
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Staudinger reaction using 2,6-dichlorophenyl azide derivatives for robust aza-ylide formation applicable to bioconjugation in living cells†
Tomohiro
Meguro
a,
Norikazu
Terashima
a,
Harumi
Ito
 ab,
Yuka
Koike
c,
Isao
Kii
bc,
Suguru
Yoshida
*a and
Takamitsu
Hosoya
*ad
ab,
Yuka
Koike
c,
Isao
Kii
bc,
Suguru
Yoshida
*a and
Takamitsu
Hosoya
*ad
aLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: s-yoshida.cb@tmd.ac.jp; thosoya.cb@tmd.ac.jp
bPathophysiological and Health Science Team, Division of Bio-Function Dynamics Imaging, Imaging Platform and Innovation Group, RIKEN Center for Life Science Technologies (CLST), 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan
cCommon Facilities Unit, Compass to Healthy Life Research Complex Program, RIKEN Cluster for Science and Technology Hub, 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan
dChemical Biology Team, Division of Bio-Function Dynamic Imaging, RIKEN Center for Life Science Technologies (CLST), 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan
First published on 14th May 2018
Abstract
Efficient formation of water- and air-stable aza-ylides has been achieved using the Staudinger reaction between electron-deficient aromatic azides such as 2,6-dichlorophenyl azide and triarylphosphines. The reaction proceeds rapidly and has been successfully applied to chemical modification of proteins in living cells.
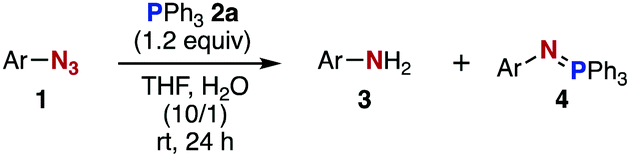
Click reactions,1 including copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC)2 and strain-promoted azide–alkyne cycloaddition (SPAAC),3 have been recognized as epoch-making methods for reliable conjugations of molecules over a broad range of research fields within chemistry and biology. In particular, click reactions resulting in efficient formation of stable covalent bonds have been widely utilized for chemical modification of biomolecules in chemical biology and drug discovery research.4 However, several problems using conventional methods, such as non-specific in-cell click labeling by SPAAC, have been reported; thus, a new method is required to address these issues.5
Staudinger–Bertozzi ligation using triarylphosphines bearing an ortho ester moiety in conjugation with aliphatic azides has emerged as an early bioorthogonal reaction (Fig. 1A).6 The method forms a robust amide bond and has been demonstrated to be useful for the chemical modification of various biomolecules. Nevertheless, Bertozzi and coworkers developed a SPAAC reaction to achieve a faster and more efficient bioconjugation. Thereafter, a number of methods using cyclooctynes with improved characteristics have been reported.7 In the course of our recent studies regarding phosphorus chemistry8 and molecular conjugation chemistry,9 we revisited the Staudinger reaction between aromatic azides and various phosphines.8b These studies gave us an idea of preparing an aza-ylide that would be stable toward hydrolysis and oxidation. We considered that this type of aza-ylide would be useful for chemical modification of biomolecules. Herein, we report a new method for molecular conjugation using the Staudinger reaction to form robust aza-ylides. This chemistry has been found to be applicable to efficient bioconjugation in living cells (Fig. 1B).10,11
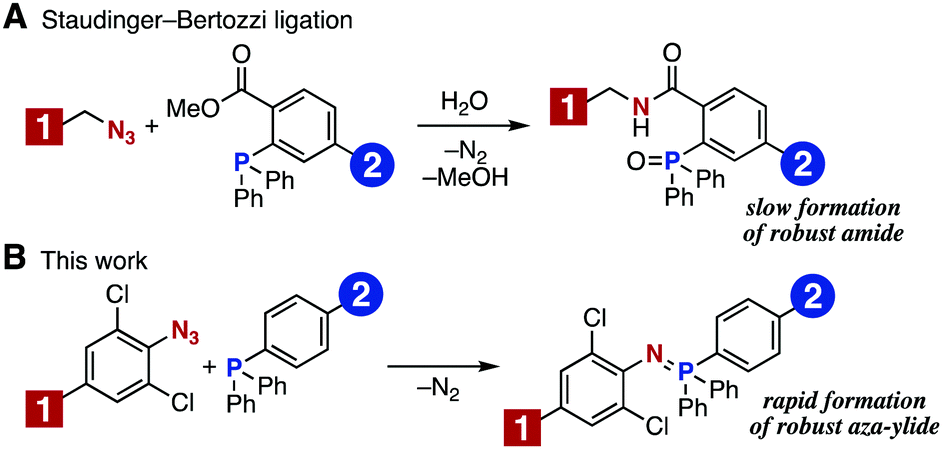 | ||
| Fig. 1 Molecular conjugation using the Staudinger reaction. (A) Staudinger–Bertozzi ligation. (B) This work, using electron-deficient aromatic azides and triarylphosphines. | ||
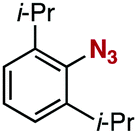
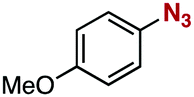
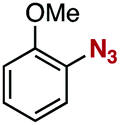
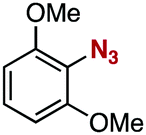
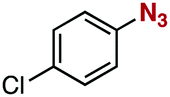
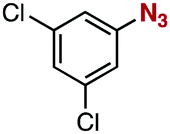
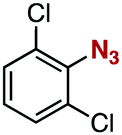
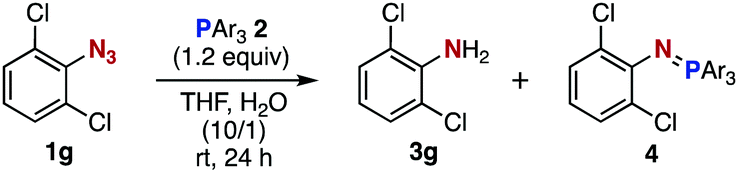
After extensive screening of aromatic azides for the Staudinger reaction with triphenylphosphine in the presence of water, we found that 2,6-dichlorophenyl azide was efficiently transformed to the corresponding aza-ylide without the formation of the aniline derivative (Table 1). An initial attempt of the reaction between sterically congested 2,6-diisopropylphenyl azide (1a) and triphenylphosphine (2a) by stirring the mixture in tetrahydrofuran (THF) and water (v/v = 10/1) at room temperature for 24 h afforded aza-ylide 4a along with a small amount of side-products (entry 1). When electron-rich aromatic azides 1b–1d were employed, the yields of anilines 3b–3d, formed from hydrolysis of aza-ylides 4b–4d, increased (entries 2–4). In contrast, the formation of anilines was suppressed using electron-deficient aromatic azides such as chloro- and dichlorophenyl azides 1e–1g (entries 5–7). In particular, the reaction of 2,6-dichlorophenyl azide (1g) and triphenylphosphine (2a) afforded aza-ylide 4g quantitatively after purification by silica-gel column chromatography (entry 7). The formation of hydrolyzed product 3g was not observed, clearly indicating that aza-ylide 4g was stable toward water and air. In addition, azide 1g was stable in the presence of an excess amount of n-dodecanethiol and in cell lysate, and aza-ylide 4g remained unchanged in solutions that contained hydrochloric acid, sodium bicarbonate, cysteine, lysine, or tyrosine, demonstrating its remarkable stability.12
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | ArN3 | 1 | 3 | Yield of 3a (%) | 4 | Yield of 4a (%) |
| a Yields were determined by 1H NMR analysis. b A small amount of unknown side-products (ca. 4%) were obtained. c Isolated yield shown in parentheses. | ||||||
| 1b |
|
1a | 3a | 0 | 4a | 95 |
| 2 |
|
1b | 3b | 20 | 4b | 75 |
| 3 |
|
1c | 3c | 34 | 4c | 65 |
| 4 |
|
1d | 3d | 18 | 4d | 81 |
| 5 |
|
1e | 3e | 8 | 4e | 92 |
| 6 |
|
1f | 3f | 2 | 4f | 97 |
| 7 |
|
1g | 3g | 0 | 4g | 98 (quant)c |
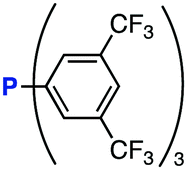
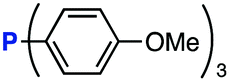
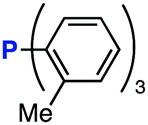
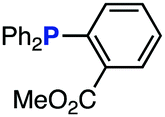
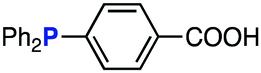
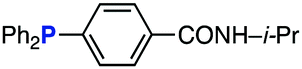
The efficiency of aza-ylide formation from 2,6-dichlorophenyl azide (1g) was greatly affected by the substituents on the phenyl groups of the triarylphosphines (Table 2). Although the reaction between 1g and electron-deficient phosphine 2b did not afford aza-ylide 4h and aniline 3g was formed instead (entry 1), the quantitative formation of aza-ylide 4i was observed when electron-rich phosphine 2c was employed (entry 2). The bulky tri(o-tolyl)phosphine (2d) did not react with 1g under the standard conditions (entry 3). Phosphine 2e, bearing an ortho ester moiety, reacted smoothly with 1g to afford aza-ylide 4k quantitatively (entry 4). No imine13 or Staudinger–Bertozzi ligation product6 was found. While the use of phosphine 2f, bearing a para carboxy group, yielded a significant amount of aniline 3gvia hydrolysis of aza-ylide 4l (entry 5), phosphine 2g, having a para amide moiety, quantitatively afforded aza-ylide 4m, showing the superior stability of 4m over 4l (entry 6). From the kinetic study of the Staudinger reaction between 2,6-dichlorophenyl azide (1g) and phosphine 2g in acetonitrile-d3, the second-order rate constant was determined to be 0.63 ± 0.02 M−1 s−1. This was 250-fold higher than that for the amide formation between benzyl azide and 2e![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6d and about two-fold higher than that for the SPAAC between benzyl azide and a bicyclo[6.1.0]non-4-yne (BCN) derivative.7e
6d and about two-fold higher than that for the SPAAC between benzyl azide and a bicyclo[6.1.0]non-4-yne (BCN) derivative.7e
Several competition experiments also demonstrated a rapid Staudinger reaction between 2,6-dichlorophenyl azide (1g) and triphenylphosphine (2a) (Fig. 2). Treatment of an equimolar mixture of phosphines 2a and 2e with azide 1g predominantly afforded 2a-derived aza-ylide 4g, showing that the ortho ester moiety of 2e significantly decreased the reactivity toward azide 1g (Fig. 2A). The treatment of an equimolar mixture of 1g and benzyl azide (5) with triphenylphosphine (2a) predominantly afforded aza-ylide 4g, demonstrating a remarkable reactivity of electron-deficient aromatic azide 1g toward phosphines (Fig. 2B). The treatment of an equimolar mixture of 2a and BCN derivative 8 with azide 1g in methanol afforded almost equal amounts of aza-ylide 4g and triazole 9, indicating that the Staudinger reaction proceeded as fast as the SPAAC reaction7e (Fig. 2C).
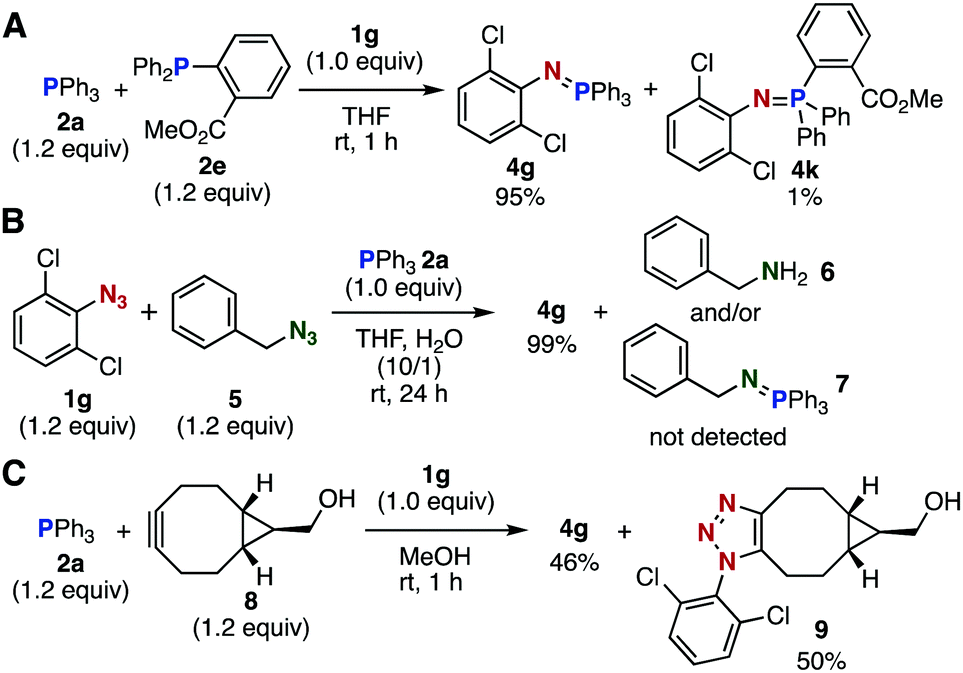 | ||
| Fig. 2 Competition experiments. (A) Phosphines. (B) Azides. (C) Staudinger reaction vs. SPAAC reaction. Isolated yields are shown. | ||
Further competition experiments using an equimolar mixture of phosphine 2a and dibenzo-fused cyclooctyne 10 exhibited a unique orthogonality (Fig. 3). While the Staudinger reaction of azide 1g with 2a proceeded significantly faster than the SPAAC reaction of 1g with 10 (Fig. 3A-1), benzyl azide (5) exclusively reacted with cyclooctyne 10 to afford triazole 11b along with a small amount of Staudinger reaction-derived product 6 (Fig. 3A-2). This orthogonality between Staudinger and SPAAC reactions enabled simultaneous bisconjugation in a site-selective manner using diazide 12 bearing 2,6-dichlorophenyl azide and alkyl azide moieties (Fig. 3B). Thus, the treatment of an equimolar mixture of phosphine 2a and cyclooctyne 10 with diazide 12 afforded a three-component coupled product 13 in high yield. This result indicated that the Staudinger reaction with 2a and SPAAC reaction with 10 proceeded selectively at the 2,6-dichlorophenyl azide and alkyl azide sites of diazide 12, respectively, which served as an efficient hinge molecule to conjugate two different types of azidophiles.
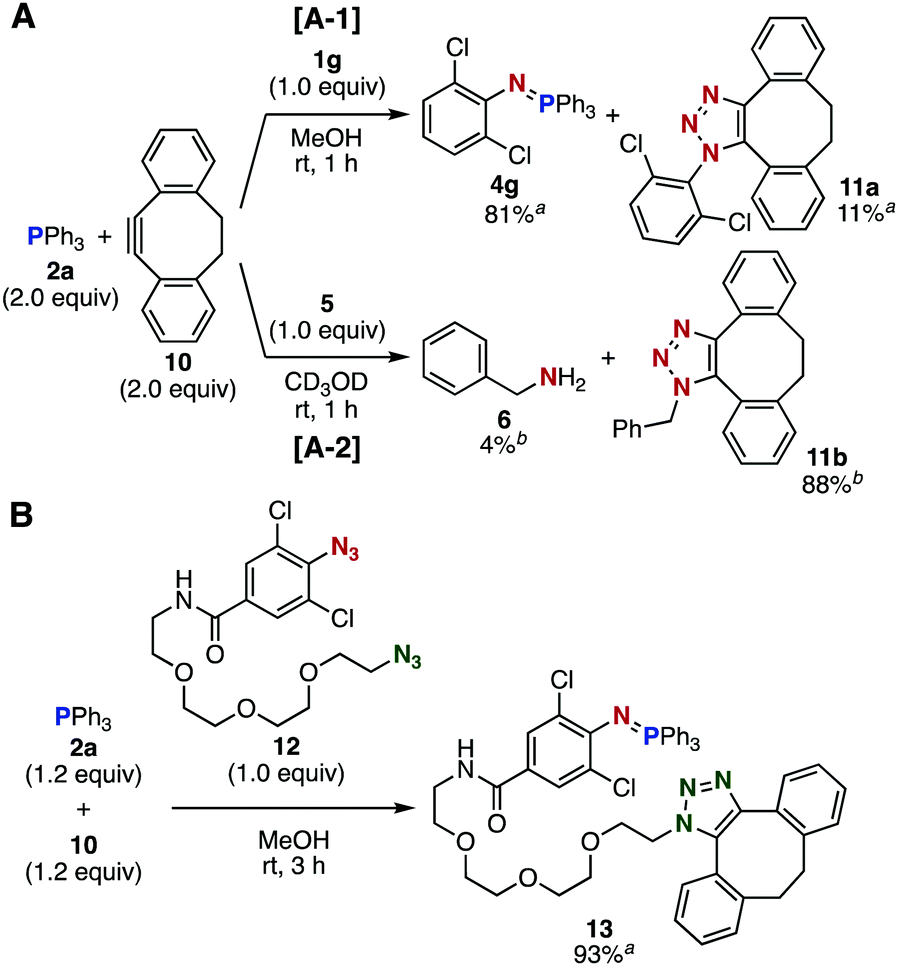 | ||
| Fig. 3 Selective conjugations. (A) Reactions of an equimolar mixture of phosphine 2a and cyclooctyne 10 with azide 1g or 5. (B) Reaction of diazide 12 with an equimolar mixture of 2a and 10. aIsolated yields are shown. bYields were determined by 1H NMR analysis. | ||
The formation of a stable aza-ylide by the Staudinger reaction between 2,6-dichlorophenyl azide and triarylphosphine was applied to the chemical modification of biomolecules (Fig. 4). According to the previous report,7f an azido-protein was prepared by conjugating GST-fused HaloTag protein (GST-HaloTag) with the azido-HaloTag ligand (14) on a GSH-conjugated resin,12,14 followed by the treatment with fluorescent TESRA-phosphine (15) (Fig. 4A). The following SDS-PAGE analysis showed that TESRA-labeled GST-HaloTag protein (51 kDa) was successfully prepared (Fig. 4B, lane 4).12 This result indicated that the aza-ylide formed from the Staudinger reaction was sufficiently stable under bioconjugation conditions, demonstrating the bioorthogonality of this method.12,14
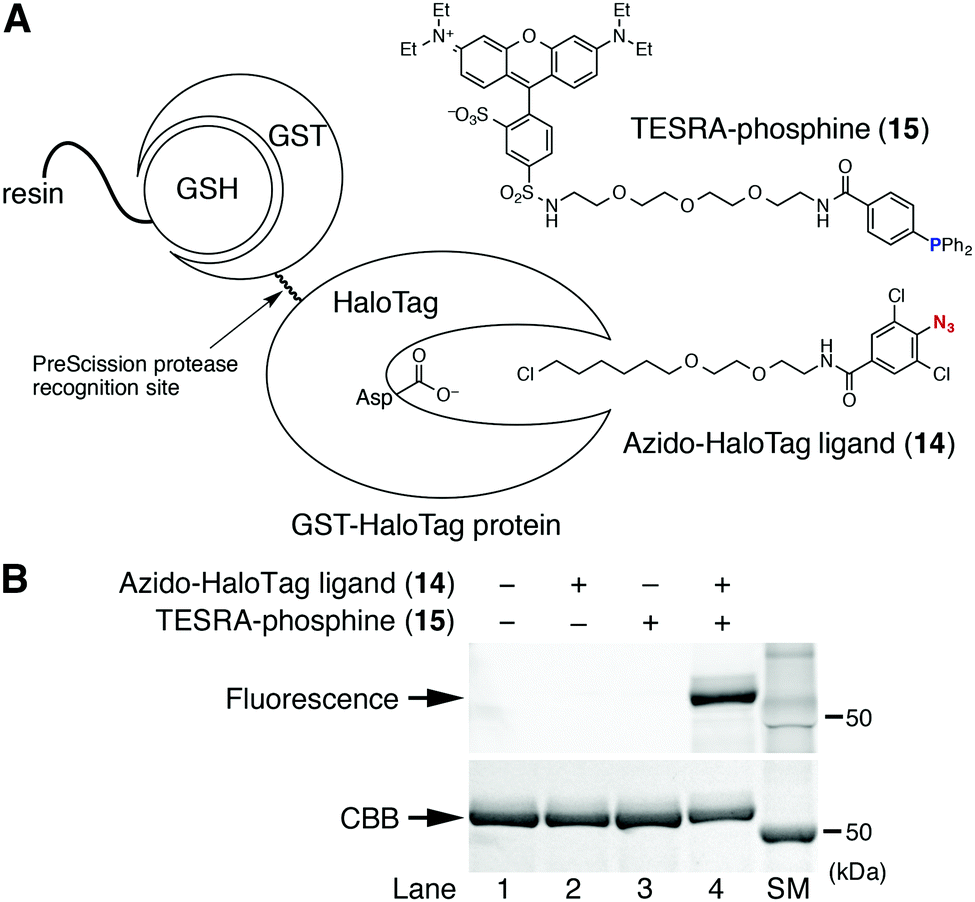 | ||
| Fig. 4 Chemical modification of an azido-protein using the Staudinger reaction. (A) GST-HaloTag protein bound on GSH-resin, azido-HaloTag ligand (14), and TESRA-phosphine (15). (B) SDS-PAGE analysis of the labeled GST-HaloTag proteins eluted from the resin. The gel was scanned using a fluorescence image analyzer and then stained with Coomassie brilliant blue (CBB). SM indicates the size marker lane. | ||
The novel Staudinger ligation was also applicable to chemical modification of proteins in living cells (Fig. 5). For example, cell surface-specific fluorescent labeling was achieved by the expression of transmembrane domain-fused HaloTag protein on the cell surface, in which HaloTag is present outside the cells, followed by the treatment with azido-HaloTag ligand (14) and TESRA-phosphine (15) (Fig. 5A). Notably, our Staudinger ligation method was also effective for fluorescent labeling of HaloTag fused with NUP133 nuclear pore complex protein (Fig. 5B). This result indicated that this method could be used for chemical modification of intracellular biomolecules. Indeed, compared with the fluorescent labeling method using SPAAC modification with a dibenzo-fused cyclooctyne possessing a TESRA moiety,12 our Staudinger ligation method showed a superior result in terms of labeling efficiency inside the cells.
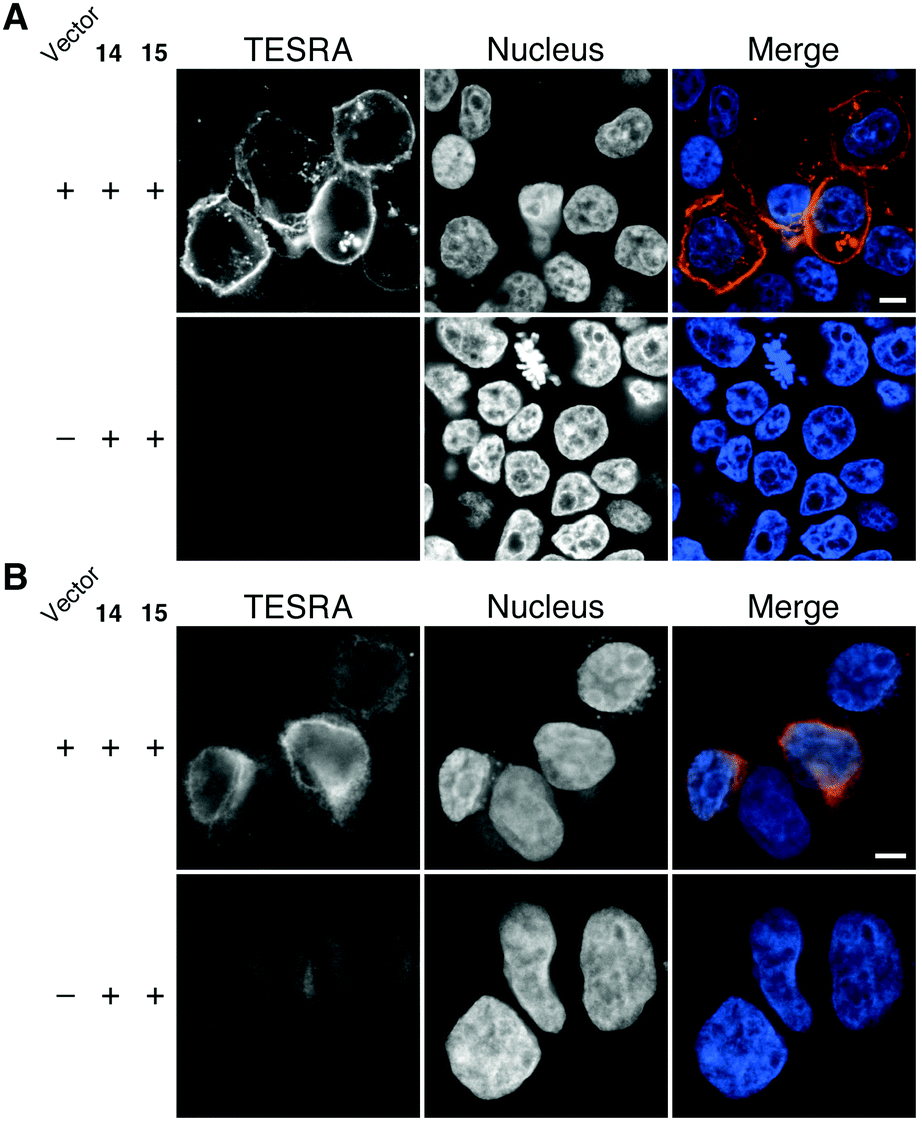 | ||
| Fig. 5 Fluorescent labeling of living cells expressed with HaloTag-fused proteins by incubation with 10 μM of azido-HaloTag ligand (14) for 30 min at 37 °C, followed by incubation with 1 μM of TESRA-phosphine (15) for 30 min at 37 °C. (A) HEK293 cells with HaloTag protein on the cell surface outside the cells. (B) HEK293 cells with HaloTag protein on the nucleus inside the cells. Vector (+) indicates the expression of the HaloTag fusion proteins, and (−) indicates no expression. Scale bar, 5 μm. | ||
In summary, we have demonstrated that the Staudinger reaction of 2,6-dichlorophenyl azide derivatives with triarylphosphines proceeds rapidly to form robust aza-ylides. The method allows for the efficient chemical modification of proteins in living cells.
This work was supported by AMED under Grant Numbers JP18am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research) and JP18am0301024 (Basic Science and Platform Technology Program for Innovative Biological Medicine); the Cooperative Research Project of Research Center for Biomedical Engineering; JSPS KAKENHI Grant Numbers 15H03118 and 18H02104 (B; T. H.), 16H01133 and 18H04386 (Middle Molecular Strategy; T. H.), 17H06414 (Organelle Zone; T. H.), 26350971 (C; S. Y.), 18H04568 (CMCB; I. K.), and 18J11113 (JSPS Research Fellow; T. M.); and the Naito Foundation (S. Y.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews, see: (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef; (b) J. Lahann, Click Chemistry for Biotechnology and Materials Science, John Wiley & Sons: West Sussex, 2009 Search PubMed; (c) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef; (d) K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16 CrossRef PubMed.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef; (c) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef PubMed.
- (a) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef PubMed; (b) J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182 CrossRef PubMed; (c) M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168 CrossRef PubMed; (d) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC.
- (a) P. V. Chang, J. A. Prescher, M. J. Hangauer and C. R. Bertozzi, J. Am. Chem. Soc., 2007, 129, 8400 CrossRef PubMed; (b) S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664 CrossRef PubMed; (c) G. Charron, M. M. Zhang, J. S. Yount, J. Wilson, A. S. Raghavan, E. Shamir and H. C. Hang, J. Am. Chem. Soc., 2009, 131, 4967 CrossRef PubMed; (d) K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317 CrossRef PubMed; (e) H. E. Murrey, J. C. Judkins, C. W. am Ende, T. E. Ballard, Y. Fang, K. Riccardi, L. Di, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, J. Am. Chem. Soc., 2015, 137, 11461 CrossRef PubMed ; For reviews, see: ; (f) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef PubMed; (g) D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592 CrossRef PubMed; (h) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764 CrossRef PubMed.
- (a) R. van Geel, G. J. M. Pruijn, F. L. van Delft and W. C. Boelens, Bioconjugate Chem., 2012, 23, 392 CrossRef PubMed; (b) T. H. Poole, J. A. Reisz, W. Zhao, L. B. Poole, C. M. Furdui and S. B. King, J. Am. Chem. Soc., 2014, 136, 6167 CrossRef PubMed; (c) H. Tian, T. P. Sakmar and T. Huber, Chem. Commun., 2016, 52, 5451 RSC.
- (a) E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef PubMed; (b) B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939 CrossRef PubMed; (c) E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141 CrossRef PubMed; (d) F. L. Lin, H. M. Hoyt, H. van Halbeek, R. G. Bergman and C. R. Bertozzi, J. Am. Chem. Soc., 2005, 127, 2686 CrossRef PubMed; (e) N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644 CrossRef PubMed; (f) R. Serwa, I. Wilkening, G. del Signore, M. Mühlberg, I. Claußnitzer, C. Weise, M. Gerrits and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2009, 48, 8234 CrossRef PubMed; (g) I. Wilkening, G. del Signore and C. P. R. Hackenberger, Chem. Commun., 2011, 47, 349 RSC; (h) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666 CrossRef PubMed; (i) M. R. J. Vallée, L. M. Artner, J. Dernedde and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2013, 52, 9504 CrossRef PubMed; (j) L. Shah, S. T. Laughlin and I. S. Carrico, J. Am. Chem. Soc., 2016, 138, 5186 CrossRef PubMed; (k) G. Ren, Q. Zheng and H. Wang, Org. Lett., 2017, 19, 1582 CrossRef PubMed ; For reviews, see: ; (l) M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106 CrossRef PubMed; (m) S. S. van Berkel, M. B. van Eldijk and J. C. M. van Hest, Angew. Chem., Int. Ed., 2011, 50, 8806 CrossRef PubMed; (n) C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840 RSC.
- (a) J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486 CrossRef PubMed; (b) X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253 CrossRef PubMed; (c) A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769 CrossRef PubMed; (d) J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688 CrossRef PubMed; (e) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422 CrossRef PubMed; (f) I. Kii, A. Shiraishi, T. Hiramatsu, T. Matsushita, H. Uekusa, S. Yoshida, M. Yamamoto, A. Kudo, M. Hagiwara and T. Hosoya, Org. Biomol. Chem., 2010, 8, 4051 RSC; (g) R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2015, 54, 1190 CrossRef PubMed; (h) K. Kaneda, R. Naruse and S. Yamamoto, Org. Lett., 2017, 19, 1096 CrossRef PubMed; (i) E. G. Burke, B. Gold, T. T. Hoang, R. T. Raines and J. M. Schomaker, J. Am. Chem. Soc., 2017, 139, 8029 CrossRef PubMed.
- (a) S. Yoshida and T. Hosoya, Chem. Lett., 2013, 42, 583 CrossRef; (b) T. Meguro, S. Yoshida and T. Hosoya, Chem. Lett., 2017, 46, 473 CrossRef; (c) Y. Nishiyama, Y. Hazama, S. Yoshida and T. Hosoya, Org. Lett., 2017, 19, 3899 CrossRef PubMed.
- (a) S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590 CrossRef PubMed; (b) T. Meguro, S. Yoshida and T. Hosoya, Chem. Lett., 2017, 46, 1137 CrossRef; (c) S. Yoshida, K. Kanno, I. Kii, Y. Misawa, M. Hagiwara and T. Hosoya, Chem. Commun., 2018, 54, 3705 RSC.
- During prepartion of this manuscript, a rapid Staudinger reaction between perfluoroaryl azides and aryl phosphines forming stable aza-ylides was reported. See: M. Sundhoro, S. Jeon, J. Park, O. Ramström and M. Yan, Angew. Chem., Int. Ed., 2017, 56, 12117 CrossRef PubMed.
- A part of this work was presented in March, 2017; see: N. Terashima, T. Meguro, S. Yoshida and T. Hosoya, 97th Annual Meeting of the Chemical Society of Japan, Hiyoshi, March 17, 2017, Abstr., No. 2F1-02.
- See the ESI† for details.
- J. A. Restituyo, L. R. Comstock, S. G. Petersen, T. Stringfellow and S. R. Rajski, Org. Lett., 2003, 5, 4357 CrossRef PubMed.
- The prolonged incubation time of labeled HaloTag proteins under the biological conditions demonstrated the sufficient stability of 2,6-dichlorophenyl azide and aza-ylide derivatives.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, and characterization for new compounds including NMR spectra. See DOI: 10.1039/c8cc00179k |
| This journal is © The Royal Society of Chemistry 2018 |