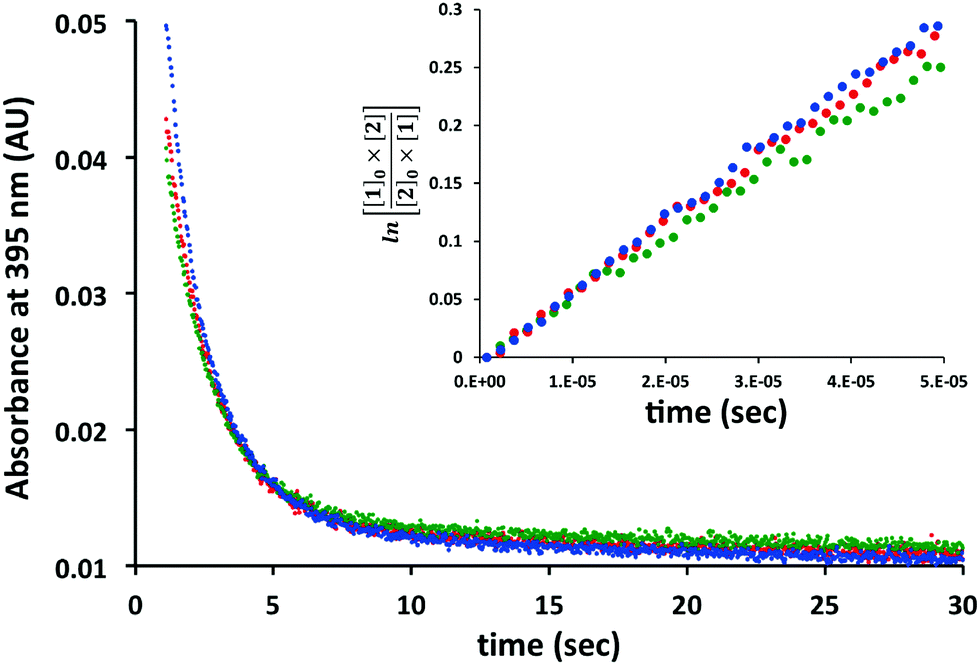 | ||
Fig. 1 Determination of the reaction rate constant of quinone 1 (100 μM) with cpTCO 2 (100 μM) in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (1:1). Three separate experiments are depicted in the graph. :MeOH (1:1). Three separate experiments are depicted in the graph. | ||
After this, we explored the suitability of cpTCO-quinone cycloaddition for protein conjugation (Fig. 2 and Fig. S6–S9, ESI†). To this end, a cpTCO-functionalized lissamine–PEG conjugate was synthesized and compared head-to-head to its BCN counterpart in labelling of a model antibody (trastuzumab), which contained an exposed G4Y-tag on each heavy chain (abbreviated as Tras[HC]G4Y). Previously, we already showed that without this G4Y-tag, labelling of a smaller protein or an antibody did not proceed.12 In analogy to our previous work, we studied the alkene–SPOCQ reaction between cpTCO–PEG–lissamine and Tras[HC]G4Y upon incubation with mushroom tyrosinase (mTyr) to oxidize the exposed tyrosine phenol ring of the G4Y-tag to ortho-quinone.19 Indeed, when Tras[HC]G4Y was mixed with mTyr in the presence of cpTCO–lissamine, rapid labelling of the antibody was observed that apparently levelled off after ∼90 minutes (Fig. 2, lanes 4–11); no labelling was observed in the absence of mTyr (Fig. 2, lane 3). In the absence of a G4Y-tag on the light chain, labelling of this part of the antibody was not observed (i.e. no fluorescent bands around 25 kDa were observed), thus demonstrating (1) the bioorthogonality of alkene–SPOCQ, and (2) the need for a G4Y-tag on the protein. Based on HPLC analysis, a yield of ∼95% may be calculated (Fig. S10–S12, ESI†), with reduced side-product formation in comparison to alkyne-SPOCQ (Fig. S9, ESI†), i.e. antibody oligomers formed by cross-linking of the ortho-quinone with endogenous protein nucleophiles (Fig. 2, lane 2). The reduced formation of protein oligomers for cpTCO versus BCN further corroborates the higher reaction rate with ortho-quinones. Since oxidation of tyrosine residues by mTyr is the rate-limiting step in either SPOCQ reaction,20 the total reaction time does not decrease despite the higher reaction rate for cpTCO. However, the short lifetime of the generated quinone favours labelling with the fast reacting cpTCO by reducing side product formation.
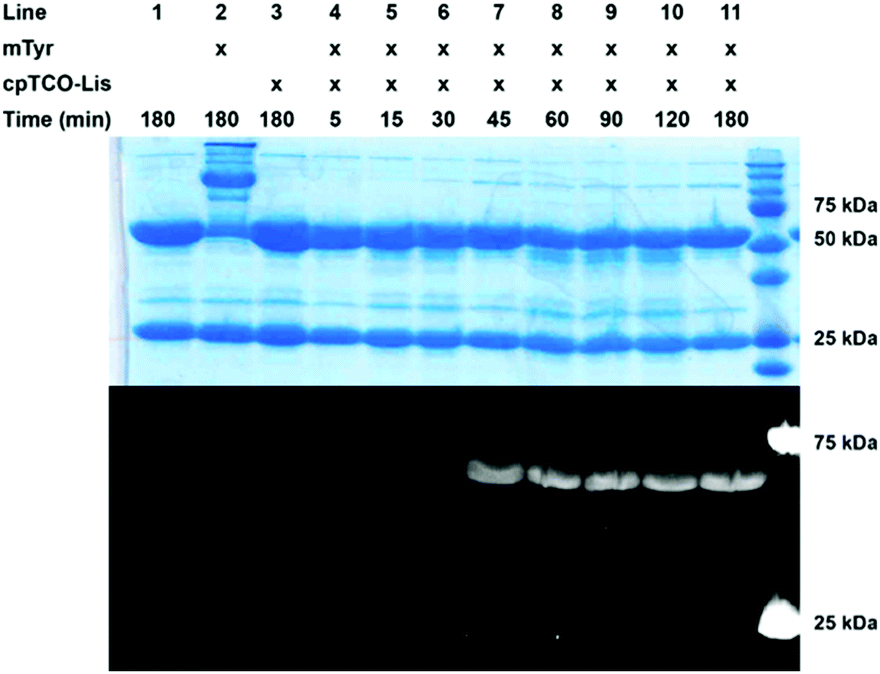 | ||
| Fig. 2 Time-resolved SDS-PAGE analysis, based on heavy chain labelling, of cpTCO–PEG–lissamine SPOCQ with Tras[HC]G4Y (lane 1) in the presence of mTyr (lanes 4–11). No reaction is apparent after 3 h in the absence of mTyr (lane 3), while the present of mTyr alone (absence of cpTCO reagent) leads to degradation/oligomer formation (lane 2). | ||
In order to exploit the lack of reactivity of cpTCO for azides, and high reaction rate with ortho-quinones, we envisioned that tandem, dual labelling of an antibody should be accessible by orthogonal reactions of BCN and cpTCO with azide and (exposed) tyrosine, respectively. To test this, we explore the possibility of tandem installation of two different functional moieties onto a glycan-remodelled monoclonal antibody24 by cpTCO–SPOCQ and BCN–SPAAC, executed in either order. We started by the cpTCO–SPOCQ reaction on the Tras[HC]G4Y C-terminus followed by SPAAC modification of azido-glycan with BCN–MMAE (method A), but also checked method B (Fig. 3A).‡
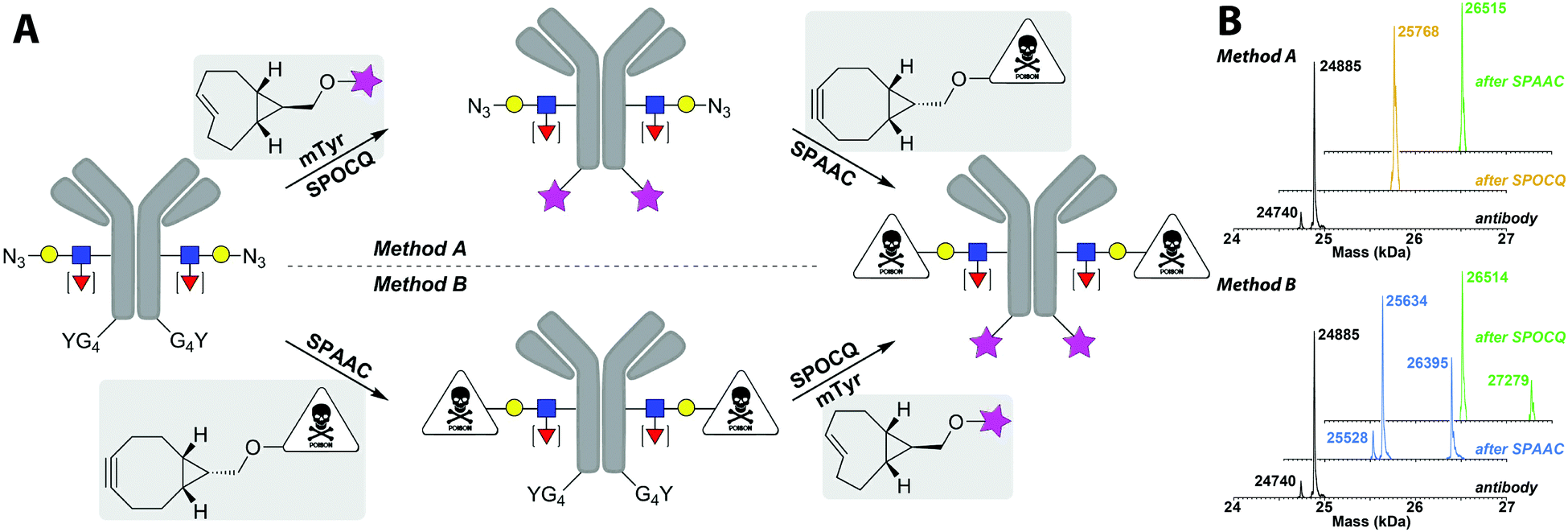 | ||
| Fig. 3 (A) Schematic depiction of the two routes for dual labelling of antibodies with lissamine and MMAE. One route employs the modification of the G4Y-tag using the cpTCO–SPOCQ reaction with cpTCO–lissamine, followed by SPAAC reaction with BCN–MMAE. The other route employs first a SPAAC reaction on the azido-glycan followed by SPOCQ on the G4Y-tag of the heavy chain. (B) The deconvoluted mass spectra (crude) of the different steps of the tandem bioconjugation process using the two methods are shown. The main peak in the black trace identifies with GlcNAc-(1,6-Fuc)-6-N3-GalNAc, the minor peak at Δ – 145: GlcNAc-6-N3-GalNAc; in-line fragmentation of the MMAF toxin is also visible. | ||
In order to obtain the same conjugate via both routes, we used cpTCO–PEG–lissamine and BCN–sulfamide–MMAE (see ESI† for structures) with a cathepsin-cleavable linker (MMAE = monomethyl auristatin E, a potent antimitotic agent that inhibits cell division). In addition, the sulfamide spacer in the BCN unit enhanced solubility and conjugation efficiency.25
Thus, clean and efficient enzymatic glycan remodelling was achieved by endoglycosidase and glycosyltransferase, to install 6-azido-GalNAc onto the core-GlcNAc of transiently expressed Tras[HC]G4Y (Fig. 3A and Fig. S13, ESI†).14 Next, a SPOCQ reaction between cpTCO–lissamine and the G4Y-tag on the antibody heavy chain was performed in the presence of mTyr (Fig. S14, ESI†). The observed mass of 25768 Da (Fig. 3B, yellow peak) corresponds to the SPOCQ product of the azido-glycan antibody and cpTCO–lissamine (expected: 25766 Da), and corroborates the complete lack of reactivity of the cpTCO moiety with the azido-group on the glycan chain. Subsequently, SPAAC modification of the azido-functionalized glycan chain with BCN–MMAE resulted in the clean formation of the dual labelled fluorescent antibody–drug conjugate. HPLC analysis showed almost complete conversion (Fig. S15, ESI†), and MS analysis identified the presence of the target compound with a FW of 26516 Da (expected: 27275 Da) (Fig. 3B, green peak in top spectrum). The difference between the observed an expected mass corresponds to loss of the C-terminal part of MMAE due to fragmentation in the mass chamber (Δ 762 Da).
Lastly, the tandem labelling steps were performed in reverse order, i.e. first SPAAC reaction on the remodelled azido-glycan chain using BCN–MMAE SPAAC (Fig. S16, ESI†), then cpTCO–lissamine SPOCQ reaction on the G4Y-tag upon tyrosinase oxidation (Fig. S17, ESI†). As expected, the same final product is obtained, albeit via a different intermediate product. We were happy to find that in this case, besides the mass of the linker-fragmented final conjugate (at 26514 Da), the mass of the desired dual labelled ADC is also observed at 27279 Da (expected: 27275 Da) (Fig. 3B, lower set of MS spectra). This difference is presumably due to some experimental variability during the MS analysis.
In conclusion, we have uncovered a useful addition to the toolbox of copper-free click chemistry, i.e. the rapid reaction of strained alkenes and ortho-quinones, and used this reaction to establish a convenient and high-yielding new strategy for the labelling of antibodies. The rate of the SPOCQ reaction between cpTCO and ortho-quinone is ∼2.5 times higher than that for BCN, ∼300 times higher than that for TCO (Fig. S30 and S31, ESI†), and >103 times higher than that for cyclopropene; the last two strained alkenes would be too slow for efficient SPOCQ labelling of proteins, due to the high reactivity of the quinone, e.g. with competing nucleophiles. This high reactivity of cpTCO enables faster and cleaner conjugation, as superior labelling of a G4Y-derivatized antibody with a fluorescent moiety was demonstrated when compared to its BCN counterpart. Ultimately, we developed a convenient protocol for the dual labelling of an antibody with a toxic payload and a dye using the orthogonal character of cpTCO and BCN, which may find application in the assessment of internalization potential of glycan-conjugated antibody–drug conjugates for targeted therapy.25 Our setup allows a convenient preparation of a glycan-remodelled azido-containing antibody that also contains a peptidic G4Y tag, and to use this in a two-step dual-labelling strategy to obtain homogeneous ADCs. We note that other methods have been reported for metal-free click conjugation to unnatural amino acids genetically encoded into monoclonal antibodies, for example p-acetylphenylalanine26 or p-azidomethylphenylalanine,27 followed by oxime ligation or SPAAC, respectively. However, low antibody titers (1 g L−1 maximum), which are significantly lower than expression titers on monoclonal antibodies based on canonical amino acids alone (3–7 g L−1), limit such applications. Similar arguments apply to genetic encoding of L-DOPA into proteins, which is also suitable for SPOCQ modification.28 We expect that our current method will find widespread application, not only in the preparation of ADCs but extending to other applications in the field of bioconjugation, such as PEGylation, immobilization, protein–protein dimer formation, and beyond. Efforts along those lines are currently ongoing in our laboratory.
Conflicts of interest
F. L. V. D. declares that he is also an employee and share-holder of Synaffix BV.Notes and references
- (a) M. D. Best, Biochemistry, 2009, 48, 6571 CrossRef PubMed; (b) R. K. V. Lim and Q. Lin, Chem. Commun., 2010, 46, 1589 RSC; (c) F. Liu, Y. Liang and K. N. Houk, Acc. Chem. Res., 2017, 50, 2297 CrossRef PubMed.
- S. S. van Berkel and F. L. van Delft, Drug Discovery Today, 2013, 10, e45 CrossRef PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef PubMed.
- V. Cassinelli, B. Oberleitner, J. Sobotta, P. Nickels, G. Grossi, S. Kempter, T. Frischmuth, T. Liedl and A. Manetto, Angew. Chem., Int. Ed., 2015, 54, 7795 CrossRef PubMed.
- E. H. M. Lempens, B. A. Helms, M. Merckx and E. W. Meijer, ChemBioChem, 2009, 10, 658 CrossRef PubMed.
- K. K.-Y. Kung, K.-F. Wong, K.-C. Leung and M.-K. Wong, Chem. Commun., 2013, 49, 6888 RSC.
- M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805 CrossRef PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518 CrossRef PubMed.
- (a) D. M. Patterson and J. A. Prescher, Curr. Opin. Chem. Biol., 2015, 28, 141 CrossRef PubMed; (b) J. A. Wagner, D. Mercadante, I. Nikic, E. A. Lemke and F. Grater, Chem. – Eur. J., 2015, 21, 12431 CrossRef PubMed.
- J. Ohata and Z. T. Ball, J. Am. Chem. Soc., 2017, 139, 12617 CrossRef PubMed.
- J. J. Bruins, A. H. Westphal, B. Albada, K. Wagner, L. Bartels, H. Spits, W. J. H. van Berkel and F. L. van Delft, Bioconjugate Chem., 2017, 28, 1189 CrossRef PubMed.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, J. Am. Chem. Soc., 2016, 138, 5978 CrossRef PubMed.
- (a) A. Borrmann, O. Fatunsin, J. Dommerholt, A. M. Jonker, D. W. P. M. Lowik, J. C. M. van Hest and F. L. van Delft, Bioconjugate Chem., 2015, 26, 257 CrossRef PubMed; (b) A. M. Jonker, A. Borrmann, E. R. H. van Eck, F. L. van Delft, D. W. P. M. Lowik and J. C. M. van Hest, Adv. Mater., 2015, 27, 1235 CrossRef PubMed.
- R. Sen, J. Escorihuela, F. van Delft and H. Zuilhof, Angew. Chem., Int. Ed., 2017, 56, 3299 CrossRef PubMed.
- H. B. Albada, F. Wieberneit, I. Dijkgraaf, J. H. Harvey, J. L. Whistler, R. Stoll, N. Metzler-Nolte and R. H. Fish, J. Am. Chem. Soc., 2012, 134, 10321 CrossRef PubMed.
- J. Ohata, M. K. Miller, C. M. Mountain, F. Vohidov and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 2827 CrossRef PubMed.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646 CrossRef PubMed.
- J. J. Bruins, B. Albada and F. L. van Delft, Chem. – Eur. J., 2018, 24, 4749 CrossRef PubMed.
- F. Garcia-Molina, A. N. P. Hiner, L. G. Fenoll, J. N. Rodriguez-Lopez, P. A. Garcia-Ruiz, F. Garcia-Canovas and J. Tudela, J. Agric. Food Chem., 2005, 53, 3702 CrossRef PubMed.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422 CrossRef PubMed.
- D. Gahtory, R. Sen, A. R. Kuzmyn, J. Escorihuela and H. Zuilhof, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201800937.
- (a) K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317 CrossRef PubMed; (b) R. Rossin, S. M. van den Bosch, W. ten Hoeve, M. Carvelli, R. M. Versteegen, J. Lub and M. S. Robillard, Bioconjugate Chem., 2013, 24, 1210 CrossRef PubMed; (c) A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770 RSC.
- R. van Geel, M. A. Wijdeven, R. Heesbeen, J. M. M. Verkade, A. A. Wasiel, S. S. van Berkel and F. L. van Delft, Bioconjugate Chem., 2015, 26, 2233 CrossRef PubMed.
- J. M. M. Verkade, M. A. Wijdeven, R. van Geel, B. M. G. Janssen, S. S. van Berkel and F. L. van Delft, Antibodies, 2018, 7, 12 CrossRef.
- F. Tian, Y. Lu, A. Manibusan, A. Sellers, H. Tran, Y. Sun, T. Phuong, R. Barnett, B. Hehli, F. Song, M. J. DeGuzman, S. Ensari, J. K. Pinkstaff, L. M. Sullivan, S. L. Biroc, H. Cho, P. G. Schultz, J. DiJoseph, M. Dougher, D. Ma, R. Dushin, M. Leal, L. Tchistiakova, E. Feyfant, H.-P. Gerber and P. Sapra, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1766 CrossRef PubMed.
- E. S. Zimmerman, T. H. Heibeck, A. Gill, X. Li, C. J. Murray, M. R. Madlansacay, C. Tran, N. T. Uter, G. Yin, P. J. Rivers, A. Y. Yam, W. D. Wang, A. R. Steiner, S. U. Bajad, K. Penta, W. J. Yang, T. J. Hallam, C. D. Thanos and A. K. Sato, Bioconjugate Chem., 2014, 25, 351 CrossRef PubMed.
- S. Kim, B. H. Sung, S. C. Kim and H. S. Lee, Chem. Commun., 2018, 54, 3002 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, synthetic protocols, HR-MS analysis of probes, MS-analysis of conjugates, results of control experiments. See DOI: 10.1039/c8cc02638f |
| ‡ This work is funded by the NWO Gravity Program Institute for Chemical Immunology (ICI). We thank Synaffix for providing the reagents and expertise to perform the glycan remodelling and metal-free conjugation chemistry, in particular Dr R. van Geel. |
| This journal is © The Royal Society of Chemistry 2018 |