
Determination of methylmercury in sediment and cyanobacteria samples: method validation and application to methylation investigation
Louise Aparecida
Mendes
 *a,
Maione Wittig
Franco
b,
Francisco Antônio
Rodrigues Barbosa
b,
Paula Iannarelli
Aires de Carvalho
b,
Jorge
Carvalho de Lena
c and
Cláudia Carvalhinho
Windmöller
a
*a,
Maione Wittig
Franco
b,
Francisco Antônio
Rodrigues Barbosa
b,
Paula Iannarelli
Aires de Carvalho
b,
Jorge
Carvalho de Lena
c and
Cláudia Carvalhinho
Windmöller
a
aDepartamento de Química, ICEx, UFMG, Antônio Carlos Av., 6627, 31270-901 – Belo Horizonte, MG, Brazil. E-mail: louisemendes@ufmg.br; louisemendes@yahoo.com.br; claucw@netuno.lcc.ufmg.br; Tel: +55-31-3409-5759
bLaboratório de Limnologia Ecotoxicologia e Ecologia Aquática (LIMNEA), ICB, UFMG, Antônio Carlos Av., 6627, 31270-901 – Belo Horizonte, MG, Brazil. E-mail: maionewf@hotmail.com; barbosa@icb.ufmg.br; paula.iannarelli27@gmail.com
cDepartamento de Geologia, DEGEO, UFOP, Morro do Cruzeiro, Escola de Minas, 354000-000 – Ouro Preto, MG, Brazil. E-mail: jorge.delena@degeo.ufop.br
First published on 21st November 2017
Abstract
The aim of this work was to validate two methods for methylmercury (CH3Hg+) determination in sediments and cyanobacteria and their application to the Hg methylation study of planktonic organisms in water from a contaminated site in Brazil. Analytical methods for the determination of CH3Hg+ include many steps, and the necessity of adaptation to different matrices is very common and not always easy. In addition, these adaptations require new optimization and validation, which are found in a few articles using the matrix sediments but in none with planktonic organisms. The methods presented here were based on the extraction of CH3Hg+ from these matrices, derivatization, trapping, thermal desorption, GC separation, pyrolysis and detection by AFS (GC-pyro-AFS). The results showed good linearity (0.994) in the range of 0 to 400 pg CH3Hg+ with a repeatability of 3%, an intermediate precision of 8%, a recovery in spiked sample tests in the range of 93 to 129%, and detection limits of 0.04 μg kg−1 for sediments and 1.3 μg kg−1 for cyanobacteria. Certified reference materials showed good recoveries. The method for the cyanobacteria matrix was used for the Hg2+ methylation assay with cultures of cyanobacteria and microalgae isolated from water samples collected in an ancient Hg-contaminated gold mining area in Brazil. The production of CH3Hg+ was detected only in the culture of the microalga Stichococcus species (0.23% of total Hg), indicating its participation in the biotransformation of Hg. The methods can be used as important tools in routine analysis and/or studies of the biogeochemical cycle of Hg.
1. Introduction
CH3Hg+ is a highly toxic species with high mobility in the environment; it is easily bioaccumulated in living organisms, and consequently, it biomagnifies in the trophic chain.1,2 This species is formed in the environment from inorganic Hg2+, usually through the activity of sulfate-reducing bacteria (SRB)3–5 and iron-reducing bacteria (IRB) in the sediments.3 The methylation of Hg2+ occurs through the transfer of a methyl group from organic compounds, a reaction that needs to be catalyzed by microorganisms or mediated by a photochemical process.6Sediment is an environmental compartment that is very important for the study of the Hg cycle. According to Shi et al., sediment is considered to be the main site of production of CH3Hg+.7 When entering aquatic ecosystems, Hg can react with different compounds or interact with particulate materials present in the water, and it can precipitate in the sediment, mainly in the insoluble form HgS.8 The sediment permits innumerable organic and inorganic reactions.9 In this matrix, Hg can interconvert to several species of low or high toxicity (for example, CH3Hg+) in a process mediated by microorganisms, which can compromise aquatic biota and human health. Studies show that specific conditions are required to favor and/or accelerate methylation, such as low pH, moderately elevated temperature,10 seasonality,1,10 a high concentration of organic matter,10–12 a low water flow rate,11 oxidant redox potential and high concentrations of sulfate.12
Short-chain organomercurial species, such as CH3Hg+, can easily penetrate the cell membrane due to the lipophilic character of the alkyl radical.13 Given that CH3Hg+ is also soluble in water and is easily absorbed by microorganisms,8 it tends to bioaccumulate, mainly due to its affinity for sulfhydryl groups of proteins.13 As a consequence, Hg biomagnifies in the food chain and can reach humans. One of the effects of Hg intoxication is irreversible damage to the central nervous system.13
Cyanobacteria are a group of photosynthetic prokaryotes capable of colonizing various habitats,14,15 and they are found in the water column (pelagic) or sediments (benthic) that receive light radiation and in humid soils.16 They are basically composed of high molecular weight proteins, glycoproteins and an outer layer of mucilage formed by polysaccharides.17 Cyanobacteria are part of the phytoplankton community,17,18 playing the role of primary producers and are one of the main entry routes of CH3Hg+ into the aquatic food chain.1 In this complex community, consisting of algae, protozoa, bacteria, fungi and other organisms, the cyanobacteria stand out for their environmental adaptations.19 In addition, cyanobacteria play a major role in the structuring of biofilms due to the production of an abundant layer of exopolysaccharides, which are the basis for the installation of a complex microbial community.20,21 A study by Lázaro et al. demonstrated that the dominance of cyanobacteria is associated with increased production of CH3Hg+.1 Therefore, the cyanobacteria play important ecological roles that support the processes of Hg bioaccumulation and biotransformation, influencing the biogeochemistry of this element.
Hg chemical speciation studies provide relevant information for the understanding of metal toxicity, mobility and availability.22,23 In this case, studying the CH3Hg+ species in sediment and cyanobacteria matrices becomes very important because they are possibly the first matrices where the methylation process occurs. In the case of cyanobacteria, bioaccumulation may also be occurring. There are few studies on the methylation of Hg and quantification of CH3Hg+ in phytoplankton samples or cyanobacteria cultures in the laboratory.
Many studies have been conducted to develop methodologies for the determination of CH3Hg+ in several different matrices. These methodologies generally use gas chromatography-pyrolysis-atomic fluorescence spectrometry (GC-pyro-AFS),24–26 gas chromatography inductively coupled plasma mass spectrometry (GC-ICP-MS),26–28 high performance liquid chromatography inductively coupled plasma mass spectrometry (HPLC-ICP-MS)25,29,30 or a gas chromatography electron capture detector (GC-ECD).10,11,31 Techniques that use ICP-MS as the detector and isotope dilution are mainly used to study artifact formation, which may occur during the extraction of the analyte.24,32–34 GC-pyro-AFS and HPLC-ICP-MS are the techniques that have been used most widely. The great advantage of the former over the latter is the low cost of the analysis.
The objective of this work was to validate methods for the determination of CH3Hg+ by the GC-pyro-AFS technique in the environmental samples of sediment and cyanobacteria that could be used as important tools for the study of the biogeochemical cycle of Hg in the environment and methylation studies specifically. An example application was shown by studying Hg2+ methylation with cultures of cyanobacteria and microalgae isolated from water samples collected in a Hg-contaminated area.
2. Methodology
2.1 Apparatus
The determination of CH3Hg+ was performed on a gas chromatograph coupled to a pyrolysis system with an atomic fluorescence detection system (GC-pyro-AFS), Merx, Mark Brooks Rand Labs, USA. Argon gas free of Hg (grade > 99.9992) – Air Products (Sao Paulo, Brazil) was used as the mobile phase of the chromatographic column. A Tenax® trap was used to adsorb the Hg species produced in the derivatization and trapping step. Purging with nitrogen gas (N2) free of Hg (high purity grade 99.997) – Oxichama (Contagem, Brazil) was used in the adsorption step.2.2 Reagents
All reagents used were of analytical grade. The standards were prepared from a CH3HgCl standard solution of 1000 μg L−1 (Brooks Rand Labs, USA), acidified with 0.5% v/v CH3COOH (J. T. Baker, USA) and 0.2% v/v HCl (Merck, Germany). KOH, methanol (gradient grade for liquid chromatography), H2SO4 and KCl solution were prepared with Merck reagents (Germany). The sodium tetraethylborate solution (NaBEt4 – 1.33 mol L−1) was prepared by dissolving NaBEt4 salt (97%, Sigma Aldrich, USA) in 2 mL of anhydrous tetrahydrofuran (THF) and completing the volume with 38 mL of 2% w/v KOH solution. The preparation of this solution requires care such as the use of an inert gas chamber. Sodium acetate buffer was obtained from Brooks Rand Labs. Ultrapure water (>18 MΩ cm) from a Milli-Q Element system (Millipore, Bedford, MA, USA) was used throughout this work.2.3 Optimization of the chromatographic system and of the derivatization and trapping step
Preliminarily to the validation of the method, some parameters of the chromatographic system and of the derivatization and scattering step were optimized in order to obtain the highest signal area value of CH3Hg+. Table 1 presents the parameters optimized with the conditions under which the parameters were tested. A mass of 400 pg CH3Hg+ was used for the optimization. The column length was unchanged, as only one column composed of phenylmethylsiloxane and packed with OV-3, 31.75 centimeters long and 2.1 mm in diameter, was commercially available (Brooks Rand Labs, USA). Another parameter that remained constant was the temperature of the pyrolytic column (750 °C), where thermal decomposition and reduction of mercury species to elemental Hg (Hg0) occur for quantification in the AFS detector.| Parameter | Condition |
|---|---|
| Argon flow (GC mobile phase) (mL min−1) | 10.0; 14.0; 16.0; 17.0; 19.0; 25.0 and 31.0 |
| Temperature GC (°C) | 30.0; 33.0; 35.0; 36.0; 38.0 and 40.0 |
| Nitrogen flow in the trapping bubbler (mL min−1) | 68; 91; 127; 160; 203; 247; 297; 344 and 394 |
| Reaction time between CH3Hg+ and NaBEt4 (min) | 0; 2; 5; 8; 11; 14; 17; 20; 23 and 26 |
| Trapping time (min) | 5; 10; 15; 20; 25 and 30 |
| Trap drying time (min) | 0; 3; 6; 9 and 12 |
| NaBEt4 volume (μL) | 25; 50; 75; 100 and 125 |
2.4 Samples used for validation
The sediment sample used in the validation of the method was collected in an area contaminated by Hg from gold mining in Descoberto-MG, Brazil. The sample was intentionally chosen with a low concentration of total Hg in order to guarantee a low concentration of CH3Hg+. Thus, a low CH3Hg+ concentration present in the sample would not influence doping with the same species required for the validation tests.Cyanobacteria biomass used in this study was obtained from cultures of the strain Nostoc paludosum BA033 (GenBank: KX423684), currently maintained in the Laboratory of Limnology, Ecotoxicology and Aquatic Ecology – LIMNEA's algae culture bank in the Institute of Biological Sciences, Universidade Federal de Minas Gerais. The samples were cultivated in Erlenmeyer flasks with 5 L capacity in a BG-11 culture medium and kept under controlled conditions of light (27 μmol cm−2) and temperature (20 ± 1 °C). After 40 days, the samples were centrifuged and lyophilized.
2.5 Extraction of CH3Hg+
The sediment sample was subjected to a distillation system (Brooks Rand Labs) following the modified procedure of Horvat et al.36 A sample mass of 0.5 g was weighed precisely in a Teflon tube to which 30 mL of ultrapure water was added together with 500 μL of 8 mol L−1 H2SO4 and 200 μL of 20% w/v KCl. Other Teflon tubes for the collection of the distillate containing 5 mL of ultrapure water were placed in an ice bath. The sample was distilled at 125 °C under a nitrogen flow of 61 mL min−1 for approximately 3 hours after which 75% of the distillate had been collected. Then, the distillate was diluted to 50 mL and transferred to a glass jar derivatization system.
2.6 Derivatization and trapping step, and the determination of CH3Hg+
Three hundred μL of sodium acetate buffer and 50 μL of sodium tetraethylborate solution (NaBEt4) were added to the test solution in a reaction vessel of the derivatization system. The setup was allowed to stand for 17 minutes (reaction time) for the reactants to contact, and then the CH3HgEt4 formed in the reaction solution was purged with nitrogen gas at 91 mL min−1 for 25 minutes and trapped on a Tenax® column. Traps were dried with nitrogen gas for 6 minutes and analyzed individually by using the thermodesorption system coupled to a chromatographic column filled with OV-3. The GC temperature was maintained at 35 °C. Argon gas was used as the mobile phase, and the flow rate used was 17 mL min−1.2.7 Method validation
To evaluate the figures of merit of the methods, the INMETRO DOQ-CGCRE-008 validation guide was used.38 The tested figures of merit were matrix effect, linearity, working range, limit of detection, limit of quantification, precision and trend/recovery tests.The analytical curve from 0 to 500 pg was prepared from a CH3HgCl standard solution of 1000 μg L−1 and acidified with 0.5% v/v CH3COOH and 0.2% v/v HCl. The analytical curves were constructed in triplicate in aqueous solution using the matrix adjustment method, doping the sediment and cyanobacteria samples. Reference materials CRM-580 (estuarine sediment) and DORM-3 (fish protein) were used, whose concentrations of CH3Hg+ were (75 ± 4) μg kg−1 and (355 ± 56) μg kg−1, respectively.
The blank of the analytical curve in aqueous solution was prepared with 0.5% v/v of CH3COOH and 0.2% v/v HCl and then derivatized for posterior analysis. The blanks for the curves for the matrix adjustment method were prepared together with sediment or cyanobacteria extraction procedures for each curve independently by the same extraction procedures used for sediment and cyanobacteria, without the addition of the CH3HgCl standard. The blanks were then derivatized for posterior analysis.
The blank of the analytical routine was subjected to the same procedure applied to the sediment and cyanobacteria samples, i.e., performing all extractions without the presence of the matrix. The analysis of 8 independent replicates of these blanks was used to calculate the limit of detection (LOD) and limit of quantification (LOQ).
The statistical treatment of the data was performed with Excel 2007, following the guidelines of Souza et al.39
2.8 Application of the methods
An application of the method for the determination of CH3Hg+ in sediment samples has already been published in previous work,40 and therefore, only the application for samples of cyanobacteria and microalgae will be addressed.Strains isolated from water samples collected in the Hg contaminated area in Descoberto, MG, Brazil, were used. Three research studies have already described the contaminated area.40–42 It is a small area (ca. 1300 m2) located on a slope descending towards a stream. Two samples were collected: one in a sedimentation box, whose function is to retain the material leached by rain, and another one in the stream. A volume of 500 mL of the samples was collected in polypropylene flasks and placed in styrofoam boxes to keep the temperature low. Two mL of each sample were enriched in aqueous BG-11 culture medium in Petri dishes maintained under controlled light and temperature conditions. Microcontrolling and solid plating methods were used to obtain the isolates. After the cultures were obtained, they were analyzed under an optical microscope for their identification based on morphological characteristics. The cyanobacteria of the genus Fischerella sp. and the microalgae of the genus Stichococcus sp. were identified. Isolation and identification of the species were carried out by researchers from the LIMNEA laboratory. To produce the samples used in the application of CH3Hg+ determination, cultures of cells aged 40 days were doped separately with a concentrated solution of Hg2+ to yield a final concentration of 20 μg L−1 in the cultures. They were cultivated under the same conditions previously described for the cyanobacteria Nostoc paludosum (item 2.4). The samples were shaken for 7 days and finally centrifuged, washed and lyophilized for the determination of CH3Hg+ by GC-pyro-AFS.
The total Hg quantification using a DMA-80 (Milestone) was performed in order to evaluate the percentage of CH3Hg+ present in the sample relative to the added Hg.
3. Results and discussion
3.1 Method validation
According to the validation guide INMETRO, validation has the following definitions: (i) validation entails confirmation by examination and providing objective evidence that the specific requirements for a particular intended use are addressed; (ii) validation entails the verification that the specified requirements are suitable for an intended use. In this work, the main parameters to validate a method were systematically studied, aiming at application in future studies on the biogeochemical cycle of Hg. The validation process was conducted using assays with standard solutions, blanks, spiked samples and certified reference materials.38| Mobile phase flow rate | ||
|---|---|---|
| Flow rate (mL min−1) | R s (Hg0/CH3Hg+) | Retention time (min) |
| a Best conditions obtained in the optimization. | ||
| 10.0 | 2.62 | 4.74 |
| 14.0 | 2.12 | 3.77 |
| 16.0 | 2.24 | 3.56 |
| 17.0 | 2.64 | 3.43 |
| 19.0 | 2.14 | 3.16 |
| 25.0 | 1.54 | 2.64 |
| 31.0 | 1.26 | 2.25 |
The value of Rs was higher than 1.5 for all the varied conditions of the mobile phase flow rate (except for the flow rate of 31.0 mL min−1, Rs = 1.26) and the chromatographic column, showing that there was a complete separation between the peaks of Hg0 and CH3Hg+. The best values of Rs were found for flow rates of 10.0 and 17.0 mL min−1 and at temperatures of 30.0 and 33.0 °C. However, at the 10 mL min−1 flow rate the full peak of Hg2+ was not obtained by setting the total analysis time to 10 minutes, since the retention time of 4.74 minutes for CH3Hg+ was considered to be long. Instability was also observed for this flow rate, which was the minimum flow rate value of the rotameter, and its use is not advisable.
For flow rates of 17.0 and 19.0 mL min−1, the total time of analysis can be reduced to 10 minutes, leading to the observation of the three main complete peaks (Hg0, CH3Hg+ and Hg2+). It was decided to use the flow rate of 17.0 mL min−1, because it presented better resolution than the flow rate of 19.0 mL min−1 and a reduction of the total analysis time and retention time of CH3Hg+ compared to the flows of 10.0, 14.0 or 16.0 mL min−1.
Although the flow rate of the mobile phase recommended by the EPA 1630 method is 30 mL min−1,43 the highest signal area value was observed with lower argon flow rates. The flow rate suggested by the EPA 1630 method43 has the advantage of reducing the analysis time to 7 minutes but does not guarantee a good separation of the peaks of Hg0 and CH3Hg+, since Rs < 1.5 was observed in the flow rate of 31 mL min−1 (Table 2). Carrasco and Vassileva used the flow rate of 17.3 mL min−1,24,25 which is similar to the flow rates used in this work.
Even though at temperatures of 30.0 and 33.0 °C the highest value of Rs was observed in relation to the others, with these values it was not possible to obtain the complete signal of Hg2+ in 10 minutes of analysis. This is due to the increase in retention time when the GC temperature is decreased. Although the signal used for analysis was the only one related to CH3Hg+, it was considered important to record the exit of all Hg species as a guarantee of the chromatographic column cleaning and to obtain some qualitative information on the presence of this species. Therefore, these temperatures were not chosen for the CH3Hg+ analyses. At temperatures of 35.0 to 40.0 °C, the complete signals of all components (Hg0, CH3Hg+ and Hg2+) were observed after 10 minutes, with CH3Hg+ retention times lower than those observed at temperatures of 30.0 and 33.0 °C. The values of Rs were better at temperatures of 35.0 and 36.0 °C (Table 2). The manufacturer, Merx, recommends the temperature of 36.0 °C for the GC, which can be adjusted. Carrasco and Vassileva used the temperature of 36.0 °C for GC in their work.24,25 In the present study, no significant difference (p > 0.05) was observed between the evaluated factors for the temperatures of 35.0 and 36.0 °C. It was therefore decided to set the temperature at 35.0 °C.
| Parameter | Condition |
|---|---|
| N2 gas flow in the trap bubbler | 91 mL min−1 |
| Derivatization reaction time | 17 min |
| Trapping time | 25 min |
| Drying time of the trap | 6 min |
| Volume of NaBEt4 | 50 μL |
With the increase of the flow of the N2 gas in the bubbler, which has the function of carrying CH3Hg+ to the Tenax® trap, the analytical signal decreases, increasing the standard deviation of the analysis. This suggested that there are two forms of CH3Hg+ signal loss due to intense bubbling: one can be caused by the water droplets that are carried into the trap, making it difficult to adsorb the Hg species; another is that the force with which the gas carries the analyte does not favor the contact time required for its adsorption. The most intense analytical signal was obtained for the flow rate of 91 mL min−1, which is lower than the one recommended by the method EPA 1630,43 which suggests a N2 flow rate of (350 ± 50) mL min−1. A difference of 21% was observed between the largest analytical signal for CH3Hg+ obtained with the N2 flow rate of 91 mL min−1 and the lowest analytical signal obtained for the flow rate close to the value recommended by the method EPA 1630,43 which is 344 mL min−1. Another advantage of using a lower flow rate is the reduction of gas consumption. Therefore, the selected N2 flow rate of the trapping step for the CH3Hg+ determination was 91 mL min−1.
The reaction time was optimized in order to guarantee the complete reaction between the derivative and the CH3Hg+. The increase of the reaction time leads to the increased value of the analytical signal, i.e., the longer the reaction time, the greater the extent of the conversion of CH3Hg+ to CH3HgCH2CH3. The highest value for the analytical signal was observed at 17 minutes. After this time, this value decreased. Based on this value, the reaction time was set at 17 minutes, corroborating the recommendation of the method EPA 1630.43
The trapping time is the time required to withdraw the analyte from the reaction vial and to adsorb it in the trap. The EPA 1630 method recommends a trapping time of 17 minutes,43 while in the studies of Carrasco and Vassileva, the time used was 15 minutes.24,25 However, the optimization showed better signals (more intense) with the time of 25 min, so this time was selected to proceed with the CH3Hg+ analyzes in this work.
The drying of the trap is necessary, because the humidity present in it affects the desorption of the Hg species and, therefore, the analytical signal. There was a difference of 54% among the CH3Hg+ signals observed between 0 and 3 min. This indicates that the peak area is reduced drastically without the drying of the trap, which confirms the desorption inefficiency. From 3 minutes on, the analytical signal had little variation, with a difference of only 4% between 3 and 12 minutes. In this case, any time above 3 minutes could be chosen for drying the trap. The time of 6 minutes was selected, since at this point a standard deviation of less than 3 minutes was observed and the total drying of the trap was guaranteed. The EPA 1630 method recommends a time of 7 minutes for drying the trap.43
The volume of the NaBEt4 solution also influences the analytical signal. As the NaBEt4 volume increases, the analytical signal of CH3Hg+ declines. The largest area of the peak was reached by using a volume of 50 μL. According to EPA 1630,43 the volume of NaBEt4 solution suggested is 40 μL. The volume of NaBEt4 selected to proceed with the CH3Hg+ analyses was 50 μL, as in the studies of Carrasco and Vassileva.24,25
3.2 Matrix effect
To evaluate the matrix effect, three analytical curves of CH3Hg+ (0, 100, 200, 300, 400 and 500 pg) were prepared in aqueous solution, in sediment, and a third in cyanobacteria biomass. The matrix effect was evaluated by comparing the angular coefficients of the analytical curves of the analyte in the absence and presence of these matrices. These angular coefficients were compared using Fisher's test to evaluate any differences between the angular coefficient variance value and Student's t-test to determine whether there was a significant difference between these values, which is indicative of the occurrence of a matrix effect. The results are presented in Table 4. The hypothesis tests were performed with a significance level of 0.05 using Excel software.Table 4 presents the angular coefficients for each analytical curve with their respective standard deviations. Test F shows that there was no significant difference between the standard deviations of the curve obtained with cyanobacteria and the curve with aqueous solution. However, a significant difference was observed between the standard deviations of the curve obtained with the sediment matrix and the curve with aqueous solution. This has an influence on the choice of the method of calculating t-Student values for comparison of the means of the angular coefficients.44 The results show that there was no significant difference between the values of angular coefficients of the curves prepared with sediment and the curves prepared with aqueous solutions, as well as between the curves prepared with cyanobacteria biomass and aqueous solutions. There was no significant matrix effect between the CH3Hg+ curve in the aqueous solution and the matrix adjustment curve for both sediment and cyanobacteria, showing that the curve in aqueous solution can be used for analytical purposes. Thus, all subsequent tests were performed with the analytical curve of CH3Hg+ in aqueous solution.
3.3 Linearity and regression
Statistical tests were used to evaluate if the analytical curve of CH3Hg+ in aqueous solution meets the premises for the use of the Ordinary Least Squares Method (OLSM). Linearity was assessed according to INMETRO and guidelines proposed by Souza.38,39 The following parameters were evaluated: visual inspection of the curve, the Jackknife method of standardized residuals (outliers), the Ryan–Joiner test for normality, the Durbin–Watson test for residual independence, homoscedasticity by the Brown–Forsythe or Levene test, and significance of the results obtained for linearity evaluation. Each of the six levels of the curve was determined in triplicate and independently, totaling 18 points.Visual inspection indicated that the analytical curve is linear, and the coefficient of determination (R2) was equal to 0.989. However, the correlation coefficient (r) and the determination coefficient (R2) alone are not sufficient for the linearity test. Therefore, other parameters were systematically analyzed.
The occurrence of discrepant values was assessed by the standardized Jackknife method, calculated for each point on the calibration curve. Values with residuals greater than the critical value were eliminated. The test was applied repeatedly until there was no outlier present, following the elimination protocol of up to 2/9 of the data. A total of four data were removed by the Jackknife test, among them the level of 500 pg and a replicate of the mass corresponding to 400 pg.
The Ryan–Joiner test was applied to the regression residuals to verify their normality. The results showed that these residuals followed the normal distribution, since the deviations of normality were not significant (p > 0.10) and the correlation coefficient (R = 0.9818) obtained was higher than the critical R (0.9481) for the analytical curve of CH3Hg+ (Table 5).
| Statistical parameter | Calibration curve |
|---|---|
| Slope | 28.11 |
| Intercept | 288.6 |
| Linear range | 100–400 pg |
| R 2 | 0.994 |
| Number of observations | 14 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Normality for p > 0.10 | |
| R | 0.9818 |
| R critical | 0.9481 |
|
|
| Independence for p > 0.05 | |
| d | 1.672 |
| There is no correlation | 1.349 < d < 2.650 |
|
|
| Homoscedasticity | |
| t L | 6.91 × 10−1 |
| p | 0.5029 |
|
|
| Linearity deviation (for α = 0.01) | |
| F | 5.96 |
| F critical | 7.00 |
|
|
| Regression (for α = 0.05) | |
| F | 2134 |
| F critical | 4.74 |
The Durbin–Watson test (d) was applied to evaluate the independence of the residuals. The applied test led to a d value of 1.672 (Table 5). In the independence test graph, the points were well distributed in the quadrants, allowing the conclusion that there was no correlation among residuals (p > 0.05).
For the homoscedasticity test of the regression residuals, the points of the curve were divided into two groups. Group 1 corresponds to the mass of 0, 100 and 200 pg of CH3Hg+, and group 2 corresponds to the mass of 300 and 400 pg of CH3Hg+. Homoscedasticity can be estimated by calculating tL (6.91 × 10−1) using the Brown–Forsythe and Levene test. The tests show that there was no significant difference between the variances in groups 1 and 2 (p > 0.05), showing that the variances are independent of the concentration for this analytical curve.
The range of 100 to 500 pg for the construction of the analytical curve of CH3Hg+ for GC-pyro-AFS showed deviation from linearity by the ANOVA test. After exclusion of the 500 pg level and one replicate of the 400 pg level, linearity was obtained for a range of 100 to 400 pg at the 99% confidence level. The regression was significant (p < 0.05), indicating that the proposed range meets the premises for OLSM use. The analytical curve data obtained are shown in Table 5.
3.4 Precision and accuracy
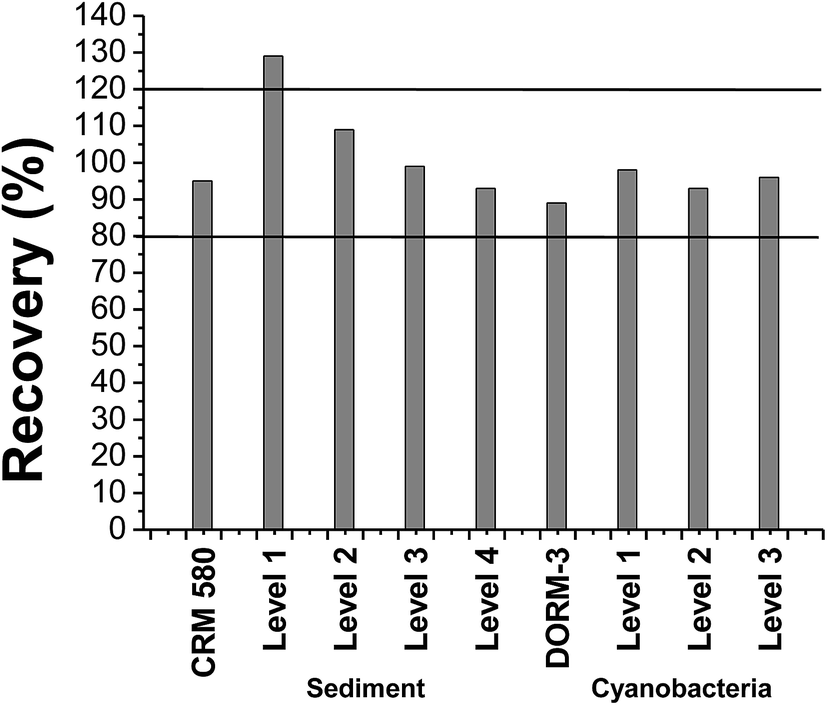
The repeatability (8 independent replicates analyzed on the same day) and intermediate precision (16 replicates in total; sets of 8 independent replicates analyzed on different days) were tested with the standard of 250 pg of CH3Hg+ in aqueous solution, whose level is located near the central point of the analytical curve. In the absence of a matrix effect, the tests mentioned above were performed in aqueous solution. The recovery rate was 103% for the repeatability test with a relative standard deviation (RSD) of 3%. For the intermediate precision, a recovery of 102% with a RSD of 8% was obtained. These results are within the criteria established by the guide of INMETRO and EPA, where recovery must be between 80% and 120% and RSD should not exceed 15%.38,45The trend/recovery was verified with spiked samples and the use of certified reference material: CRM-580 for sediment and DORM-3 for cyanobacteria. Standard sediment spiked tests were conducted with drops of CH3Hg+ solution for the final mass corresponding to 80, 175, 275, or 375 pg. For cyanobacteria the mass of CH3Hg+ for doping was 50, 250 and 350 pg. All doping was carried out in triplicate. The results of trend/recovery are shown in Fig. 1.
 | ||
| Fig. 1 Recovery percentage obtained for the tests with reference material and spiked samples. | ||
The recovery rate of the analyte obtained for the trend/recovery tests and reference materials ranged from 89% to 129% (Fig. 1). According to the acceptance criteria recommended by the INMETRO guide,38 which establish satisfactory recoveries between 80% and 120%,45,46 the proposed method presents accuracy, in terms of percentage recovery, within the acceptable range.
Only level 1, corresponding to 80 pg of CH3Hg+ in the sediment, exceeded the value of 120% (Fig. 1), suggesting a possible artifact formation. This event has been discussed in the literature as mentioned in the introduction. However, the confirmation of accidental methylation could not be carried out in this work due to the lack of accessibility to an instrument capable of performing isotope dilution tests. Considering that the positive error was not observed for the certified reference material, which has a high concentration of HgT, and in the other evaluated levels, the artifact production does not seem probable. Given that overestimation of the CH3Hg+ content was observed at the level at which the concentration was close to the detection limit of the technique, the high standard deviation and the positive error are justified.
3.5 Limit of detection and limit of quantification
The LOD and the LOQ were calculated using analytical signals of 8 independent blanks. These blank solutions were prepared using the same reagents used for extraction and derivatization of sediment samples and cyanobacteria. For these calculations, the standard deviation of the analytical signal of the blanks (s) and the slope of the analytical curve (b) were considered in the following expressions: LOD = 3.3 s b−1 and LOQ = 10 s b−1. The dilutions of each matrix were also considered for the calculation of final values.For sediments, the obtained LOD and LOQ were 0.04 and 0.13 μg kg−1, respectively. For the cyanobacteria the LOD was 1.3 μg kg−1 and LOQ was 3.9 μg kg−1. Faucheur et al. reported that phytoplankton accumulates between 2 and 30 μg of CH3Hg+ per kg of seaweed in dry weight.47 The concentration ranges of CH3Hg+ in sediment studies were (4.1–43.4 μg kg−1),10 (8–68 μg kg−1),11 (1–40 μg kg−1),48 and (0.046–5.2 μg kg−1).10,11,48,49 These LOD and LOQ values therefore allow the methods to be applied for Hg methylation studies in sediments and cyanobacteria.
3.6 Application of the methods
The validated methods were applied to samples of sediments and cultures of cyanobacteria and microalgae obtained from water samples collected in an area in Brazil (Descoberto-MG) contaminated with Hg, due to the use of this metal for the exploitation of gold. A research study on sediments of this area was published in 2016 by Mendes et al. The authors quantified CH3Hg+ in river sediments in the contaminated area and in sedimentation boxes at concentrations ranging from <0.11 to 8.0 μg kg−1.As described in the methodology, two water samples were collected in the same area of the sediment sampling sites: one in the sedimentation box and another in a stream in the contaminated area. The cultures of living cells were doped with Hg2+ solution in order to evaluate the production of CH3Hg+ by the culture of the microorganisms found in the samples.
After the assay, the cyanobacterium Fischerella sp. showed a concentration of CH3Hg+ below the LD (1.3 μg kg−1) of the method, which corresponds to values below 0.003% of HgT (dry weight), whose concentration was determined by DMA-80. The microalga presented a production of this organomercurial species at a concentration of (25 ± 2) μg kg−1 after the test, corresponding to 0.23% of HgT (dry weight). These data show that the microalgae may be one of the microorganisms responsible for the methylation of Hg2+ at the studied site, since it was found in the same sedimentation box where the concentration of CH3Hg+ was 8.0 μg kg−1 (the highest one observed in the contaminated area by Mendes et al.).40
Although it is clear that there was methylation by the microalgae culture, this proportion of CH3Hg+ was very low, and it is important that other experiments be performed to better understand under what conditions this microorganism promotes the methylation of Hg and whether other microorganisms present in the contaminated site are also contributing to this important process of the Hg cycle.
4. Conclusions
The validation of the methods for the determination of CH3Hg+ in sediment samples and cyanobacteria biomass using GC-pyro-AFS showed good results. The cyanobacteria, as well as other phytoplanktonic organisms, are among the first to participate in the methylation process of Hg. It is important to understand this process and there is still much to be researched. This is the first detailed description for optimization/validation/application of a methodology for the cyanobacteria matrix using these techniques. Among the parameters evaluated in the optimization, most noteworthy are the increase of the time and the reduction of the N2 gas flow in the scattering stage, conditions that yield the highest signal area. The matrix effect was not observed for the methodology applied in both matrices, making possible the use of the analytical curve of CH3Hg+ in aqueous solution without prejudice to the reliability of the results obtained. Linearity, precision and trend/recovery tests were adequate for the proposed method. Low LOD and LOQ were obtained for both cyanobacteria and sediment analyses. Although there is no consolidated Brazilian legislation regarding the concentration of CH3Hg+ in these matrices, the LOD and LOQ values are appropriate for a possible comparison and discussion of CH3Hg+ contamination in environmental samples. These methodologies are an important tool to be used in routine analyses of Hg methylation studies of cyanobacteria and sediment studies in areas vulnerable to environmental contamination by Hg.The study of methylation by microorganisms found in the contaminated area in Descoberto showed that the cyanobacteria Fischerella sp. was not able to produce CH3Hg+ under the studied conditions. The microalgae Stichococcus sp. presented mild methylation ability. However, these are the first results of methylation studies of Hg in living organisms from this contaminated area. It would be important to carry out other studies with changes in some parameters considered important for the biosynthesis of Hg, such as pH, concentration of organic matter, temperature and sulfide concentration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank CNPq, CAPES and FAPEMIG for the financial support; Limnologia Ecotoxicologia e Ecologia Aquática Laboratory for the partnership.References
- W. L. Lázaro, J. R. D. Guimarães, A. R. A. Ignácio, C. J. Silva and S. Díez, Cyanobacteria enhance methylmercury production: a hypothesis tested in the periphyton of two lakes in the Pantanal floodplain, Brazil, Sci. Total Environ., 2013, 456–457, 231–238 CrossRef PubMed.
- L. Carrasco, C. Barata, E. Gárcia-Berthou, A. Tobias, J. M. Bayona and S. Díez, Patterns of mercury and methylmercury bioaccumulation in fish species downstream of a long-term mercury-contaminated site in the lower Ebro River (NE Spain), Chemosphere, 2011, 84, 1642–1649 CrossRef CAS PubMed.
- S. Jonsson, U. Skylberg, M. B. Nilsson, P. Westlund, E. L. Shchukarev and E. Björn, Mercury methylation rates for geochemically relevant Hg(II) species in sediments, Environ. Sci. Technol., 2012, 46, 11653–11659 CrossRef CAS PubMed.
- D. Achá, H. Hintelmann and J. Yee, Importance of sulfate reducing bacteria in mercury methylation and demethylation in periphyton from Bolivian Amazon region, Chemosphere, 2011, 82, 911–916 CrossRef PubMed.
- G. C. Compeau and R. Bartha, Sulfate-reducing bacteria: principal methylators of mercury in anoxic estuarine sediment, Appl. Environ. Microbiol., 1985, 50, 498–502 CAS.
- F. M. M. Morel, A. M. L. Kraepiel and M. Amyot, The chemical cycle and bioaccumulation of mercury, Annu. Rev. Ecol. Syst., 1998, 29, 543–566 CrossRef.
- J. Shi, L. Liang, G. Jiang and X. Jin, The speciation and bioavailability of mercury in sediments of Haihe River, China, Environ. Int., 2005, 31, 357–365 CrossRef CAS PubMed.
- T. Tomiyasu, A. Naganoa, N. Yonehara, H. Sakamoto, R. K. Oki and H. Akagi, Mercury contamination in the Yatsushiro Sea, south-western Japan: spatial variations of mercury in sediment, Sci. Total Environ., 2000, 257, 121–132 CrossRef CAS PubMed.
- C. Green-Ruiz, J. Ruelas-Inzunza and F. Páez-Osuna, Mercury in surface sediments and benthic organisms from Guaymas Bay, east coast of the Gulf of California, Environ. Geochem. Health, 2005, 27, 321–329 CrossRef CAS PubMed.
- J. Pinedo-Hernández, J. Marrugo-Negrete and S. Díez, Speciation and bioavailability of Mercury in sediments impacted by gold mining in Colombia, Chemosphere, 2015, 119, 1289–1295 CrossRef PubMed.
- J. Marrugo-Negrete, J. Pinedo-Hernández and S. Díez, Geochemistry of Mercury in tropical swamps impacted by gold mining, Chemosphere, 2015, 134, 44–51 CrossRef CAS PubMed.
- C. S. Eckley, T. P. Luxton, J. L. Mckernan, J. Goetz and J. Goulet, Influence of reservoir water level fluctuations on sediment methylmercury concentrations downstream of the historical Black Butte Mercury mine, OR, Appl. Geochem., 2015, 61, 284–293 CrossRef CAS.
- R. C. C. M. Micaroni, M. I. M. S. Bueno and W. F. Jardim, Compostos de mercúrio. Revisão de métodos de determinação, tratamento e descarte, Quim. Nova, 2000, 23, 487–495 CrossRef.
- K. Kosek, Z. Polkowska, B. Zyszka and J. Lipok, Phytoplankton communities of polar regions–diversity depending on environmental conditions and chemical anthropopressure, J. Environ. Manage., 2016, 171, 243–259 CrossRef CAS PubMed.
- W. F. Vicent. Cyanobacterial Dominance in the Polar Regions, in The Ecology of Cyanobacteria, B. A. Whitton and M. Potts, Kluwer Academic Publishers, New York, Boston, Dordrecht, London, Moscow, 2002, pp. 321–340 Search PubMed.
- L. Hoffmann, Marine cyanobacteria in tropical regions: diversity and ecology, Eur. J. Phycol., 1999, 34, 371–379 CrossRef.
- R. E. Lee, Phycology, Cambridge University Press, Colorado, USA, 4th edn, 2008 Search PubMed.
- A. Pełechata, M. Pelechaty and A. Pukacz, Factors influencing cyanobacteria community structure in Chara-lakes, Ecol. Indic., 2016, 71, 477–490 CrossRef.
- C. C. Carey, B. W. Ibelings, E. P. Hoffmann, D. P. Hamilton and J. D. Brookes, Eco-physiological adaptations that favour freshwater cyanobacteria in a changing climate, Water Res., 2012, 46, 1394–1407 CrossRef CAS PubMed.
- F. Rossi and R. Philipps, Role of Cyanobacterial Exopolysaccharides in Phototrophic Biofilms and in Complex Microbial Mats, Life, 2015, 5, 1218–1238 CrossRef PubMed.
- N. Noffke, G. Gerdes and T. Klenke, Benthic cyanobacteria and their influence on the sedimentary dynamics of peritidal depositional systems (siliciclastic, evaporitic salty, and evaporitic carbonatic), Earth-Sci. Rev., 2003, 62, 163–176 CrossRef CAS.
- M. Leermakers, W. Baeyens, P. Quevauviller and M. Horvat, Mercury in environmental samples: speciation, artifacts and validation, Trends Anal. Chem., 2005, 24, 383–392 CrossRef CAS.
- NRC (National Research Council), Bioavailability of contaminants in soils and sediments: Processes, Tools and Applications, The National Academies Press, Washington, DC, 2003 Search PubMed.
- L. Carrasco and E. Vassileva, Determination of methylmercury in marine sediment samples: method validation and occurrence data, Anal. Chim. Acta, 2015, 853, 167–178 CrossRef CAS PubMed.
- L. Carrasco and E. Vassileva, Determination of methylmercury in marine biota samples: method validation, Talanta, 2014, 122, 106–114 CrossRef CAS PubMed.
- J. J. B. Nevado, R. C. R. Martin-Doimeadios, E. M. Krupp, F. J. G. Bernardo, N. R. Fariñas, M. Moreno, D. Wallace and M. J. P. Ropero, Comparison of gas chromatographic hyphenated techniques for mercury speciation analysis, J. Chromatogr. A, 2011, 1218, 4545–4551 CrossRef PubMed.
- H. Pietilä, P. Perämäki, J. Piispanen, M. Starr, T. Nieminen, M. Kantola and L. Ukonmaanaho, Determination of low methylmercury concentrations in peat soil samples by isotope dilution GC-ICP-MS using distillation and solvent extraction methods, Chemosphere, 2015, 124, 47–53 CrossRef PubMed.
- B. F. Beck and N. W. Johnson, Geochemical factors influencing the production and transport of methylmercury in St. Louis River Estuary sediment, Appl. Geochem., 2014, 51, 44–54 CrossRef CAS.
- R. Jagtap, F. Krikowa, W. Maher, S. Foster and M. Ellwood, Measurement of methyl Mercury(I) and Mercury(II) in fish tissues and sediments by HPLC-ICPMS and HPLC-HGAAS, Talanta, 2011, 85, 49–55 CrossRef CAS PubMed.
- J. S. Santos, M. Guardia, A. Pastor and M. L. P. Santos, Determination of organic and inorganic mercury species in water and sediment samples by HPLC on-line coupled with ICP-MS, Talanta, 2009, 80, 207–211 CrossRef PubMed.
- T. Tomiyasu, A. Matsuyama, R. Imura, H. Kodamatani, J. Miyamoto, Y. Kono, D. Kocman, J. Kotnik, V. Fajon and M. Horvat, The distribution of total and methylmercury concentrations in soils near the Idrija mercury mine, Slovenia, and the dependence of the mercury concentrations on the chemical composition and organic carbon levels of the soil, Environ. Earth Sci., 2012, 65, 1309–1322 CrossRef CAS.
- M. L. Avramescu, J. Zhu, E. Yumvihoze, H. Hintelmann, D. Fortin and D. R. S. Lean, Simplified sample preparation procedure for measuring isotope-enriched methylmercury by gas chromatography and inductively coupled plasma mass spectrometry, Environ. Toxicol. Chem., 2010, 29, 1256–1262 CAS.
- G. M. M. Rahman and H. M. Kingston, Application of speciated isotope dilution mass spectrometry to evaluate extraction methods for determining mercury speciation in soils and sediments, Anal. Chem., 2004, 76, 3548–3555 CrossRef CAS PubMed.
- L. Lambertsson, E. Lundberg, M. Nilsson and W. Frech, Applications of enriched stable isotope tracers in combination with isotope dilution GC-ICP-MS to study mercury species transformation in sea sediments during in situ ethylation and determination, J. Anal. At. Spectrom., 2001, 16, 1296–1301 RSC.
- Brooks Rand Labs, Application note: extraction of methylmercury from sediments and soils, 2013a Search PubMed.
- M. Horvat, N. S. Bloom and L. Liang, Comparison of distillation with other current isolation methods for the determination of methyl mercury compounds in low level environmental samples, Anal. Chim. Acta, 1993, 281, 135–152 CrossRef CAS.
- Brooks Rand Labs, Application Note: Alkaline Digestion of Biological Tissue for Methylmercury Analysis, 2013 Search PubMed.
- INMETRO – DOQ-CGCRE-008, Orientações sobre Validação de Métodos Analíticos, Revisão 05, 2016, p. 19 Search PubMed.
- S. V. C. Souza, C. T. Pinto and R. G. Junqueira, In-house method validation: application in arsenic analysis, J. Food Compos. Anal., 2007, 20, 241–247 CrossRef.
- L. A. Mendes, J. C. Lena, C. M. Valle, P. M. Fleming and C. C. Windmöller, Quantification of methylmercury and geochemistry of mercury in sediments from a contaminated area of Descoberto (MG), Appl. Geochem., 2016, 75, 32–43 CrossRef CAS.
- C. C. Windmoller, W. A. Durão Júnior, A. Oliveira and C. M. Valle, The redox processes in Hg-contaminated soils from Descoberto (Minas Gerais, Brazil): implications for the mercury cycle, Ecotoxicol. Environ. Saf., 2015, 112, 201–211 CrossRef CAS PubMed.
- W. A. Durão Jr, H. E. L. Palmiere, M. C. Trindade, O. E. A. Branco, C. A. Carvalho Filho, P. M. Fleming, J. B. B. Silva and C. C. Windmöller, Speciation, distribution, and transport of mercury in contaminated soils from Descoberto, J. Environ. Monit., Minas Gerais, Brazil, 2009, vol. 11, pp. 1056–1063 Search PubMed.
- EPA 1630, Methyl Mercury in Water by Distillation, Aqueous Ethylation, Purge and Trap, and CVAFS, U. S. Environmental Protection Agency, Washington DC, 2001, p. 49 Search PubMed.
- D. H. Sanders and R. K. Smidt, Statistics a First Course, Mc Graw Hill, Boston, 6th edn, 2000, p. 736 Search PubMed.
- EPA, Guidance for Methods Development and Methods Validation for the RCRA Program SW-846 Methods, U. S. Environmental Protection Agency, Washington DC, 1992, p. 32 Search PubMed.
- J. M. Green, Doing a thorough method validation can be tedious, but the consequences of not doing it right are wasted time, money, and resources: a practical guide to analytical method validation, Anal. Chem., 1996, 68, 305–309 CrossRef.
- S. L. Faucheur, P. G. C. Campbell, C. Fortin and V. I. Slaveykova, Interactions between mercury and phytoplankton: speciation, bioavailability, and internal handling, Environ. Toxicol. Chem., 2014, 33, 1211–1224 CrossRef PubMed.
- L. Spada, C. Annicchiarico, N. Cardellicchio, S. Giandomenico and A. D. Leo, Mercury and methylmercury concentrations in Mediterranean seafood and surface sediments, intake evaluation and risk for consumers, Int. J. Hyg. Environ. Health, 2012, 215, 418–426 CrossRef CAS PubMed.
- S. Wang, Y. Jia, S. Wang, X. Wang, H. Wang, Z. Zhao and B. Liu, Total mercury and monomethylmercury in water, sediments, and hydrophytes from the rivers, estuary, and bay along the Bohai Sea coast, northeastern China, Appl. Geochem., 2009, 24, 1702–1711 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |