
Arylsilylation of aryl halides using the magnetically recyclable bimetallic Pd–Pt–Fe3O4 catalyst †
Jisun
Jang‡
a,
Sangmoon
Byun‡
 bc,
B. Moon
Kim
*b and
Sunwoo
Lee
*a
bc,
B. Moon
Kim
*b and
Sunwoo
Lee
*a
aDepartment of Chemistry, Chonnam National University, 77 Yongbong-ro, Buk-gu, Gwangju, 61186, Republic of Korea. E-mail: sunwoo@chonnam.ac.kr
bDepartment of Chemistry, College of Natural Sciences, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul, 08826, Republic of Korea. E-mail: kimbm@snu.ac.kr
cThe Research Institute of Basic Sciences, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul, 08826, Republic of Korea
First published on 14th March 2018
Abstract
Transition metal-catalyzed silylations have typically involved the use of homogeneous non-recyclable catalytic systems. In this work, the first example of a recyclable catalytic system for the synthesis of arylsilanes has been reported, which utilizes the bimetallic complex, Pd–Pt–Fe3O4 nanoparticles. Various arylsilanes were prepared by the reaction of aryl iodides (or bromides) with hydrosilanes. This methodology showed good functional group tolerance toward ester, ketone, aldehyde, nitro, and cyano groups. The bimetallic Pd–Pt–Fe3O4 catalytic system showed better activity than monometallic Pt–Fe3O4 and Pd–Fe3O4 catalysts. In addition, the bimetallic Pd–Pt–Fe3O4 catalytic system could be easily recovered and reused for over twenty cycles.
Owing to their unique properties, arylsilanes are valuable synthetic building blocks in pharmaceutical and materials chemistry.1 These compounds have also received significant attention owing to their role in electrophosphorescent devices.2 In addition, they have been widely used as a coupling partner in the Hiyama coupling reaction.3 A number of synthetic methods have been developed and widely used. Classically, silyl groups have been introduced in organic molecules through the use of organolithium or Grignard reagents and a silicon electrophile. However, this method lacks functional group tolerance toward base-sensitive groups.4 To overcome this shortcoming, transition metal-catalyzed coupling reactions of aryl halides with disilanes or hydrosilanes have been widely reported.5 Several coupling reactions of aryl halides with organosilanes catalyzed by Pd-,6 Rh-,7 and Pt-8 based complexes have been reported. However, coupling reactions with hydrosilanes as the silylating reagent have not been studied as much as those with other nucleophiles because of the strong reducing power of hydrosilanes.9 Apart from aryl halides, transition metal-catalyzed silylations with pivalates,10 phenolic esters,11 or cyano arenes12 have also been reported.
The direct C–H silylation reaction is an ideal methodology and an attractive alternative to the abovementioned strategies because it is the best atom-economical tool. Although direct C–H silylation has been reportedly achieved with Rh-,13 Ru-,14 Pt-,15 and Ir-16 based catalysts, the substrate scope is limited to heteroarenes and arenes bearing an ortho-directing group. Moreover, all these catalysts are based on precious metals. Generally, transition metal-catalyzed silylations of aryl halides or C–H activations proceed through homogeneous catalytic systems.
To the best of our knowledge, a reusable catalytic system has not been developed for the silylation reaction even though most of the catalysts utilized for this transformation are expensive metals. Recently, a few metal-free silylation methods have been developed, however they can be applied only to a limited number and type of substrates.17
Bimetallic catalysis has received significant attention for the synthesis of target molecules that are difficult to prepare through the use of conventional monometallic catalysts.18 Recently, we have reported the synthesis of bimetallic Pd–Pt–Fe3O4 nanoflake-shaped alloy nanoparticles as catalysts for the reduction of nitroarenes.19 The magnetically recoverable nanoparticles provided nearly quantitative conversions and yields in the aforementioned reaction, and could be reused for up to 250 catalytic cycles. It has also been reported that bimetallic nanoparticles show extraordinary catalytic activities better than their parent metals.
Therefore, it was envisioned that the bimetallic Pd–Pt–Fe3O4 catalyst might exhibit better activity than a monometallic catalytic system for the silylation reaction. Homogeneous palladium and platinum-catalyzed silylation reactions suffer from several drawbacks. Palladium-based catalytic systems are known to require specific phosphine ligands in some cases. These ligands are generally unstable and quite expensive. Similarly, the use of platinum is undesirable as it is an expensive metal even though it may not require special ligands. Therefore, the employment of the bimetallic Pd–Pt–Fe3O4 nanoparticles as a catalyst is expected to address the abovementioned issues and those of reusability and easy recovery.
Pd–Pt–Fe3O4 nanocatalysts were synthesized via the one-pot solution phase reduction process. To obtain a better morphology for better catalytic activity compared to the previously reported Pd–Pt–Fe3O4 NPs, we prepared the Pd–Pt–Fe3O4 nanocatalysts from Fe3O4 NPs possessing a sphere morphology and decorated more well-dispersed Pd–Pt NPs on the Fe3O4 support by changing the amount of polyvinylpyrrolidone (PVP). As shown in Fig. S1 (ESI†), various PVP conditions were tried, and good morphology and dispersity of the NPs were obtained at 4 times PVP equivalent to Fe3O4 NPs. The detailed structure and morphology of the Pd–Pt–Fe3O4 NPs were characterized using high resolution transmission electron microscopy (HR-TEM), scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS), high bright-field scanning TEM (BF-STEM), high-angle annular dark-field scanning TEM (HAADF-STEM) and elemental analysis mapping by Cs-STEM-EDS (Fig. 1 and Fig. S2–S7, ESI†). The Pd–Pt alloy NPs were well immobilized and distributed on the Fe3O4 NP surface (Fig. 1 and Fig. S4, ESI†). The EDS mapping image of Pd–Pt–Fe3O4 showed that Pd(red) and Pt(blue) points excellently decorated the Fe(yellow) surface (Fig. S4, ESI†). As shown in the HAADF-STM and BF-STEM images (Fig. S5, ESI†), it was confirmed that the Pd–Pt alloy nanocrystals well on the Fe3O4 support. A randomly homogeneous Pd–Pt alloy phase was confirmed by Cs-STEM-EDS (Fig. S6 and S7, ESI†). The Pd–Pt alloy NPs were very evenly dispersed on the Fe3O4 NPs with an average size of 4.8 nm (Fig. S8, ESI†).
 | ||
| Fig. 1 (a) and (b) HR-TEM images of fresh Pd–Pt–Fe3O4 NPs. | ||
The Pd–Pt–Fe3O4 NPs are composed of 4.10 wt% palladium and 9.60 wt% platinum according to inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis. The alloy status of the Pd–Pt on Fe3O4 NPs was confirmed by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) analysis (Fig. S9–S11, ESI†). The Pd–Pt–Fe3O4 NPs can be effectively separated from the reaction solution through the use of an external magnet and collected within 1 minute (Fig. S15, ESI†).
To find the optimal conditions for the silylation reaction using the Pd–Pt–Fe3O4 NPs, we chose methyl 4-iodobenzoate (1a) and triethylsilane (2a) as substrates (Table 1). Based on the previously reported conditions for the Pt-catalyzed silylation reaction, sodium acetate (NaOAc) was employed as a base. First, various solvents were tested for the coupling reaction and it was found that using NMP (N-methyl-2-pyrrolidone) resulted in the best yield of the product (entry 5). Next, with NMP as the solvent of choice, different bases were tested. Metal acetates such as KOAc and CsOAc employed as bases in the silylation reaction showed higher yields of the desired product, 75% and 74%, respectively (entries 6 and 8), compared to other acetates. The reaction with Cs2CO3 showed 75% yield (entry 9). When organic amine bases were employed, 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) and 1,5-diazabicyclo(4.3.0)non-5-ene (DBN) afforded the desired products in 53% and 71% yields, respectively (entries 10 and 11). Finally, the reaction with i-Pr2EtN resulted in 88% yield of the desired product (entry 12). When the catalyst was reduced to 1 mol%, the yield of the product also decreased to 45% (entry 13). When the reaction was conducted with monometallic Pt–Fe3O4 or Pd–Fe3O4, the obtained yields were lower than those obtained from the reaction employing the bimetallic Pd–Pt–Fe3O4 catalyst (entries 14 and 15). Moreover, the reaction employing a combination of Pd–Fe3O4 and Pt–Fe3O4 afforded a lower yield of the product than that with the bimetallic catalytic system (entry 16). Instead of Pd–Pt–Fe3O4 as a catalyst, several Pd- and Pt-based homogeneous catalysts were employed. These Pd and Pt catalysts were ineffective in yielding the desired product (entries 17 and 18). The electronic effect20 could be evidence for the high catalytic activity of the Pd–Pt–Fe3O4 alloy NPs in the hydrosilylation of aryl halides. For the understanding of the unique effects in the Pd–Pt–Fe3O4 NPs, it is necessary to compare the electronic structures of monometallic NPs (Pd– or Pt–Fe3O4) and the bimetallic ones (Pd–Pt–Fe3O4) by using XPS. It can be confirmed that both the Pd 3d (340.2 eV and 334.9 eV) and Pt 4f (73.6 eV and 70.3 eV) peaks slightly shifted toward a lower binding energy in the Pd–Pt–Fe3O4 NPs compared with both the monometallic Pd–Fe3O4 (340.3 eV and 335.0 eV) and Pt–Fe3O4 NPs (74.6 eV and 71.3 eV) (Fig. S10–S12, ESI†). Surprisingly, about 1.0 eV of negative shift was detected for the Pd–Pt–Fe3O4 NPs in the Pt 4f peaks, as Pt gains electrons from Pd. The electronic shift effect induced by Pd–Pt metal alloying, electronically rich Pt can be responsible for the improved hydrosilylation reaction reactivity.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | 1a Conv.b (%) | 3a Yieldb (%) |
| a Reaction conditions: a mixture of 1a (0.2 mmol), 2a (0.3 mmol), Pd–Pt–Fe3O4 (20 mg, 5 mol% of Pd–Pt) and base (0.6 mmol) was stirred in a 1.0 mL solvent at 70 °C for 15 h. b Determined by 1H NMR using an internal standard. c 1 mol% catalyst was employed. d Homogeneous palladium catalysts include Pd(OAc)2, Pd(CH3CN)2Cl2, Pd(PPh3)4, and Pd(PPh3)2Cl2. e Homogeneous platinum catalysts include PtCl2 and PtO2. f DMI = N,N-dimethylimidazolin-2-one. | |||||
| 1 | Pd–Pt–Fe3O4 | NaOAc | DMIf | 86 | 39 |
| 2 | Pd–Pt–Fe3O4 | NaOAc | DMF | 66 | 4 |
| 3 | Pd–Pt–Fe3O4 | NaOAc | DMSO | 67 | 10 |
| 4 | Pd–Pt–Fe3O4 | NaOAc | toluene | 40 | 6 |
| 5 | Pd–Pt−Fe3O4 | NaOAc | NMP | 94 | 44 |
| 6 | Pd–Pt–Fe3O4 | KOAc | NMP | 90 | 75 |
| 7 | Pd–Pt–Fe3O4 | LiOAc | NMP | 94 | 18 |
| 8 | Pd–Pt–Fe3O4 | CsOAc | NMP | 81 | 74 |
| 9 | Pd–Pt–Fe3O4 | Cs2CO3 | NMP | 92 | 75 |
| 10 | Pd–Pt–Fe3O4 | DBU | NMP | 75 | 53 |
| 11 | Pd–Pt–Fe3O4 | DBN | NMP | 100 | 71 |
| 12 | Pd–Pt–Fe3O4 | i-Pr2EtN | NMP | 94 | 88 |
| 13c | Pd–Pt–Fe3O4 | i-Pr2EtN | NMP | 67 | 45 |
| 14 | Pt–Fe3O4 | i-Pr2EtN | NMP | 91 | 70 |
| 15 | Pd–Fe3O4 | i-Pr2EtN | NMP | 63 | 21 |
| 16 | Pt–Fe3O4/Pd–Fe3O4 | i-Pr2EtN | NMP | 84 | 62 |
| 17 | Homo Pdd | i-Pr2EtN | NMP | 0–20 | 0 |
| 18 | Homo Pte | i-Pr2EtN | NMP | 21–23 | Trace |
To optimize the reaction time, the standard reaction was monitored by 1H NMR. As shown in Fig. S16 (ESI†), when the reaction was conducted at 70 °C, the yield of the product reached 51% and 88% in 12 h and 15 h, respectively, and thereafter did not show any increase. The reaction performed at lower temperatures of 25 °C and 50 °C did not give satisfactory results even after 24 h, and the maximum yields at these temperatures were 13% and 36%, respectively. The reaction carried out at 100 °C gave 83% yield in 10 h, although this yield decreased to 67% at 13 h and slowly decreased further to 52% as a result of the thermal decomposition of the arylsilane substrate. From these results, it was concluded that this catalytic system showed optimal yields at 70 °C and the reaction was complete in 15 h.
Under these optimized reaction conditions, we employed a variety of aryl iodides and bromides to evaluate the substrate scope (Scheme 1). Aryl iodides (1) having carbonyl groups such as methyl ester, acetyl, and aldehyde reacted with triethylsilane in the presence of Pd–Pt–Fe3O4 NPs to provide the corresponding aryl silanes 3a, 3b, and 3c in 88%, 84%, and 81% yields, respectively.
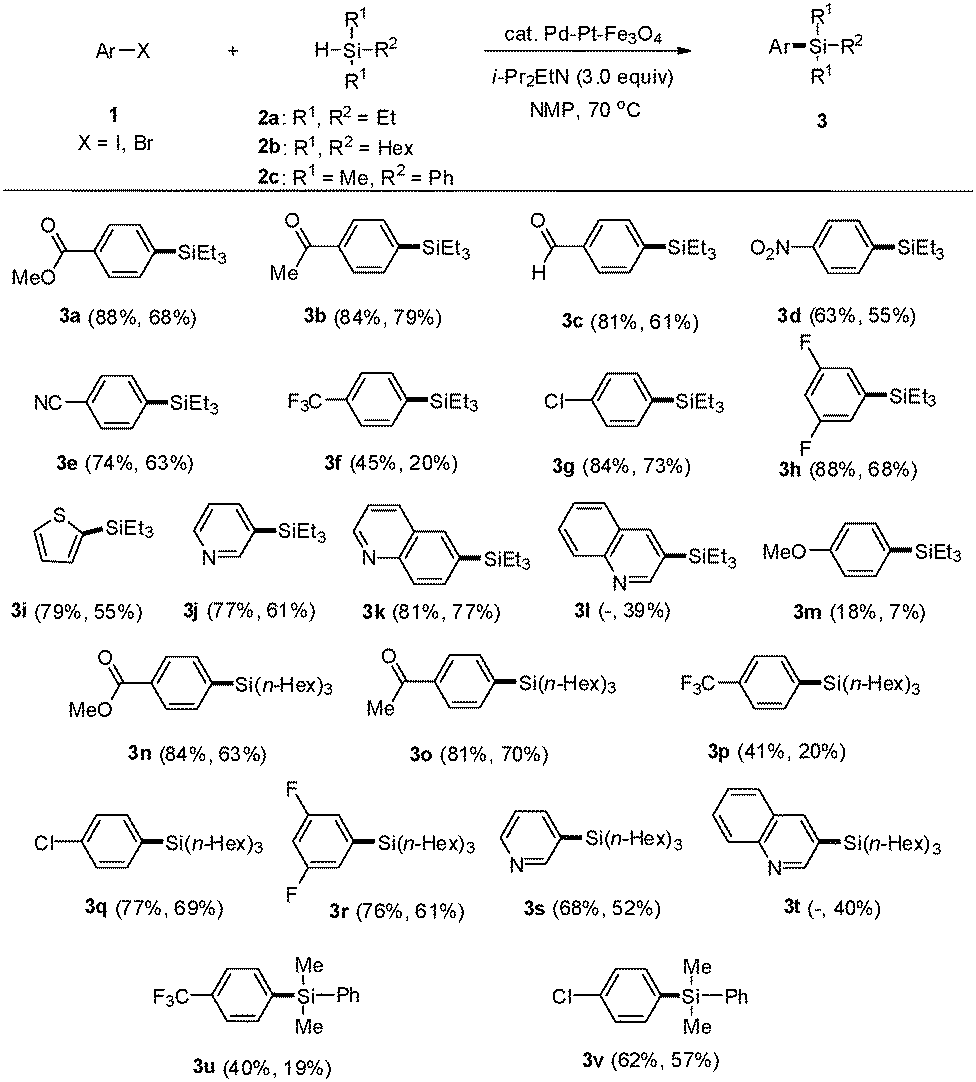 | ||
| Scheme 1 Arylsilylation of aryl iodides and bromides. Reaction conditions: a mixture of 1 (0.7 mmol), 2 (1.05 mmol), i-Pr2EtN (1.05 mmol) and Pd–Pt–Fe3O4 (70 mg) was stirred in NMP (4.0 mL) at 70 °C for 15 h. Yields from aryl iodides are at the left and yields from aryl bromides are at the right in parentheses. | ||
Aryl iodides substituted with electron-withdrawing groups such as 1-iodo-4-nitrobenzene and 4-iodobenzonitrile gave the corresponding products 3d and 3e in 63% and 74% yields, respectively. 1-Iodo-4-(trifluoromethyl)benzene produced 3f in a slightly lower yield. However, the chloro- and fluoro-substituted iodobenzenes formed 3g and 3h in good yields. Heteroaryl iodides such as 2-iodothiophene, 3-iodopyridine, and 6-iodoquinoline afforded the corresponding heteroaryl silanes 3i, 3j, and 3k in 79%, 77%, and 81% yields, respectively. As 3-iodoquinoline is not commercially available, 3-bromoquinoline was employed as a coupling partner instead to yield, unexpectedly, 3-(triethylsilyl)quinoline (3l) in 39% yield. This result prompted an investigation of aryl bromides as potential substrates for this reaction methodology. As expected, all aryl bromides were suitable substitutes for the tested aryl iodides and yielded the desired arylsilanes in good to moderate yields, which were slightly lower than those obtained from aryl iodides. However, 4-haloanisoles afforded the desired product 3m with low yields. These results implied that aryl halides bearing electron-donating groups showed low activity in this arylsilylation. Trihexylsilane (2b) was also coupled with aryl iodides and bromides to give the desired products 3n, 3o, 3p, 3q, 3r, 3s and 3t in moderate to good yields. Dimethylphenylsilane (2c) was coupled with aryl iodides and bromides to provide 3u and 3v in moderate yields.
To evaluate the recyclability of the Pd–Pt–Fe3O4 catalyst in the silylation of methyl 4-iodobenzoate, we recovered the catalyst and reused after each run. After the reaction was complete, the catalyst was separated by using an external magnet and reused for the silylation of the next batch of aryl iodides and triethylsilanes in the presence of fresh i-Pr2EtN. This procedure was repeatedly conducted with the recovered catalyst and the yield of the product was monitored in every cycle. As shown in Fig. 2, the products were isolated in 84–90% yields consistently for the reactions performed over twenty times. In contrast, the yields obtained from the reaction with the monometallic Pt–Fe3O4 catalyst slowly decreased after the tenth recycle. Similarly, the reaction employing the monometallic Pd–Fe3O4 catalyst afforded the desired product in only 12% yield after recycling nine times and its catalytic activity did not recover in the tenth run. From these results, it was concluded that the bimetallic catalyst showed pronouncedly better catalytic activity and recyclability in the hydrosilylation of aryl substrates compared to the corresponding monometallic catalysts.
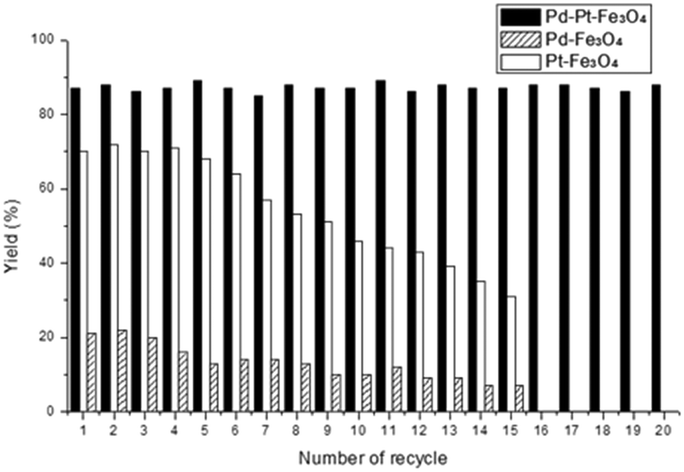 | ||
| Fig. 2 Recyclability of Pd–Pt–Fe3O4, Pd–Fe3O4, and Pt–Fe3O4 catalysts in the arylsilylation reaction. | ||
In the case of Pd–Pt–Fe3O4, the catalytic activity was maintained even after 20 times of reuse, however, the catalytic activity of Pd–Fe3O4 or Pt–Fe3O4 reduced in the 15th recycle. For the comparison of the two different cases, HR-TEM, SEM-EDS, ICP-AES, XRD and XPS of the fresh and spent nanocatalysts were performed. The oxidation state of the nanocatalysts after the reactions did not change much (Fig. S17 and S18, ESI†). However, the HR-TEM image of the Pd–Pt–Fe3O4 NPs after 20 recycles showed some agglomeration of the Pd–Pt NPs (Fig. S19, ESI†). Some detachment of the Pd–Pt NPs from the Fe3O4 support and the inclusion of 0.80 wt% Si were confirmed by the SEM-EDS mapping images and patterns (Fig. S20–S22, ESI†). In addition, metal contents of spent Pd–Pt–Fe3O4 decreased from their fresh state, e.g. from 3.30 wt% to 2.70 wt% for Pd, and from 5.54 wt% to 3.94 wt% for Pt (Table S1, ESI†). As a result, after 20 recycles of Pd–Pt–Fe3O4, it was estimated that on average 0.03 wt% of Pd and 0.08 wt% of Pt were lost upon each cycle. On the other hand, in the cases after 15 recycles of the monometallic catalysts, the Pd–Fe3O4, Pt–Fe3O4 NPs were shown to be highly aggregated and a large degree of metal loss was confirmed (Fig. S23–S26 and Tables S2 and S3, ESI†). The remaining Pd and Pt contents after 15 recycles of Pd–Fe3O4 and Pt–Fe3O4 catalysts were greatly reduced, i.e. 3.44 wt% from 7.47 wt% (fresh) and 2.51 wt% from 10.76 wt% (fresh), respectively. In case of Pd–Fe3O4 and Pt–Fe3O4 recycle tests, on average 0.26 wt% of Pd and 0.55 wt% of Pt leached out upon each run. It is surprising to note that the Pd–Pt–Fe3O4 NPs showed considerably less liberation of the transition metal contents compared to the monometallic catalysts. On the other hand, the XRD peaks of the spent nanocatalysts were not precisely identified due to the leaching of a transition metal (Fig. S29, ESI†). The Pd–Pt alloy has excellent crystallinity as shown in the BF-STEM image of Pd–Pt–Fe3O4 (Fig. S30, ESI†). We may think that the Pd–Pt alloy is structurally and morphologically very stable, and its durability in catalytic reactions is also outstanding. Thus, the bimetallic Pd–Pt–Fe3O4 catalyst exhibits an excellent synergistic effect in both the reactivity of silylation reactions and catalyst durability. Based on the previous report6a and our experimental results, we proposed the reaction mechanism (Fig. S31, ESI†).
In summary, a recyclable bimetallic catalytic system for the silylation of aryl halides has been developed. The reaction with aryl iodides (or bromides) and hydrosilanes in the presence of i-Pr2EtN and the Pd–Pt–Fe3O4 catalyst provided the corresponding aryl silanes in good to moderate yields. In addition, this catalytic system showed good tolerance toward the ester, ketone, aldehyde, nitro, and nitrile functional groups. This is the first report of a recyclable catalytic system for the arylsilylation reaction. The bimetallic Pd–Pt–Fe3O4 catalyst can be readily recovered and reused, and shows better activity and durability compared to the monometallic Pd–Fe3O4 and Pt–Fe3O4 catalysts. The catalyst was recycled up to twenty times and the product yields were found to be consistently good.
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2015R1A4A1041036) and the Nano Material Development Program (NRF-2012M3A7B4049655) through the NRF funded by the Ministry of Education, Science and Technology. The spectral data were obtained from the Gwangju center and the HRMS data from the Daegu center of Korea Basic Science Institute.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) W. Bains and R. Tacke, Curr. Opin. Drug Discovery Dev., 2003, 6, 526 CAS; (b) X.-M. Liu, C. He, J. Haung and J. Xu, Chem. Mater., 2005, 17, 434 CrossRef CAS; (c) Y. You, C.-G. An, J.-J. Kim and S. Y. Park, J. Org. Chem., 2007, 72, 6241 CrossRef CAS PubMed; (d) D. Sun, Z. Ren, M. F. Bryce and S. Yan, J. Mater. Chem. C, 2015, 3, 9496 RSC.
- (a) X. Ren, J. Li, R. Holmes, P. Djurovich, S. Forrest and M. Thompson, Chem. Mater., 2004, 16, 4743 CrossRef CAS; (b) X.-M. Liu, C. He, J. Huang and J. Xu, Chem. Mater., 2005, 17, 434 CrossRef CAS.
- (a) Y. Hatanaka and T. Hiyama, Synlett, 1991, 845 CrossRef CAS; (b) K. A. Horn, Chem. Rev., 1995, 95, 1317 CrossRef CAS; (c) Z. Qureshi, C. Toker and M. Lautens, Synthesis, 2017, 1 CAS.
- (a) S. Lulinski and J. Serwatowski, J. Org. Chem., 2003, 68, 9384 CrossRef CAS PubMed; (b) M. Schlosser and C. Heiss, Eur. J. Org. Chem., 2003, 4618 CrossRef CAS; (c) M. Oestreich, G. Auer and M. Keller, Eur. J. Org. Chem., 2004, 184 Search PubMed.
- Z. Xu, W.-S. Huang, J. Zhang and L.-W. Xu, Synthesis, 2015, 3645 CrossRef.
- (a) Y. Yamanoi, J. Org. Chem., 2005, 70, 9607 CrossRef CAS PubMed; (b) E. McNeill, T. E. Barder and S. L. Buchwald, Org. Lett., 2007, 9, 3785 CrossRef CAS PubMed; (c) Y. Kurihara, M. Nishikawa, Y. Yamanoi and H. Nishihara, Chem. Commun., 2012, 48, 11564 RSC; (d) A. Sinai, A. Mészáros, A. Balogh, M. Zwillinger and Z. Novák, Synthesis, 2017, 2374 CAS.
- (a) M. Murata, M. Ishikura, M. Nagata, S. Watanabe and Y. Masuda, Org. Lett., 2002, 4, 1843 CrossRef CAS PubMed; (b) Y. Yamanoi and H. Nishihara, J. Org. Chem., 2008, 73, 6671 CrossRef CAS PubMed; (c) H. Fang, L. Guo, Y. Zhang, W. Yao and Z. Huang, Org. Lett., 2016, 18, 5624 CrossRef CAS PubMed.
- A. Hamze, O. Provot, M. Alami and J.-D. Brion, Org. Lett., 2006, 8, 931 CrossRef CAS PubMed.
- R. Boukherroub, C. Chatgilialoglu and G. Manuel, Organometallics, 1996, 15, 1508 CrossRef CAS.
- C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236 CrossRef CAS PubMed.
- X. Pu, J. Hu, Y. Zhao and Z. Shi, ACS Catal., 2016, 6, 6692 CrossRef CAS.
- (a) M. Tobisu, R. Nakamura, Y. Kita and N. Chatani, Bull. Korean Chem. Soc., 2010, 31, 582 CrossRef CAS; (b) M. Tobisu, Y. Kita, Y. Ano and N. Chatani, J. Am. Chem. Soc., 2008, 130, 15982 CrossRef CAS PubMed.
- (a) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853 CrossRef CAS PubMed; (b) P. I. Djurovich, A. R. Dolich and D. H. Berry, J. Chem. Soc., Chem. Commun., 1994, 1897 RSC.
- (a) F. Kakiuchi, M. Matsumoto, K. Tsuchiya, K. Igi, T. Hayamizu, N. Chatani and S. Murai, J. Organomet. Chem., 2003, 686, 134 CrossRef CAS; (b) H. Ihara and M. Suginome, J. Am. Chem. Soc., 2009, 131, 7502 CrossRef CAS PubMed.
- (a) N. Tsukada and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 5022 CrossRef CAS PubMed; (b) M. Murata, N. Fukuyama, J.-I. Wada, S. Watanabe and Y. Masuda, Chem. Lett., 2007, 36, 910 CrossRef CAS.
- (a) T. Ishiyama, K. Sato, Y. Nishio and N. Miyaura, Angew. Chem., Int. Ed., 2003, 42, 5346 CrossRef CAS PubMed; (b) G. Choi, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2013, 135, 13149 CrossRef CAS PubMed; (c) E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70 CrossRef CAS PubMed.
- (a) L. Wang, H. Zhu, S. Guo, J. Cheng and J.-T. Yu, Chem. Commun., 2014, 50, 10864 RSC; (b) M. Sasaki and Y. Kondo, Org. Lett., 2015, 17, 848 CrossRef CAS PubMed; (c) W.-B. Liu, D. P. Schuman, Y.-F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stolz, J. Am. Chem. Soc., 2017, 139, 6867 CrossRef CAS PubMed; (d) A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80 CrossRef CAS PubMed.
- (a) Y. Misumi, Y. Ishii and M. Hidai, Organometallics, 1995, 14, 1770 CrossRef CAS; (b) M. Sankar, N. Dimitratos, P. J. MIedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099 RSC; (c) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC; (d) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed; (e) H. Miura, K. Endo, R. Ogawa and T. Shishido, ACS Catal., 2017, 7, 1543 CrossRef CAS.
- S. Byun, Y. Song and B. M. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 14637 CAS.
- (a) H. Zhang, M. Jin and Y. Xia, Chem. Soc. Rev., 2012, 41, 8035–8049 RSC; (b) F.-M. Li, X.-Q. Gao, S.-N. Li, Y. Chen and J.-M. Lee, NPG Asia Mater., 2015, 7, 219–225 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data and 1H and 13C NMR spectra. See DOI: 10.1039/c7cc09926f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |