
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon dioxide-based facile synthesis of cyclic carbamates from amino alcohols†
Teemu
Niemi
a,
Israel
Fernández
 *b,
Bethany
Steadman
a,
Jere K.
Mannisto
a and
Timo
Repo
*a
*b,
Bethany
Steadman
a,
Jere K.
Mannisto
a and
Timo
Repo
*a
aDepartment of Chemistry, University of Helsinki, P.O. Box 55, 00014, Finland. E-mail: timo.repo@helsinki.fi
bDepartamento de Quimica Orgánica I and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria, 28040 Madrid, Spain. E-mail: israel@quim.ucm.es
First published on 5th March 2018
Abstract
We report herein a straightforward general method for the synthesis of cyclic carbamates from amino alcohols and carbon dioxide in the presence of an external base and a hydroxyl group activating reagent. Utilizing p-toluenesulfonyl chloride (TsCl), the reaction proceeds under mild conditions, and the approach is fully applicable to the preparation of various high value-added 5- and 6-membered rings as well as bicyclic fused ring carbamates. DFT calculations and experimental results indicate a SN2-type reaction mechanism with high regio-, chemo-, and stereoselectivity.
Cyclic carbamate core structures are present in a plethora of valuable chemicals. In particular, 2-oxazolidinones are found in chiral auxiliaries (with 4-substitution) and in superantibiotics such as Linezolid and Tedizolid (with 3-aryl,5-alkyl substitution pattern), whereas aryl-fused 6-membered rings are present in some HIV-battling antiretrovirals and N-methyl-D-aspartate (NMDA) receptor antagonists.1–5 These compounds are commonly synthesized by utilizing hazardous or expensive reagents such as isocyanides and phosgene.1,4,6,7 As such, the replacement of these starting materials with the relatively cheap and safer carbon dioxide offers a unique opportunity to enhance the sustainability and greenness of the synthesis of these high value-added chemicals.
Several strategies for CO2-based preparation of cyclic carbamates have been reported, such as CO2's cycloaddition to aziridines, oxetanes, or amino epoxides,8–12 and the transesterification of amines with cyclic organic carbonates, which are formed in situ in the cycloaddition of carbon dioxide and epoxides.13,14 More recently, approaches utilizing the addition of CO2 to acyclic unsaturated compounds such as alkenes, alkynes, and propargylic amines or alcohols, have garnered attention.15–26 Meanwhile, our group has studied the synthesis of cyclic carbamates utilizing haloalkylamines,27 and the multicomponent reaction between anilines, dihaloalkanes, and carbon dioxide.28 While certainly more efficient than conventional non-CO2-based carbamate syntheses, all these methods have one or more drawbacks, namely the lack of stereoselectivity,29 toxicity, and commercial unavailability of the starting materials, or the inability to easily access the substitution patterns required for pharmaceuticals.
Amino alcohols (AAs) are often seen as ideal nitrogen sources in the synthesis of cyclic carbamates from CO2 due to their inexpensiveness, nontoxicity, and ready accessibility. However, the cyclization of AAs with carbon dioxide usually requires high temperatures and pressures even when catalysts, dehydrating agents, or electrochemical methods are employed.8,30–40 As a result of these harsh conditions, side reactions (such as a second dehydration to yield the corresponding cyclic urea) are commonplace.30,32,34,35,41 Furthermore, previous reports are mainly limited to the synthesis of alkyl substituted oxazolidinones, and additional functionalization is required to prepare high value-added chemicals.
Herein, we present the synthesis of cyclic carbamates from carbon dioxide and amino alcohols. Utilizing p-toluenesulfonyl chloride (TsCl) as an activating reagent, the reaction proceeds under mild conditions with good yields and high enantiomeric excess, giving 2-oxazolidinones with the 3-aryl-5-alkyl substitution pattern desired for pharmaceutical applications in a single step. This straightforward method offers predictable regio-, chemo-, and stereoselectivity, and the scope is expandable to the syntheses of 6-membered rings and fused bicyclic compounds, making the process unprecedently versatile. In addition, the reaction mechanism was elucidated by DFT calculations.
Our one-pot approach involves the formation of a carbamate salt from CO2 and AAs in the presence of an external base, followed by the tosylation of the alcohol group to improve its leaving group character, and the subsequent ring-closing via intramolecular substitution (Fig. 1). ROH-tosylation has been previously employed in the synthesis of cyclic carbonates from 1,3-diols;42 here, however, our obvious challenge is the competing N-tosylation reaction, which also prevents the formation of the carbamate moiety and subsequently inhibits the cyclization reaction. Thus, we started our study by finding the optimal reaction conditions in which the formation of the N-tosylated product is minimized (Table 1). Using the simple, inexpensive and commercially available 2-(phenylamino)ethan-1-ol 1a as a model substrate, we first investigated the reaction conditions we had previously employed successfully in the synthesis of N-aryl-2-oxazolidinones from anilines and dibromoalkanes (Table 1, entry 1).28 However, N-tosylation is heavily favored under these conditions, as the carbamate species is too labile to sufficiently protect the nitrogen from a reaction with TsCl. Indeed, decreasing the temperature and increasing the CO2 pressure to shift the equilibrium towards carbamate formation (Table 1, entry 2) leads to a clear increase in selectivity. Lowering the temperature or raising the pressure any further gives no significant improvement, so the rest of the experiments were conducted at RT and 5 bar CO2.
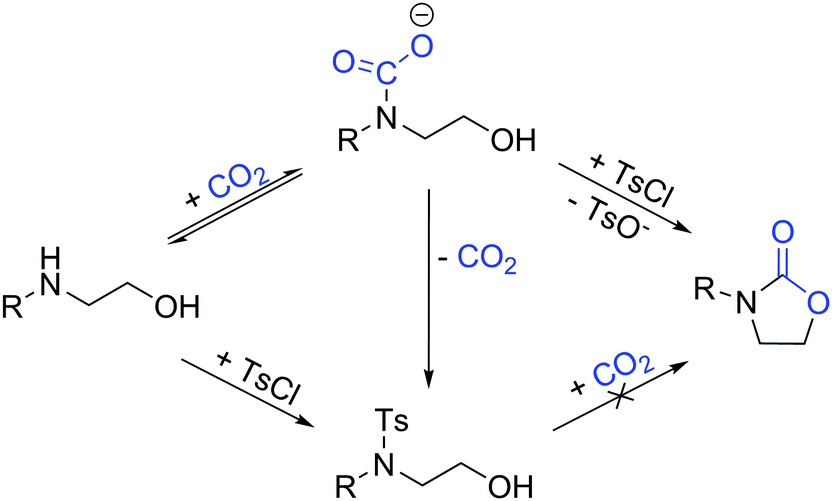 | ||
| Fig. 1 Tosyl-assisted cyclization of carbon dioxide and amino alcohols in the presence of an external base. The carbamate species must be formed first and stabilized to avoid the competing N-tosylation reaction, which prevents the formation of the desired product. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base | p (bar) | T (°C) | Conversiona (%) | Selectivityb (%) |
| a Based on total GC yield of 2a and 3a with mesitylene as external standard. b Defined as yield(2a)/yield(2a + 3a). c TMG = 1,1,3,3-tetramethylguanidine. | ||||||
| 1 | MeCN | Cs2CO3 | 1 | 60 | 72 | 16 |
| 2 | MeCN | Cs2CO3 | 5 | RT | 41 | 65 |
| 3 | EtOH | Cs2CO3 | 5 | RT | 25 | 92 |
| 4 | DCM | Cs2CO3 | 5 | RT | 5 | 70 |
| 5 | DMF | Cs2CO3 | 5 | RT | 10 | 100 |
| 6 | Acetone | Cs2CO3 | 5 | RT | 50 | 100 |
| 7 | Acetone | Et3N | 5 | RT | 18 | 47 |
| 8 | Acetone | TMGc | 5 | RT | 17 | 100 |
| 9 | Acetone | KOH | 5 | RT | 12 | 14 |
| 10 | Acetone | K2CO3 | 5 | RT | 27 | 40 |

Next, we screened different combinations of solvents and bases. Polar protic solvents (e.g.Table 1, entry 3) react with TsCl and are thus not suitable for this reaction, while nonpolar solvents are not capable of solvating all the reagents and, as a result, generally give poor conversions (e.g.Table 1, entry 4). Polar aprotic solvents were found to offer the highest selectivity towards 2a, with acetone also significantly enhancing the total conversion (Table 1, entry 6).
Among the Brønstedt bases screened, Cs2CO3 proved to be the most efficient. Et3N (Table 1, entry 7), which is commonly used in sulfonation reactions shows here low reactivity and poor selectivity. Like protic solvents, even slightly nucleophilic bases such as 1,1,3,3-tetramethylguanidine (TMG), and KOH (Table 1, entries 8 and 9) favor the reaction with the sulfonating agent, therefore leading to low conversion of 1a. Finally, other carbonate bases (Table 1, entry 10) are inferior to Cs2CO3 due to their lower solubility in the reaction medium. Thus, the highest conversion accompanied with good selectivity is achieved when the reaction is carried out in acetone under 5 bar CO2 at room temperature, with Cs2CO3 as the base (Table 1, entry 6).
With amino alcohols where the OH group is primary, the reaction stops at 50% very likely due to the formation of a salt between the ionic byproducts, the remaining starting material, and possibly carbon dioxide. Although the exact composition of this salt was not elucidated, the remaining amino alcohol can be quantitatively recovered upon dissolving the solid reaction phase into H2O, indicating that a reversible salt-forming reaction indeed takes place. Attempts to overcome this plateau by increasing base loading or CO2 pressure were not successful.
As we turned our attention to other substrates (Fig. 2), we discovered that amino alcohols with secondary hydroxyl groups are more reactive, as 2b was isolated in 83% yield, which is only slightly lower than what is achieved with isocyanate-based methods.43,44 Tertiary hydroxyl groups, meanwhile, are sterically too hindered to readily undergo cyclization, and a significant decrease in yield is observed for 2c. In the synthesis of carbamate 6-membered rings from the corresponding amino propanols, a similar trend can be seen: the formation of 6-ring 4a from the commercially available 3-(phenylamino)propan-1-ol stops at 50%, while a more substituted amino alcohol yields 57% of 4b. Attempts to prepare larger 7- or 8-membered rings were not successful.
 | ||
| Fig. 2 Conversions of various amino alcohols and the isolated yields (in parentheses) of the corresponding cyclic carbamates. Reaction conditions: amino alcohol 1 (0.5 mmol), TsCl (1.1 equiv.), Cs2CO3 (3 equiv.), CO2 (5 bar), acetone (5 mL), room temperature, 20 h. | ||
Aromatic ring substituents affect the reaction outcome, as well. The presence of a weakly activating alkyl group leads to the slightly enhanced yield observed for 2d, whereas the more strongly activating alkoxy moiety affords quantitative conversion at the cost of selectivity, and 2e and the corresponding N-tosylated amino alcohol are isolated in a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This marks the only occasion in which any N-tosylation was detected under the optimized reaction conditions. A deactivating halogen substituent, meanwhile, leads to a decrease in conversion, but full selectivity is maintained in the synthesis of 2f.
:1 ratio. This marks the only occasion in which any N-tosylation was detected under the optimized reaction conditions. A deactivating halogen substituent, meanwhile, leads to a decrease in conversion, but full selectivity is maintained in the synthesis of 2f.
The reaction can also be extended to the synthesis of fused rings 5 from 2-aminobenzyl alcohols with yields up to 80%. Like N-aryl-2-oxazolidinones, these structures are omnipresent in pharmaceuticals, and can be found, for instance, in Efavirenz, an antiretroviral used to treat HIV,4 and in NMDA receptor antagonists, useful for treating stroke, cerebral ischemia, and depression.5 To our knowledge, the present work represents the first example of a CO2-based synthesis of these compounds, and is in fact as efficient as the phosgene-based method.45
To further establish the generality and applicability of this methodology in synthesis, we probed the chemo- and stereoselectivity of the reaction by screening difunctionalized and enantiopure amino alcohols as starting materials (Fig. 3). We have previously shown that the cyclization of carbon dioxide and 2,3-dichloropropan-1-amine strongly favors the formation of the more stable 5-ring (81% 5-ring, 17% 6-ring):271i, on the other hand, gives a mixture of the two possible products in a ca. 5:6 ratio, as the thermodynamic stability of the 5-ring is offset by the better leaving group nature of the –Cl. The easily synthesizable amino diol 1j, meanwhile, expectedly shows preference towards the formation of the 5-ring 2h, which can be isolated in 85% yield, and intriguingly with a higher selectivity towards the 5-ring than when the dihaloalkylamine was employed.
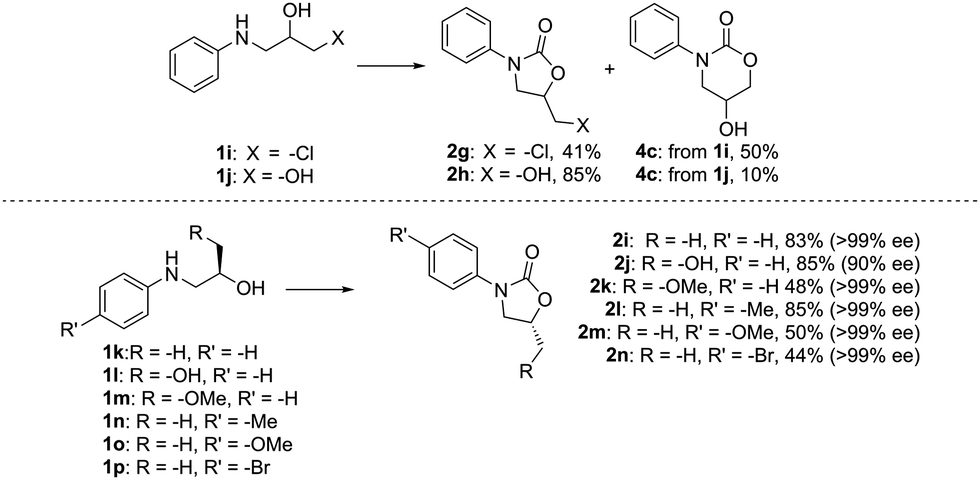 | ||
| Fig. 3 Chemo- and stereoselectivity of the reaction. All yields are isolated yields. Reaction conditions: amino alcohol 1 (0.5 mmol), TsCl (1.1 equiv.), Cs2CO3 (3 equiv.), CO2 (5 bar), acetone (5 mL), room temperature, 20 h. | ||
As we envision our method applicable to the synthesis of pharmaceutical compounds and chiral auxiliaries, predictable stereochemistry is of utmost importance. To our delight, the reaction also proceeds with high stereoselectivity, and >99% ee is observed for most substrates. In fact, only the amino diol 1l undergoes some racemization, but the corresponding oxazolidinone 2j is still isolated in a good yield and 90% ee. The products have inverted configuration at the stereocenter, which hints at a SN2-type mechanism during the ring-closing step. To shed some further light on the involved reaction mechanism, the transformation was next studied in silico.
The computed reaction profile for the reaction of the parent amino alcohol 1a and CO2 in the presence of mesylchloride (MsCl), as a model of the experimentally used TsCl, is shown in Fig. 4, which gathers the corresponding relative Gibbs free energies (ΔG298, at 298 K) in acetone computed at the PCM(acetone)-M062X/def2-TZVPP//PCM(acetone)-M062X/6-31+G(d) level.46 Our DFT calculations indicate that the process begins with the formation of zwitterionic intermediate INT1via transition state TS1, a saddle point associated with the nucleophilic attack of the amino group of 1a to the electrophilic CO2. The alternative reaction of 1a with the activating agent MsCl, leading to INT1′viaTS1′, is not competitive in view of the much higher activation barrier (ΔΔG‡ = 12.8 kcal mol−1) and endergonicity (ΔΔGR = 11.4 kcal mol−1) associated with this process, which is fully consistent with the complete regioselectivity observed for the transformation (see above). Although the formation of INT1 is endergonic (ΔGR = 17.3 kcal mol−1), the subsequent base-mediated deprotonation of this species is highly exergonic (ΔGR = −46.2 kcal mol−1) and drives the transformation forward. This step leads to the formation of the highly stabilized anionic intermediate INT2, which readily reacts with MsCl to produce, in an exergonic process (ΔGR = −3.2 kcal mol−1), the corresponding mesyl derivative INT3. This step occurs viaTS2 (activation barrier of 21.4 kcal mol−1) in a typical SN2 reaction thus releasing HCl, which is then neutralized by the excess of base. The transformation ends up with the formation of the experimentally observed carbamate 2aviaTS3 in a highly exergonic process (ΔGR = −30.1 kcal mol−1) with a reaction barrier of 18.1 kcal mol−1 (fully compatible with a process occurring at room temperature). This saddle point is associated with the intermolecular synchronous displacement of the OMs leaving group by the nucleophilic (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )CO− moiety of INT3, thus forming the new C–O bond of the carbamate. According to the geometry of TS3, this final ring closure can be viewed as an intramolecular backside SN2 reaction, which nicely agrees with the inversion of configuration observed in the processes involving the chiral substrates 1k–p (see Fig. 3).
)CO− moiety of INT3, thus forming the new C–O bond of the carbamate. According to the geometry of TS3, this final ring closure can be viewed as an intramolecular backside SN2 reaction, which nicely agrees with the inversion of configuration observed in the processes involving the chiral substrates 1k–p (see Fig. 3).
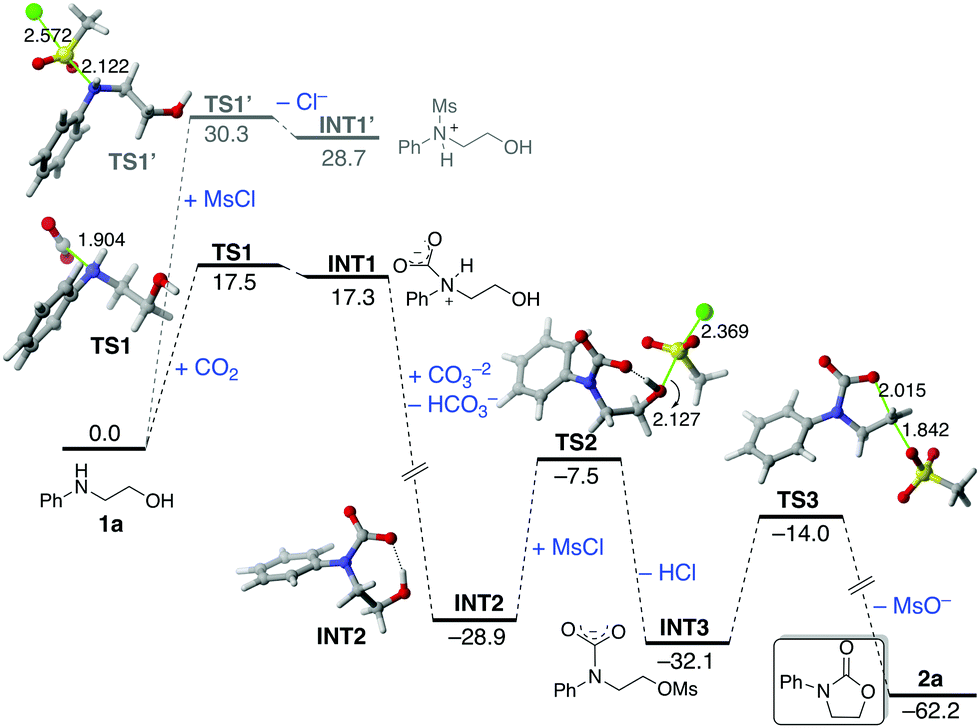 | ||
| Fig. 4 Computed reaction profile for the reaction of amino alcohol 1a and CO2 in the presence of MsCl. Relative Gibbs free energies and bond distances are given in kcal mol−1 and angstroms, respectively. All data have been computed at the PCM(acetone)-M062X/def2-TZVPP//PCM(acetone)-M062X/6-31+G(d) level. | ||
In conclusion, we have developed a facile and general method for the stereoselective synthesis of cyclic carbamates from carbon dioxide and amino alcohols. The reaction is applicable to the preparation of 3-aryl,5-alkyl substituted 2-oxazolidinones found in Linezolid and other superantibiotics, cyclic fused rings found in antiretrovirals and NMDA receptor antagonists, as well as 6-membered rings. Under the optimized reaction conditions, the process has high regio-, chemo-, and stereoselectivity. Enantiopure amino alcohols undergo inversion of configuration at the stereocenter, which hints at a SN2-type reaction mechanism during the key ring-closing step; this observation was later supported by DFT calculations.
This work was partially funded by the Academy of Finland (project #310767). TN is grateful for the University of Helsinki's doctoral programme in chemistry and molecular sciences (CHEMS) for funding. IF acknowledges financial support from the Spanish MINECO-FEDER (Grants CTQ2016-78205-P and CTQ2016-81797-REDC).
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. J. Brickner, Curr. Pharm. Des., 1996, 2, 175–194 CAS.
- H. Ucar, K. Van derpoorten, S. Cacciaguerra, S. Spampinato, J. P. Stables, P. Depovere, M. Isa, B. Masereel, J. Delarge and J. H. Poupaert, J. Med. Chem., 1998, 41, 1138–1145 CrossRef CAS PubMed.
- R. Schaadt, D. Sweeney, D. Shinabarger and G. Zurenko, Antimicrob. Agents Chemother., 2009, 53, 3236–3239 CrossRef CAS PubMed.
- V. Famiglini and R. Silvestri, Molecules, 2016, 21, 1–18 CrossRef PubMed.
- Y. Chen, C. A. Hone, B. Gutmann and C. O. Kappe, Org. Process Res. Dev., 2017, 21, 1080–1087 CrossRef CAS.
- M. S. Newman and A. Kutner, J. Am. Chem. Soc., 1951, 73, 4199–4204 CrossRef CAS.
- M. E. Dyen and D. Swern, Chem. Rev., 1967, 67, 197–246 CrossRef CAS PubMed.
- P. Tascedda and E. Dunach, Chem. Commun., 2000, 449–450 RSC.
- J. Rintjema, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2015, 21, 10754–10762 CrossRef CAS PubMed.
- V. B. Saptal and B. M. Bhanage, ChemSusChem, 2016, 9, 1980–1985 CrossRef CAS PubMed.
- H. Wang, J. C. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2017, 139, 8428–8431 CrossRef CAS PubMed.
- J. Rintjema, R. Epping, G. Fiorani, E. Martín, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2016, 55, 3972–3976 CrossRef CAS PubMed.
- B. Wang, E. H. M. Elageed, D. Zhang, S. Yang, S. Wu, G. Zhang and G. Gao, ChemCatChem, 2014, 6, 278–283 CrossRef CAS.
- U. R. Seo and Y. K. Chung, Green Chem., 2017, 19, 803–808 RSC.
- W.-Z. Zhang, X. Ren and X.-B. Lu, Chin. J. Chem., 2015, 33, 610–613 CrossRef CAS.
- B.-B. Cheng, B. Yu and C.-W. Hu, RSC Adv., 2016, 6, 87179–87187 RSC.
- K.-i. Fujita, A. Fujii, J. Sato, S.-y. Onozawa and H. Yasuda, Tetrahedron Lett., 2016, 57, 1282–1284 CrossRef CAS.
- H. Liu and R. Hua, Tetrahedron, 2016, 72, 1200–1204 CrossRef CAS.
- Q.-W. Song, Z.-H. Zhou, M.-Y. Wang, K. Zhang, P. Liu, J.-Y. Xun and L.-N. He, ChemSusChem, 2016, 9, 2054–2058 CrossRef CAS PubMed.
- B. Yu, B.-B. Cheng, W.-Q. Liu, W. Li, S.-S. Wang, J. Cao and C.-W. Hu, Adv. Synth. Catal., 2016, 358, 90–97 CrossRef CAS.
- P. Brunel, J. Monot, C. E. Kefalidis, L. Maron, B. Martin-Vaca and D. Bourissou, ACS Catal., 2017, 7, 2652–2660 CrossRef CAS.
- X. Liu, M.-Y. Wang, S.-Y. Wang, Q. Wang and L.-N. He, ChemSusChem, 2017, 10, 1210–1216 CrossRef CAS PubMed.
- M.-Y. Wang, Y. Cao, X. Liu, N. Wang, L.-N. He and S.-H. Li, Green Chem., 2017, 19, 1240–1244 RSC.
- B. Yu, D. Kim, S. Kim and S. H. Hong, ChemSusChem, 2017, 10, 1080–1084 CrossRef CAS PubMed.
- P. H. Dixneuf, Catal. Lett., 2015, 145, 360–372 CrossRef CAS.
- J. Fournier, C. Bruneau and P. H. Dixneuf, Tetrahedron Lett., 1990, 31, 1721–1722 CrossRef CAS.
- T. Niemi, J. E. Perea-Buceta, I. Fernández, S. Alakurtti, E. Rantala and T. Repo, Chem. – Eur. J., 2014, 20, 8867–8871 CAS.
- T. Niemi, J. E. Perea-Buceta, I. Fernández, O.-M. Hiltunen, V. Salo, S. Rautiainen, M. T. Räisänen and T. Repo, Chem. – Eur. J., 2016, 22, 10355–10359 CrossRef CAS PubMed.
- J. Vaitla, Y. Guttormsen, J. K. Mannisto, A. Nova, T. Repo, A. Bayer and K. H. Hopmann, ACS Catal., 2017, 7, 7231–7244 CrossRef CAS.
- E. S. Streng, D. S. Lee, M. W. George and M. Poliakoff, Beilstein J. Org. Chem., 2017, 13, 329–337 CrossRef CAS PubMed.
- S. W. Foo, Y. Takada, Y. Yamazaki and S. Saito, Tetrahedron Lett., 2013, 54, 4717–4720 CrossRef CAS.
- M. Tamura, M. Honda, K. Noro, Y. Nakagawa and K. Tomishige, J. Catal., 2013, 305, 191–203 CrossRef CAS.
- M. Kodaka, T. Tomohiro and H. Okuno, J. Chem. Soc., Chem. Commun., 1993, 81–82 RSC.
- S.-I. Fujita, H. Kanamaru, H. Senboku and M. Arai, Int. J. Mol. Sci., 2006, 7, 438 CrossRef CAS.
- S. Pulla, C. M. Felton, Y. Gartia, P. Ramidi and A. Ghosh, ACS Sustainable Chem. Eng., 2013, 1, 309–312 CrossRef CAS.
- Y. P. Patil, P. J. Tambade, S. R. Jagtap and B. M. Bhanage, J. Mol. Catal. A: Chem., 2008, 289, 14–21 CrossRef CAS.
- M. Feroci, M. Orsini, G. Sotgiu, L. Rossi and A. Inesi, J. Org. Chem., 2005, 70, 7795–7798 CrossRef CAS PubMed.
- M. A. Casadei, F. M. Moracci, G. Zappia, A. Inesi and L. Rossi, J. Org. Chem., 1997, 62, 6754–6759 CrossRef CAS.
- B. R. Buckley, A. P. Patel and K. G. U. Wijayantha, Synlett, 2014, 197–200 CAS.
- M. Feroci, A. Gennaro, A. Inesi, M. Orsini and L. Palombi, Tetrahedron Lett., 2002, 43, 5863–5865 CrossRef CAS.
- B. M. Bhanage, S.-i. Fujita, Y. Ikushima and M. Arai, Green Chem., 2003, 5, 340–342 RSC.
- G. L. Gregory, M. Ulmann and A. Buchard, RSC Adv., 2015, 5, 39404–39408 RSC.
- R. L. Paddock, D. Adhikari, R. L. Lord, M.-H. Baik and S. T. Nguyen, Chem. Commun., 2014, 50, 15187–15190 RSC.
- P. Wang, J. Qin, D. Yuan, Y. Wang and Y. Yao, ChemCatChem, 2015, 7, 1145–1151 CrossRef CAS.
- G.-J. Griffiths, M. Lorenzi and A. Warm, US 20110172419, 2011.
- See computational details in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, spectroscopic data. See DOI: 10.1039/c8cc00636a |
| This journal is © The Royal Society of Chemistry 2018 |