
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable dynamic covalent hydrogels of boronic acid polymers cross-linked by bioactive plant-derived polyphenols†
Zhuojun
Huang‡
 a,
Peyman
Delparastan‡
a,
Patrick
Burch
b,
Jing
Cheng
b,
Yi
Cao
c and
Phillip B.
Messersmith
*abd
a,
Peyman
Delparastan‡
a,
Patrick
Burch
b,
Jing
Cheng
b,
Yi
Cao
c and
Phillip B.
Messersmith
*abd
aDepartment of Materials Science and Engineering, University of California, Berkeley, Berkeley, CA 94720-1760, USA. E-mail: philm@berkeley.edu
bDepartment of Bioengineering, University of California, Berkeley, Berkeley, CA 94720-1760, USA
cDepartment of Physics, Nanjing University, Nanjing, 210093, China PR
dMaterials Science Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
First published on 2nd August 2018
Abstract
We report here the development of hydrogels formed at physiological conditions using PEG (polyethylene glycol) based polymers modified with boronic acids (BAs) as backbones and the plant derived polyphenols ellagic acid (EA), epigallocatechin gallate (EGCG), tannic acid (TA), nordihydroguaiaretic acid (NDGA), rutin trihydrate (RT), rosmarinic acid (RA) and carminic acid (CA) as linkers. Rheological frequency sweep and single molecule force spectroscopy (SMFS) experiments show that hydrogels linked with EGCG and TA are mechanically stiff, arising from the dynamic covalent bond formed by the polyphenol linker and boronic acid functionalized polymer. Stability tests of the hydrogels in physiological conditions revealed that gels linked with EA, EGCG, and TA are stable. We furthermore showed that EA- and EGCG-linked hydrogels can be formed via in situ gelation in pH 7.4 buffer, and provide long-term steady state release of bioactive EA. In vitro experiments showed that EA-linked hydrogel significantly reduced the viability of CAL-27 human oral cancer cells via gradual release of EA.
Introduction
Plant polyphenols are a family of naturally derived compounds that contain a high concentration of phenolic hydroxyl groups and are linked to diverse biological functions such as chemical defense, pigmentation, structural support, and prevention of radiation damage.1,2 In addition to their recent emergence as building blocks for functional materials,3–6 plant polyphenols are highly celebrated for their complex bioactivities and antioxidant behavior. A high dietary intake of polyphenols has been linked to a reduced incidence of a number of diseases, including cancer, diabetes, osteoporosis as well as cardiovascular and neurodegenerative diseases, and several polyphenols have been investigated as therapeutics.7–10 For instance, ellagic acid (EA) has anti-proliferative properties and has been shown effective against prostatic, breast, brain, and oral cancers in a number of in vitro and small-animal models.11–16 In addition to antiproliferative effects, many polyphenols like epigallocatechin gallate (EGCG) and tannic acid (TA) have been found to have antioxidant, antibacterial, and anti-inflammatory effects.7 However, despite their impressive therapeutic effects, polyphenols suffer from a number of drawbacks which limit their clinical translation, including poor bioavailability, rapid metabolism, and poor membrane permeability. Therefore, development of an efficient delivery method for these beneficial therapeutic molecules would be a milestone in translation into the clinic.Recently, dynamic covalent chemistry has been explored to develop hydrogels with predictable degradation and drug release kinetics that exploit the stimuli-responsive nature of the complexes formed via dynamic covalent chemistry.17–19 One example is given by boronic esters formed between boronic acids and cis-1,2 or cis-1,3 diol containing molecules.20–24 Boronic acids have been proved to be stable in biological conditions and have no or extremely little toxicity on the human body.25–27 Moreover, the equilibrium between boronic acid and boronic ester is sensitive towards pH, temperature, and competing hydroxylated compounds present in the media,20,21,28,29 enabling the formation of novel local drug delivery systems and hydrogels with mechanical properties that are responsive to changes in pH.18,22,25,30–35 Although boronate ester complexes are generally favored at alkaline pH, the equilibrium can be tuned by modification of the chemical structure of the boronic acid group.36–39 Using this approach it is possible in principle to shift the equilibrium in favor of the boronate ester to neutral pH, affording the ability to form hydrogels from boronic acid derivatized polymers under physiological conditions.36 Since the polyphenols are susceptible to oxidation and oligomerization at highly alkaline pH,3 lowering the complexation pH can further result in sustained bioactivity of these molecules due to limited oxidation.
Although the high hydroxyl content and rich bioactivity spectrum of polyphenols presents a unique opportunity to form therapeutically active dynamic covalent hydrogels with boronic acid polymers, these molecules have not been widely investigated in this context. Herein we exploit the pH dependence and dynamic nature of complexes between boronic acid functionalized PEG and polyphenols as bioactive injectable hydrogels (Fig. 1). In this concept, the polyphenol functions both as cross-linker and therapeutic, forming a hydrogel by injection of a precursor solution of polymer and polyphenol that solidifies through formation of boronate ester cross-links upon neutralization to physiologic pH. The dynamic nature of the boronate ester bond provides a mechanism for latent hydrogel disassembly and release of therapeutic polyphenols into the surrounding environment.37,38,40 Numerous combinations of natural polyphenols, substituted boronic acids and PEG polymers of different architecture were screened by rheological analysis, revealing several formulations that gave rise to rapid gelation at physiologic pH. Insight into the molecular mechanics of boronic acid–polyphenol interactions was provided by measuring bond rupture forces in a pH dependent manner using single molecule force spectroscopy (SMFS). Finally, we studied the kinetics of EA release from a PEG–boronic acid hydrogel and demonstrated the in vitro cytotoxic effects of released EA on cancer cells.
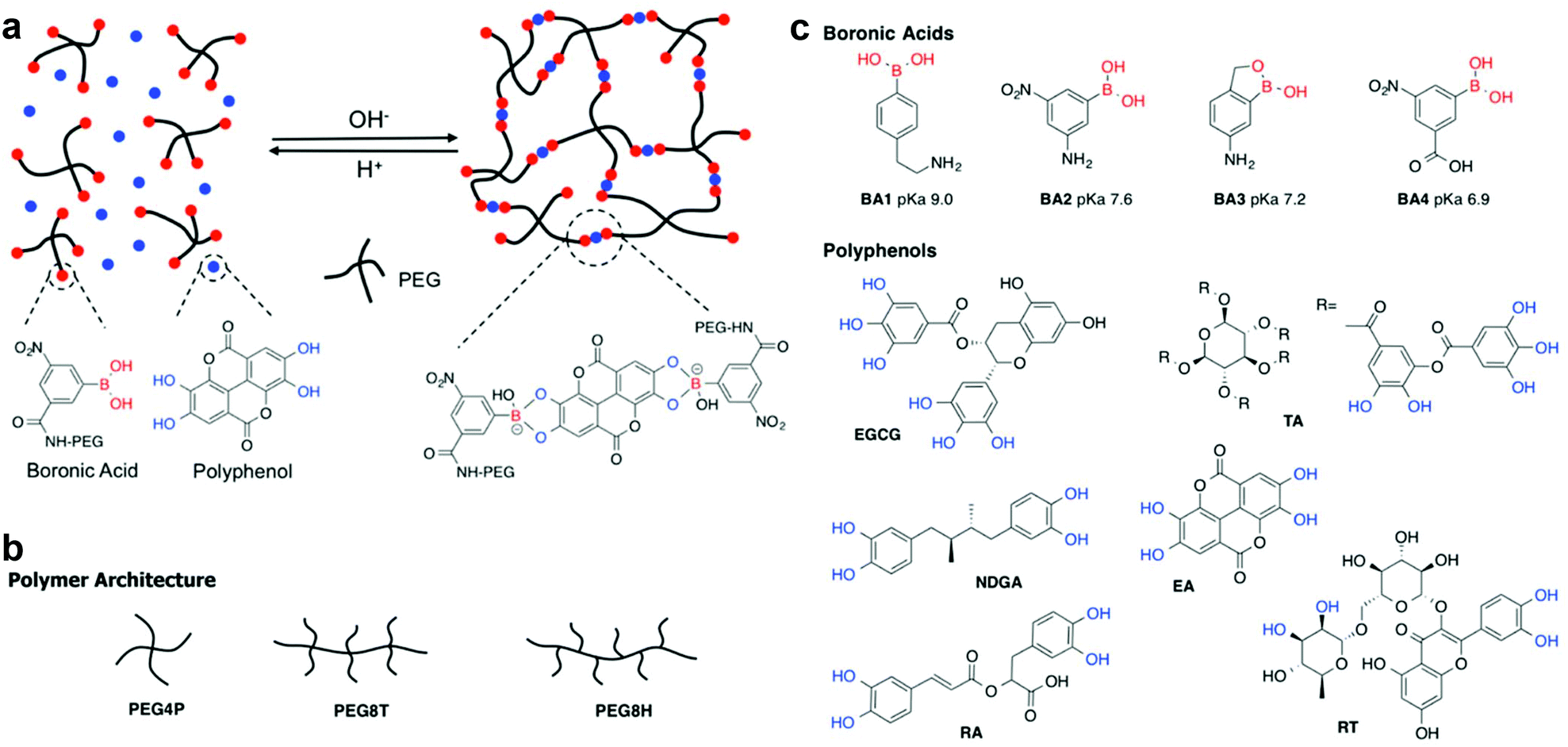 | ||
| Fig. 1 (a) Schematic illustration of pH responsive complexation between boronic acid functionalized PEG and polyphenols resulting in formation of a hydrogel network. The specific boronate ester formed between BA4 and EA is shown as an example only. Architectures of PEG polymers (b), and chemical structures of boronic acids and polyphenols (c) used in the experiments. Vicinal diols capable of boronate ester formation are shown in blue. | ||
Experimental
Materials
4-Arm PEG (pentaerythritol core, M.W. 10 kDa) (PEG4P) and 8-arm PEG (tripentaerythritol core, M.W. 20 kDa) (PEG8T) with hydroxyl endgroups were purchased from Creative PEGWorks. PEG8T and 8-arm PEG-NH2 (hexaglycerol core, M.W. 20 kDa) (PEG8H) with amine endgroups were obtained from JenKem Technology. (4-(2-Aminoethyl) phenyl) boronic acid (BA1) and 3-carboxy-5-nitrophenylboronic acid (BA4) were obtained from Sigma-Aldrich. 3-Amino-5-nitrophenylboronic acid (BA2) was purchased from Alfa Aesar and 6-nitrobenzo[c][1,2] oxaborol-1(3H)-ol was from Ark Pharm. 2,2,6,6-Tetramethyl-1-piperidinyloxy, free radical, 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), NaClO, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), triethylamine (TEA), Pd on charcoal, N,N-diisopropylethylamine (DIPEA), N,N-diisopropylcarbodiimide (DIC), hydroxybenzotriazole (HOBt), EA, EGCG, TA, NDGA, rutin trihydrate and other polyphenols and chemicals used for PEG modification were acquired from Sigma-Aldrich. Fetal bovine serum (FBS), phosphate buffered saline (PBS) buffer, Dulbecco's Modified Eagle Medium (DMEM) with glutamine and pyruvate were purchased from Invitrogen. CAL-27 human squamous cell carcinoma cells were purchased from American Type Culture Collection (ATCC).Polymer synthesis
A modular approach was adopted whereby PEGs of different molecular weights and architectures were functionalized with four different phenylboronic acids (Fig. 1c). The pKa values of the BAs used range from 6.9 to 9.0 based on previous studies.39,41–45 Detailed synthesis methods and NMR spectra of all PEG products can be found in ESI (Fig. S1–S10†). PEG-OH was converted to PEG-COOH by TEMPO and NaClO and then reacted with the primary amine on BA1, BA2 and BA3 using HBTU and TEA. BA4 was linked to PEG-NH2 using DIPEA, DIC and HOBt.Hydrogel formation
A typical procedure used to form hydrogels was as follows: 50 mg of polymer was dissolved in pH 7.4 PBS buffer (10 mM PO43−, 137 mM NaCl, and 2.7 mM KCl) and mixed with solutions of polyphenol in the PBS buffer. In a few cases for polyphenols with limited solubility in water, DMSO was used to dissolve the polyphenol (Table 2). The molecular ratio between boronic acid groups on the polymer chain and active diol moiety on polyphenol linker was kept constant at 1 to 1 ratio in all the experiments (we note that in the case of TA the stoichiometry is an estimate only because the structure shown in Fig. 1c is an idealized one). For selected cases, in situ gelation by injection was demonstrated by preparing buffer solutions of 100 mM sodium acetate/acetic acid at pHs 3.2, 4, 4.5, and 5 or 100 mM H2CO3/NaHCO3 (pH 8.4 and 9.9). A precursor solution of 8.3 mg PEG8H-BA4 and polyphenol (0.5 mg EA or 0.7 mg EGCG) in 25 μL DMSO was injected into 10 mL of the buffers listed above and the gelation process was recorded in videos.| Hydrogel composition | Crossover frequency (rad s−1) | G′ at 30 rad s−1 (Pa) |
|---|---|---|
| PEG4P-BA1-EGCG | 19.67 | 2.3 × 104 |
| PEG4P-BA2-EGCG | 2.72 | 1.1 × 104 |
| PEG4P-BA3-EGCG | 7.70 | 1.4 × 104 |
| PEG8T-BA3-EGCG | 3.52 | 4.8 × 104 |
| PEG8T-BA4-EGCG | 0.90 | 2.6 × 104 |
| PEG8H-BA4-EGCG | 0.86 | 5.2 × 104 |
| Crosslinker | Solventa | Stability | ω (rad s−1) | G′ atc (Pa) |
|---|---|---|---|---|
| a Limited solubility of some linkers required the use of DMSO or a H2O/DMSO cosolvent. b Crossover frequency. c G′ at 30 rad s−1. d PEG8H-BA4-EA formed through in situ gelation. | ||||
| Epigallocatechin gallate (EGCG) | H2O | >60 days | 0.86 | 5.2 × 104 |
| Ellagic acid (EA) | DMSOd | >60 days | 4.07 | 1.3 × 104 |
| Tannic acid (TA) | H2O | >60 days | 0.70 | 5.5 × 104 |
| Nordihydroguaiaretic acid (NDGA) | H2O/DMSO | 24 hours | 5.49 | 2.2 × 104 |
| Rutin trihydrate (RT) | H2O/DMSO | 24 hours | 22.5 | 1.6 × 104 |
| Rosmarinic acid (RA) | H2O/DMSO | 24 hours | 4.44 | 2.5 × 104 |
| Carminic acid (CA) | H2O/DMSO | 24 hours | 3.51 | 5.1 × 104 |
Hydrogel characterization
Rheological properties of hydrogels were measured with a frequency sweep test (0.6 rad s−1 to 60 rad s−1 at 25 °C) using a MCR 302 Modular Compact Rheometer (Anton Paar, Ashland, VA) with a parallel plate geometry (8 mm diameter). The crossover frequencies of storage (G′) and loss (G′′) moduli, as well as the G′ values at 30 rad s−1 of each hydrogel was recorded. Qualitative stability of the gels in buffer was measured by immersing them in excess pH 7.4 PBS buffer at 37 °C. The buffer was replaced every day and the persistence of the hydrogel was confirmed visually.AFM single molecule force spectroscopy
Glass coverslips (VWR) were cleaned in Piranha solution (H2SO4/H2O2 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 v/v, caution: care must be taken when using Piranha solution) at room temperature for 1 hour and then rinsed extensively with Milli-Q water followed by rinsing with ethanol and dried under a stream of nitrogen gas. AFM cantilevers were also cleaned using a similar method.46 Polyphenols were dissolved and mixed with a solution of PEG-BA in buffer (pH 8.4 or 9.9); several drops (50–100 μL) of the mixture were pipetted onto glass coverslips and allowed to adsorb at room temperature for 30 minutes. The coverslips were then rinsed with buffered water and used immediately in SMFS experiments. Measurements were carried out using a JPK ForceRobot 300 (JPK Instruments AG, Germany) with a tip velocity of 2000 nm s−1 over a z-piezo distance of 400 nm. Cantilevers (MLCT from Bruker Nano Inc.) of typical spring constant of 30–60 pN nm−1 were used for all experiments and calibrated using the equipartition theorem.47,48 The force-extension traces were recorded and analyzed using data processing software from JPK.
:30 v/v, caution: care must be taken when using Piranha solution) at room temperature for 1 hour and then rinsed extensively with Milli-Q water followed by rinsing with ethanol and dried under a stream of nitrogen gas. AFM cantilevers were also cleaned using a similar method.46 Polyphenols were dissolved and mixed with a solution of PEG-BA in buffer (pH 8.4 or 9.9); several drops (50–100 μL) of the mixture were pipetted onto glass coverslips and allowed to adsorb at room temperature for 30 minutes. The coverslips were then rinsed with buffered water and used immediately in SMFS experiments. Measurements were carried out using a JPK ForceRobot 300 (JPK Instruments AG, Germany) with a tip velocity of 2000 nm s−1 over a z-piezo distance of 400 nm. Cantilevers (MLCT from Bruker Nano Inc.) of typical spring constant of 30–60 pN nm−1 were used for all experiments and calibrated using the equipartition theorem.47,48 The force-extension traces were recorded and analyzed using data processing software from JPK.
Drug release measurement
50 mg of PEG and 3 mg of EA were dissolved in 150 μL of DMSO and 50 μL of this solution was then injected into 40 mL of pH 7.4 PBS buffer to form a hydrogel. At each time point the container was centrifuged at 4000 rpm for 5 minutes and the supernatant was removed, saved for analysis, and replaced with an equal volume of the fresh PBS buffer. This procedure was carried out after the first hour and on a daily basis afterwards. The amount of EA released was measured by High Performance Liquid Chromatography (HPLC) using an Agilent Technologies 1260 Infinity instrument. The standard curve of EA was measured in solutions of known EA concentrations in 10% DMSO, 90% acetonitrile (Fig. S11†).In vitro cytotoxicity
Human oral cancer cell line CAL-27 was used to test the in vitro anticancer properties of PEG8H-BA4-EA hydrogel. The measured LC50 value of EA for CAL-27 cells was 2.89 ± 0.3 μg mL−1 (Fig. S12†). CAL-27 cells were seeded onto 6-well plates and cultured in DMEM buffer with 10% fetal bovine serum (FBS) to reach a concentration of 105 cells per ml. Then the cell culture media was replaced with 3 ml 5% FBS DMEM solution and a transwell was added on top of each well. Each of the following formulations was analyzed by the transwell experiment: 50 μL DMSO solution of 17 mg PEG8H-BA4 and 1 mg EA; 50 μL DMSO solution of 1 mg EA; 50 μL DMSO solution of 17 mg PEG8H-BA4 and 50 μL DMSO. An untreated control group was also performed by placing an empty transwell in the well. The cells were incubated in 5% CO2 at 37 °C incubator for two days before they were stained with ReadyProbes® cell viability image kit blue/green (Molecular Probes) for 15 minutes. After the staining, the cells were imaged using life technologies EVOS FL fluorescence microscope at DAPI (blue, live cells) and GFP (green, dead cells) channels (Fig. S13†). The survival rate of the cells was calculated based on image analysis using ImageJ.Results and discussion
Boronic acid polymer design
The equilibrium between boronic acid and diol is highly influenced by the pH.36–39,49 Formation of boronate ester bonds is dependent on the pKas of both boronic acid and diol molecules and is usually facilitated at higher pH values, often above pH 8. Through the use of chemically modified boronic acids,39,41,43–45 it is possible to shift the pKa to lower values and therefore drive complex formation and hydrogel stability at neutral or even slightly acidic pH. Here we exploited this strategy with branched PEGs modified with boronic acids of variable pKa values (Fig. 1), combining these with naturally derived polyphenols to form hydrogels.Hydrogel formation
A structural requirement guiding the selection of cross-linkers used in this study was that they should contain at least two sites for boronate ester formation, i.e. a combination of aromatic ortho-dihydroxyl groups and alkyl cis-diol (Fig. 1c and S14, Table S1†). Only compounds that meet this criterion would be expected to form networks by cross-linking polymers via boronate ester complex formation. Studies of PEG-BAs and polyphenols utilized visual inspection to quickly screen for compositions that formed gels at pH 7.4. We initially surveyed six PEG-BAs consisting of both four-arm (PEG4P) and eight-arm (PEG8T and PEG8H) polymers terminated with the four phenyl boronic acids (BA1–BA4) as shown in Fig. 1. These polymers were mixed with over 20 polyphenols, saccharides, antibiotics and dyes (Table S1 and Fig. S14†) and examined for evidence of gel formation. Visual surveys of these combinations revealed EGCG, EA, TA, RT, RA and CA as gel-forming linkers under these conditions. Other compounds listed in Table S1† did not show evidence of gel formation and were not investigated further.Hydrogel characterization
The influence of polymer architecture and boronic acid pKa was revealed in rheology experiments using EGCG as a model cross-linker in pH 7.4 buffer (Table 1, Fig. 2 and S15†). Typically, a low crossover frequency along with high plateau G′ values are indicators of a stiffer hydrogel. The rheology results reveal the dynamics of boronate ester complexes and expected trends in hydrogel stability when the pKa value of boronic acid is changed. For example, in the case of the 4-arm PEG system, boronic acid BA1 (pKa 9.0) had a significantly higher crossover frequency when compared to BA2 (pKa 7.6) and BA3 (pKa 7.2).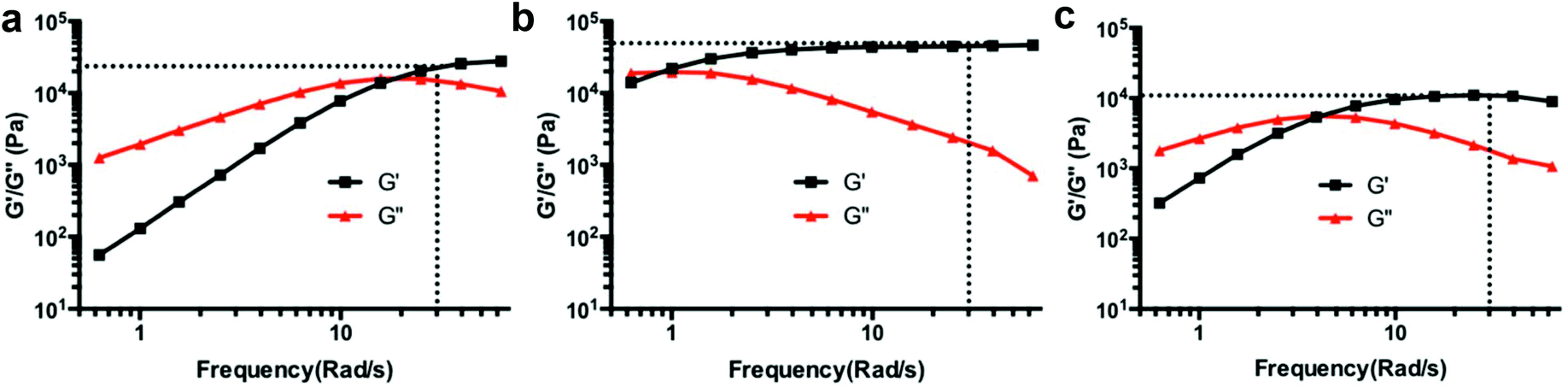 | ||
| Fig. 2 Results of rheology frequency sweep test for hydrogels cross-linked with EGCG and EA at pH 7.4. (a) PEG4P-BA1-EGCG; (b) PEG8H-BA4-EGCG; (c) PEG8H-BA4-EA. G′ and G′′ represent storage and loss moduli, respectively. The storage moduli at 30 rad s−1 are marked. | ||
Additionally, the effect of polymer architecture is apparent from the low crossover frequency and high plateau storage modulus of 8-arm PEG polymers compared to 4-arm PEGs with equivalent boronic acid composition, likely due to higher crosslinking density. The influence of hexaglycerol (H) versus tripentaerythritol (T) core in 8-arm PEG-BAs was more subtle, as PEG8H-BA4-EGCG and PEG8T-BA4-EGCG hydrogels both had similar rheological behavior.
Most importantly, PEG8H-BA4 resulted in the lowest crossover frequencies and highest plateau G′ value among all of the hydrogels tested. Therefore, we selected PEG8H-BA4 for further characterization. Using PEG8H-BA4 as a polymer platform, we then conducted a more in-depth study of polyphenol cross-linkers by comparing the rheological properties and physiologic stability of hydrogels made from PEG8H-BA4 with EGCG, EA, TA, RT, RA, NDGA and CA (Table 2 and Fig. S16†). Hydrogels formed with EGCG, TA, or EA were stable for more than two months in excess pH 7.4 PBS buffer, while gels formed with other linkers were completely dissolved over the course of several days. In the case of EA, attempts to make hydrogels by mixing solutions of PEG8H-BA4 and EA in the PBS buffer were confounded by the poor solubility of EA at pH 7.4,50 forming precipitates of EA. We therefore employed DMSO as a solvent for EA; gels formed by injecting a DMSO solution of EA into a PBS solution of PEG8H-BA4 at pH 7.4 maintained their integrity in physiological condition for several months.
Single molecule force spectroscopy
The SMFS technique has been recognized as a unique tool that enables researchers to study molecular features of noncovalent and dynamic covalent bond interactions in macromolecules.51–53 Previous SMFS reports have shown the power of this method to study mechanical properties of proteins such as unfolding events of globular modules at the molecular level.54–57 SMFS was performed here in order to provide more insight into molecular phenomena underlying the observed macroscopic behavior. To the best of our knowledge no molecular scale SMFS studies on the strength, reversibility, and pH dependence of these interactions have been reported. We applied the recently developed “multiple-fishhook” approach,58,59 which is based on establishing strong interactions between the polymer backbone and the substrate through physisorption and randomly picking up molecules by an unmodified cantilever, as shown schematically in Fig. 3. Using this method, we were able to probe the molecular mechanics of the boronate ester complexes formed between PEG8H-BA4 and polyphenol linker molecules at neutral and alkaline pH as indicated by a “sawtooth” pattern, reminiscent of what is observed upon unfolding of a globular protein under stretch,54–57 in the F–D curves (Fig. 3 and S17†). Nonspecific adsorption of polymer to the cantilever followed by retraction revealed multiple sequential dissociation events due to the multivalent nature of the polymers.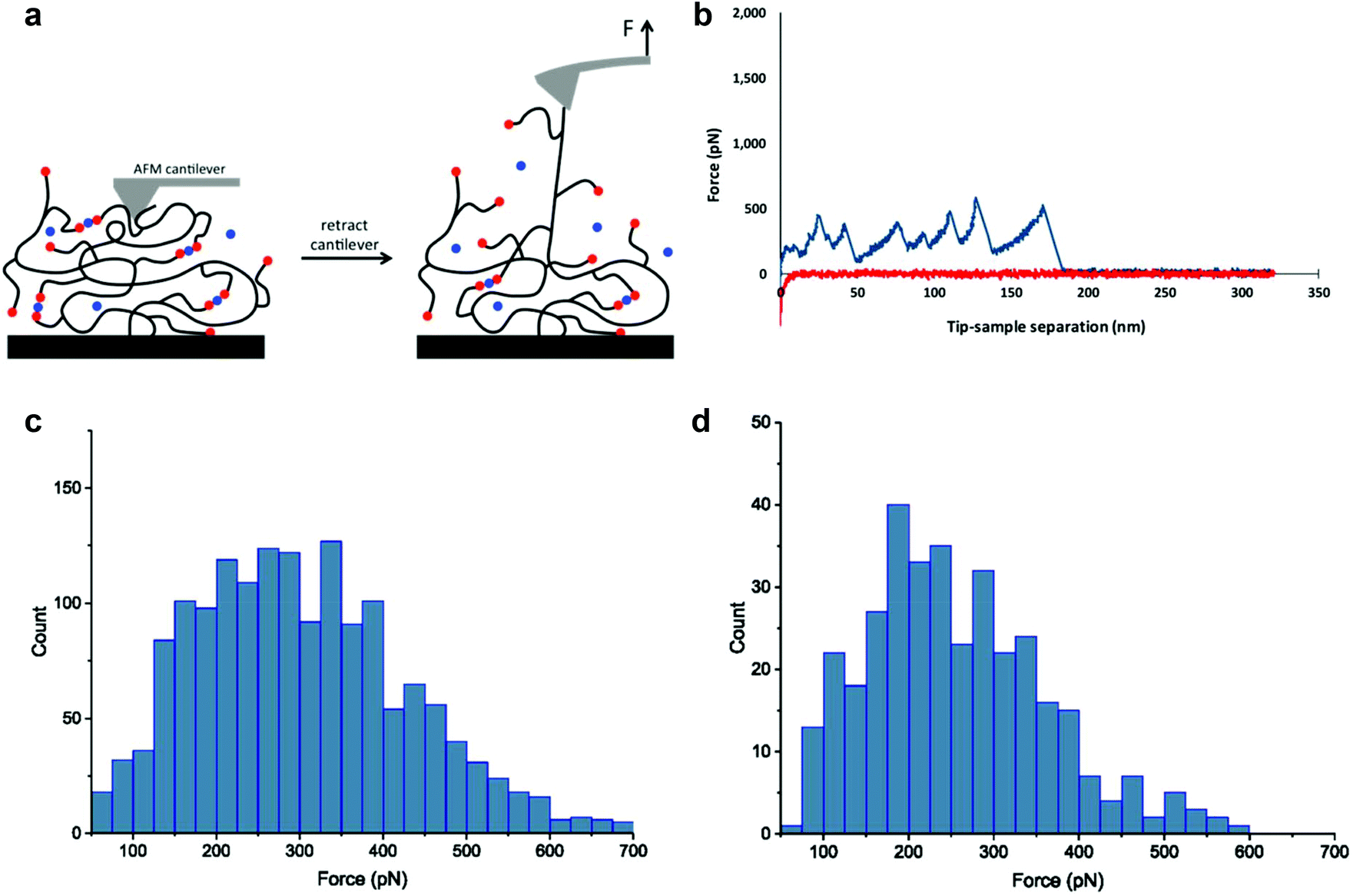 | ||
| Fig. 3 Single molecule force spectroscopy experiments. Schematic of the experiments is shown in (a) with boronic acids in red and polyphenols in blue. Polymer chains are picked up through physical adsorption to the cantilever tip. Retraction of the cantilever from the surface results in sequential rupture of boronate ester cross-links which gives rise to a sawtooth-like pattern in the force–distance curve. A representative force-separation curve of PEG8H-BA4-EGCG is shown in (b), with approach curve shown in red and retraction curve in blue. The force histograms of PEG8H-BA4-EGCG and PEG8H-BA4-EA interactions are shown in (c) and (d), respectively. | ||
Each individual dissociation event is preceded by force-induced polymer chain extension, culminating in boronate ester bond rupture, which in turn reveals previously unloaded polymer chain segments (the so-called “hidden length”), followed by further chain extension and subsequent rupturing of additional boronate ester linkages. A worm-like chain model was used to fit the unbinding events (Fig. 3b and S17†), yielding a persistence length of ∼0.4 nm, consistent with the value for a single PEG chain reported in literature.60 This suggests that the rupture forces measured for those peaks corresponded to single molecule interactions; the histograms of these values have been plotted in Fig. 3c and d. Clearly, the mechanical stability of the dynamic covalent boronate ester bonds is higher than most noncovalent interactions, such as streptavidin–biotin61 and antibody–antigen interactions,62 similar to many metal chelation bonds,59,63,64 but weaker than typical covalent bonds.65 Therefore, boronate ester bonds are expected to provide strong intermolecular cross-linking of branched polymers to form mechanically stable hydrogels.
In scatter plots of distance to rupture versus rupture force (Fig. S18†) representing the accumulation of many experiments, we observed an interesting feature that has not been previously reported in the literature for SMFS of branched polymer systems. Inspection of the data clearly shows evidence of clustering of rupture events at separation distances of 42, 84, and 136 nm (calculated values corresponding to Fig. S18a†). Interestingly, the approximately 42 nm separation is essentially the same as the calculated average contour length of the PEG8H-BA4 polymer used in the SMFS experiments (details in ESI†). Within each cluster there can be seen a distribution of pull-off distances reflecting the statistical nature of the polymer. We therefore interpret the clustering of SMFS data as consistent with the molecular weight and architecture of the polymer chain that is being pulled by the cantilever, where increments of one or more PEG8H-BA4 molecules cross-linked by polyphenol bridge the cantilever and the flat surface. These data further confirm the single molecule nature of the measurements of polymer-polyphenol dissociation.
Among all the linker molecules used in the SMFS experiments, EGCG resulted in the highest mean dissociation force in the histogram of force distribution, followed by NDGA and EA (Table S3†). In addition, the probability of observing multiple rupture events was higher for EGCG compared to NDGA and EA. As discussed above and shown in Fig. S16,† hydrogels formed with EGCG have the lowest crossover frequency whereas larger values are associated with hydrogels formed with NDGA and EA. When considering the force values obtained in SMFS and taking into account the probability of observing rupture of interactions, which in fact correlates to extent of formation of such interactions, results obtained in the single molecule force measurements were in agreement with the data collected in rheology tests performed on the hydrogels formed with these linkers.
Consistent with the equilibrium described above and our general observations that aqueous mixtures of PEG-BAs and polyphenols are liquids at low pH due to a shift in the equilibrium to the dissociated state, we expected a low observation rate of boronate ester in SMFS experiments conducted at low pH values. Indeed, the pH-dependence of these interactions was confirmed in SMFS experiments, where at low pH no sawtooth pattern was observed for either EA or EGCG in thousands of force–distance curves analyzed, indicating that the complexes were not formed (data not shown). This pH dependent behavior was exploited for in situ gelation by injecting a slightly acidic PEG8H-BA4 and polyphenol solution into pH 7.4 PBS buffer (Fig. 4, gelation video in ESI†). We found both EA and EGCG mixed with PEG8H-BA4 can gelate upon injection into the same PBS buffer (Table S2†), which offers the opportunity to use an injectable hydrogel for localized delivery of bioactive polyphenols.
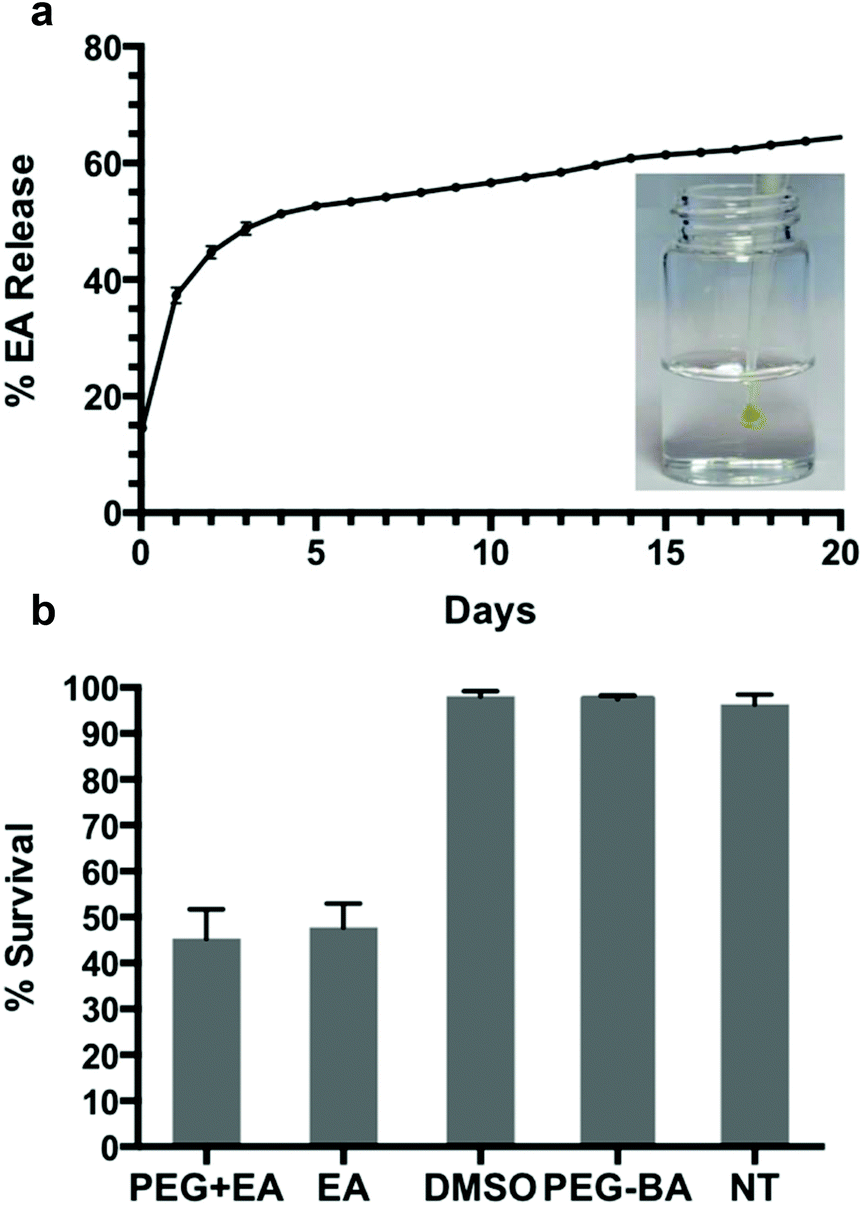 | ||
| Fig. 4 The release of EA from PEG8H-BA4-EA hydrogel (a); a steady state release of 0.8% per day was observed after day 6. Inset shows a photograph of in situ gel formation upon injection of an acidic precursor solution of PEG8H-BA4 and EA into excess buffer at pH 7.4. CAL-27 cell survival rate upon exposure to PEG8H-BA4-EA gel (b). Similar survival rates were observed for both PEG8H-BA4-EA and EA groups, whereas nearly 100% survival was found for the DMSO, PEG8H-BA4, and untreated (NT) groups. | ||
Drug release and in vitro cytotoxicity
Aside from playing a crucial role in pH dependent in situ gelation, the dynamics of boronate ester covalent bonds allow for self-healing of the hydrogel matrix,36,66 and the opportunity for gradual release of bioactive linker and dissolution of the hydrogel. To further explore the possibility of using PEG8H-BA4 based hydrogel for drug delivery, drug release and in vitro cytotoxicity experiments were performed on PEG8H-BA4-EA hydrogel. One advantage of EA as a hydrogel cross-linker is its high stability toward oxidation, as EGCG and TA are more susceptible to oxidation resulting in difficulties in maintaining structural integrity and bioactivity of the hydrogel.67–69 Additionally, the antiproliferative properties of EA have led to in vitro and in vivo investigations for cancer treatment,12–16,70 however its poor solubility in aqueous solution limits its bioavailiability and makes delivery challenging.50 In our system, the poor solubility of EA was overcome by complexation with boronic acid functionalized polymer, thus providing a means for rapid formation of a polymer hydrogel that acts as a depot for gradual delivery of EA.The release of EA from the hydrogel made by in situ gelation of PEG8H-BA4 and EA was measured over a 20-day period (Fig. 4). After 20 days of release, 65% of initial EA was released into the environment, 24% was found to remain in the hydrogel matrix, and the remaining 11% EA was unrecovered, possibly due to small fragments of hydrogel being washed away when the buffer solution was exchanged every day. Following an initial period of burst release, steady state release of approximately 0.8% every day was observed after day 6. We suspect that two general mechanisms play roles in the kinetic release of EA from the hydrogel matrix: the burst release of unbound EA molecules that are physically trapped in the hydrogel matrix, and the dissociation of dynamic covalent boronate ester bonds.37,38 During the fast in situ gelation, the viscosity of the solution increases rapidly, causing some free EA molecules to become physically trapped in the gel matrix without reacting with BA. We suspect that these unbound molecules are responsible for the initial rapid discharge of EA. After several days of immersion in pH 7.4 PBS buffer, most of the free EA molecules are cleared and the mode governing discharge of EA switches to steadfast dynamic boronate ester bond dissociation (Fig. 4). Since our experiments were conducted under sink conditions, the low concentration of EA in the surrounding environment drives a continuous release of the EA molecules from the matrix through slow boronate ester bond dissociation.
Finally, the anticancer bioactivity of EA released from in situ formed gels was assayed by exposing CAL-27 cells to PEG8H-BA4-EA gel in a transwell format. Fluorescence microscopy images of stained cells under different treatment conditions are shown in Fig. S13.† Five experimental groups were established to examine the cytotoxicity of the hydrogels developed. For each group, images were taken at different spots in the sample and cell survival was calculated by image analysis (Fig. 4 and S13†). Based on the results, hydrogel-bound and free EA molecules behave quite similarly in terms of cytotoxicity, resulting in CAL-27 oral cancer cell survival rate of approximately 45% after exposure to either free EA or PEG8H-BA4-EA hydrogel. Conversely, in other experiments using only DMSO or PEG8H-BA4, no apparent cell death was observed. Due to the promising injectable physical characteristics and the biological response of cancer cells to EA released from the hydrogels, we believe that injectable boronate polymer-EA hydrogels are candidates for further study in the context of cancer therapy.
Conclusions
The PEG-BA polymers synthesized here can be utilized in combination with several natural polyphenols as in situ forming injectable hydrogels. The polymer gel is held together by dynamic boronate ester bonds formed spontaneously at physiologic pH. Among the polyphenol linkers investigated in this study, ellagic acid (EA), epigallocatechin gallate (EGCG) and tannic acid (TA) were capable of forming stable hydrogels in physiological condition, and PEG8H-BA4-EA hydrogel in particular displayed potential for in situ gelation for oral cancer treatment. In addition to reports on bioactivity of EA, there have been many studies on the possible therapeutic effects of TA and EGCG. Thus, the concept of hydrogel networks exploiting these molecules as dual cross-linker and therapeutic may lead to additional possibilities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from NIH grant R37 DE014193 and NSFC grant 21522402. P. B. acknowledges postdoctoral fellowship support from the Swiss National Science Foundation (Project 151860). We also thank Dr Xi Xie of Hainan University for her help with in vitro cell studies.References
- E. Haslam, Practical polyphenolics : from structure to molecular recognition and physiological action, Cambridge University Press, Cambridge, UK, New York, NY, USA, 1998 Search PubMed.
- S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouysegu, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef PubMed.
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Angew. Chem., Int. Ed., 2013, 52, 10766–10770 CrossRef PubMed.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef PubMed.
- H. Ejima, J. J. Richardson and F. Caruso, Nano Today, 2017, 12, 136–148 CrossRef.
- D. G. Barrett, T. S. Sileika and P. B. Messersmith, Chem. Commun., 2014, 50, 7265–7268 RSC.
- M. Kampa, A. P. Nifli, G. Notas and E. Castanas, in Reviews of Physiology, Biochemistry and Pharmacology, ed. S. G. Amara, E. Bamberg, B. Fleischmann, T. Gudermann, S. C. Hebert, R. Jahn, W. J. Lederer, R. Lill, A. Miyajima, S. Offermanns and R. Zechner, Springer, Berlin Heidelberg, 2007, pp. 79–113 Search PubMed.
- F. D. Domenico, C. Foppoli, R. Coccia and M. Perluigi, Biochim. Biophys. Acta, Mol. Basis Dis., 2012, 1822, 737–747 CrossRef PubMed.
- D. G. Nagle, D. Ferreira and Y.-D. Zhou, Phytochemistry, 2006, 67, 1849–1855 CrossRef PubMed.
- B. N. Singh, S. Shankar and R. K. Srivastava, Biochem. Pharmacol., 2011, 82, 1807–1821 CrossRef PubMed.
- D. A. Vattem and K. Shetty, J. Food Biochem., 2005, 29, 234–266 CrossRef.
- V. Arulmozhi, K. Pandian and S. Mirunalini, Colloids Surf., B, 2013, 110, 313–320 CrossRef PubMed.
- C. Bell and S. Hawthorne, J. Pharm. Pharmacol., 2008, 60, 139–144 CrossRef PubMed.
- B. A. Narayanan, O. Geoffroy, M. C. Willingham, G. G. Re and D. W. Nixon, Cancer Lett., 1999, 136, 215–221 CrossRef PubMed.
- N. P. Seeram, L. S. Adams, S. M. Henning, Y. Niu, Y. Zhang, M. G. Nair and D. Heber, J. Nutr. Biochem., 2005, 16, 360–367 CrossRef PubMed.
- N. Wang, Z.-Y. Wang, S.-L. Mo, T. Y. Loo, D.-M. Wang, H.-B. Luo, D.-P. Yang, Y.-L. Chen, J.-G. Shen and J.-P. Chen, Breast Cancer Res. Treat., 2012, 134, 943–955 CrossRef PubMed.
- M. C. Roberts, M. C. Hanson, A. P. Massey, E. A. Karren and P. F. Kiser, Adv. Mater., 2007, 19, 2503–2507 CrossRef.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- J. A. Burdick and W. L. Murphy, Nat. Commun., 2012, 3 Search PubMed , 1269.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef PubMed.
- Y. Dong, W. Wang, O. Veiseh, E. A. Appel, K. Xue, M. J. Webber, B. C. Tang, X.-W. Yang, G. C. Weir, R. Langer and D. G. Anderson, Langmuir, 2016, 32, 8743–8747 CrossRef PubMed.
- A. P. Bapat, D. Roy, J. G. Ray, D. A. Savin and B. S. Sumerlin, J. Am. Chem. Soc., 2011, 133, 19832–19838 CrossRef PubMed.
- J. L. Guo, H. L. Sun, K. Alt, B. L. Tardy, J. J. Richardson, T. Suma, H. Ejima, J. W. Cui, C. E. Hagemeyer and F. Caruso, Adv. Healthcare Mater., 2015, 4, 1796–1801 CrossRef PubMed.
- S. H. Hong, S. Kim, J. P. Park, M. Shin, K. Kim, J. H. Ryu and H. Lee, Biomacromolecules, 2018 DOI:10.1021/acs.biomac.8b00144.
- J. N. Cambre and B. S. Sumerlin, Polymer, 2011, 52, 4631–4643 CrossRef.
- R. J. Weir and R. S. Fisher, Toxicol. Appl. Pharmacol., 1972, 23, 351–354 CrossRef PubMed.
- C. H. Linden, A. H. Hall, K. W. Kulig and B. H. Rumack, J. Toxicol., Clin. Toxicol., 1986, 24, 269–279 CrossRef PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- J. Su, F. Chen, V. L. Cryns and P. B. Messersmith, J. Am. Chem. Soc., 2011, 133, 11850–11853 CrossRef PubMed.
- P. C. Trippier and C. McGuigan, Med. Chem. Commun., 2010, 1, 183–198 RSC.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef PubMed.
- W. Yang, X. Gao and B. Wang, Med. Res. Rev., 2003, 23, 346–368 CrossRef PubMed.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef PubMed.
- C. Kim, H. Ejima and N. Yoshie, RSC Adv., 2017, 7, 19288–19295 RSC.
- Y. Guan and Y. Zhang, Chem. Soc. Rev., 2013, 42, 8106–8121 RSC.
- C. C. Deng, W. L. A. Brooks, K. A. Abboud and B. S. Sumerlin, ACS Macro Lett., 2015, 4, 220–224 CrossRef.
- S. Lascano, K.-D. Zhang, R. Wehlauch, K. Gademann, N. Sakai and S. Matile, Chem. Sci., 2016, 7, 4720–4724 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- A. Adamczyk-Woźniak, K. M. Borys, I. D. Madura, A. Pawełko, E. Tomecka and K. Żukowski, New J. Chem., 2013, 37, 188–194 RSC.
- D. G. Hall, Structure, properties, and preparation of boronic acid derivatives. Overview of their reactions and applications, John Wiley & Sons, Weinheim, Germany, 2006 Search PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef.
- Y. Kim, S. A. Hilderbrand, R. Weissleder and C. H. Tung, Chem. Commun., 2007, 2299–2301 RSC.
- J. W. Tomsho, A. Pal, D. G. Hall and S. J. Benkovic, ACS Med. Chem. Lett., 2012, 3, 48–52 CrossRef PubMed.
- Y.-S. Lo, N. D. Huefner, W. S. Chan, P. Dryden, B. Hagenhoff and T. P. Beebe, Langmuir, 1999, 15, 6522–6526 CrossRef.
- J. Lübbe, M. Temmen, P. Rahe, A. Kühnle and M. Reichling, Beilstein J. Nanotechnol., 2013, 4, 227–233 CrossRef PubMed.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef.
- I. Bala, V. Bhardwaj, S. Hariharan and M. R. Kumar, J. Pharm. Biomed. Anal., 2006, 40, 206–210 CrossRef PubMed.
- A. Janshoff, M. Neitzert, Y. Oberdörfer and H. Fuchs, Angew. Chem., Int. Ed., 2000, 39, 3212–3237 CrossRef PubMed.
- X. Zhang, C. Liu and Z. Wang, Polymer, 2008, 49, 3353–3361 CrossRef.
- H. Clausen-Schaumann, M. Seitz, R. Krautbauer and H. E. Gaub, Curr. Opin. Chem. Biol., 2000, 4, 524–530 CrossRef PubMed.
- M. S. Z. Kellermayer, C. Bustamante and H. L. Granzier, Biochim. Biophys. Acta, Bioenerg., 2003, 1604, 105–114 CrossRef.
- M. Carrion-Vazquez, A. F. Oberhauser, S. B. Fowler, P. E. Marszalek, S. E. Broedel, J. Clarke and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3694–3699 CrossRef.
- Y. Cao and H. Li, Nat. Mater., 2007, 6, 109–114 CrossRef PubMed.
- D. Sharma, O. Perisic, Q. Peng, Y. Cao, C. Lam, H. Lu and H. Li, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9278–9283 CrossRef PubMed.
- X. Han, M. Qin, H. Pan, Y. Cao and W. Wang, Langmuir, 2012, 28, 10020–10025 CrossRef PubMed.
- Y. Li, M. Qin, Y. Li, Y. Cao and W. Wang, Langmuir, 2014, 30, 4358–4366 CrossRef PubMed.
- W. Ott, M. A. Jobst, M. S. Bauer, E. Durner, L. F. Milles, M. A. Nash and H. E. Gaub, ACS Nano, 2017, 11, 6346–6354 CrossRef PubMed.
- F. Rico and V. T. Moy, J. Mol. Recognit., 2007, 20, 495–501 CrossRef PubMed.
- P. Hinterdorfer, W. Baumgartner, H. J. Gruber, K. Schilcher and H. Schindler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 3477–3481 CrossRef.
- K. M. Im, T. W. Kim and J. R. Jeon, ACS Biomater. Sci. Eng., 2017, 3, 628–636 CrossRef.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef PubMed.
- J. Ribas-Arino and D. Marx, Chem. Rev., 2012, 112, 5412–5487 CrossRef PubMed.
- S. Kim, S. K. Nishimoto, J. D. Bumgardner, W. O. Haggard, M. W. Gaber and Y. Yang, Biomaterials, 2010, 31, 4157–4166 CrossRef PubMed.
- H. Barbucci, Biological Properties and Applications, Springer, 2009 Search PubMed.
- E. M. Daniel, A. S. Krupnick, Y.-H. Heur, J. A. Blinzler, R. W. Nims and G. D. Stoner, J. Food Compos. Anal., 1989, 2, 338–349 CrossRef.
- N. Li, L. S. Taylor, M. G. Ferruzzi and L. J. Mauer, Food Res. Int., 2013, 53, 909–921 CrossRef.
- S. Kim, S. K. Nishimoto, J. D. Bumgardner, W. O. Haggard, M. W. Gaber and Y. Yang, Biomaterials, 2010, 31, 4157–4166 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm00453f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |