
Molybdenum isotope fractionation in plants measured by MC-ICPMS
Dmitry
Malinovsky
 *ab and
Nikolay A.
Kashulin
*c
*ab and
Nikolay A.
Kashulin
*c
aDivision of Applied Geology, Luleå University of Technology, S-97187 Luleå, Sweden
bLGC Limited, Queens Road, Teddington, TW11 0LY, UK. E-mail: dmitriy.malinovskiy@lgcgroup.com
cInstitute of North Industrial Ecology Problems, Kola Science Centre, Apatity, 184209, Russia. E-mail: nikolay@inep.ksc.ru
First published on 12th December 2017
Abstract
A new method was developed for precise and accurate Mo isotope ratio measurements in plant materials by multi-collector inductively coupled plasma mass spectrometry (MC-ICPMS). It is based on the use of anion-exchange chromatography to isolate Mo from concomitant matrix elements in sample digests, a desolvating Apex-Q sample inlet system as a means of Mo signal enhancement and on-line normalisation to an admixed internal standard (Pd) to correct for instrumental mass bias. Mo isotope ratios were determined in sample solutions with Mo concentrations as low as 10 ng g−1. The developed method was successfully applied to the determination of natural variations in the isotopic composition of Mo in different anatomical parts of plants. We show for the first time that Mo isotope fractionation can occur during long-distance transport of Mo in plants. Our data also show that the magnitude of Mo isotope fractionation during translocation of Mo is different for different plant species. Mo isotope ratio data obtained by MC-ICPMS can therefore be used as a sensitive probe of processes controlling transport and distribution of molybdenum in plants.
Introduction
Molybdenum is a metallic element which is distributed in the upper continental crust with an average concentration of about 1.4 mg kg−1.1 It has seven stable isotopes, namely 92Mo, 94Mo, 95Mo, 96Mo, 97Mo, 98Mo and 100Mo, with the average natural abundances of 14.7%, 9.2%, 15.9%, 16.6%, 9.5%, 24.1% and 9.6%, respectively.2 Natural variations in the isotopic composition of molybdenum were shown to be a very useful tracer in the geosciences. These data have been used for interpreting genesis of hydrothermal and igneous ore deposits and provided valuable insight into redox conditions of seawater existed in the past.3–11Although molybdenum is one of the scarcest elements in biological systems it is an important micronutrient in all kingdoms of life.12,13 The element itself seems to have no biochemical activity but once molybdenum is complexed by a unique pterin compound to form molybdenum cofactor, known as Moco, it can combine with diverse proteins to form Mo enzymes. The latter play an important role in many biochemical redox reactions, including nitrogen assimilation, sulphur metabolism, phytohormone biosynthesis and stress reactions.14,15 However, as pointed out in many recent publications, despite its importance as a cofactor in essential biochemical processes, relatively little is known about the regulation, uptake, transport and mechanisms of actions of molybdenum in biological systems.16,17 It is also worth noting that stable isotope data of many mineral micronutrients (e.g., Cu, Zn, Fe) have been successfully used to improve understanding of relevant biological processes.18 By analogy, Mo isotope data hold a great potential as a tool for deciphering biochemical reactions of molybdenum as the element displayed a wide range of Mo isotope fractionation concomitant with basic physicochemical processes. Yet very little has been done so far to explore this potential. Up to the best knowledge of the authors, no publication has reported determination of natural variations in Mo isotope abundances in plants.
A major analytical challenge for Mo isotopic analysis in biological systems is its low concentrations. In most plants, Mo concentrations range from 0.1 to 1.0 μg g−1 of dry weight.19 Somewhat elevated Mo concentrations were documented in terrestrial flora in the areas affected by anthropogenic pollution.20 Large sample aliquots and pre-concentration are required for reliable Mo isotope ratio measurements at the aforementioned typical concentration level. However, recent advances in ICP-MS and peripheral devices, including improved desolvating inlet systems and enhanced transmission skimmer cones, can be used to alleviate degree of pre-concentration needed and make the isotopic analysis more straightforward.
This paper reports, for the first time, a measurement method suitable for obtaining precise and accurate Mo isotope ratios in plants. We show that the range of Mo isotope fractionation observed in plant species can be sufficient for discriminating specific pathways of molybdenum metabolism. Findings of our study reinforce expectations that Mo isotope ratio data can be a useful probe in studies relating to Mo biochemistry and environmental chemistry.
Experimental section
Material and reagents
Analytical reagent grade nitric acid was acquired from Merck (65%, Merck, Darmstadt, Germany) and additionally purified in-house by sub-boiling distillation in a quartz still. High purity deionised water (18 MΩ cm) from a Milli-Q water purification system (Millipore Milli-Q, Bedford, USA) was used. All calibration solutions used in ICP-MS measurements were prepared by diluting single element ICP standard solutions (SPEX Plasma Standards, NJ, USA). The anion-exchange resin, Bio-Rad AG® 1-X8, 100–200 mesh size was obtained from Bio-Rad Laboratories AB. High purity ethylenediaminetetraacetic acid (EDTA) was acquired from Sigma Aldrich.Sample collection
Four types of plants – European blueberry (Vaccinium myrtillus), lingonberry (Vaccinium vitis-idaea), the common juniper (Juniperus communis L.) and rosebay willowherb (Chamerion angustifolium) – were collected in south-western part of the Kola Peninsula, Russia, on the areas with minimum exposure to anthropogenic pollution. Plant sampling was performed with scissors following general guidelines described elsewhere.21 Different anatomical parts of the plants (roots, stems, and leaves) were separated on a sampling site, placed in Ziploc bags and returned to the laboratory. Each individual sample for measurements was a composite one and comprised 20 to 100 pieces of appropriate plant parts collecting from a sampling location with size of about 10 × 10 meters. Root sampling to a depth of 40 cm was performed by digging soil profile pits and pooling root parts together to provide sufficient material for analysis. Replicate composite samples were collected on each sampling site.Sample preparation
Samples were cleaned with deionised water, placed in a force-draft oven and dried at 65 °C for at least 24 hours. They were then ground in agate mortar to pass through a 1 mm mesh size sieve. Approximately 2 g of sample was dry-ashed in a porcelain crucible for 6 hours at 500 °C in a muffle furnace. Once the ash was cooled it was transferred into a 100 ml Teflon beaker with a lid followed by adding 8 ml of 16 M HNO3 and heating with reflux at 120 °C for 2 hours. The solution was evaporated to near dryness, after which the residue was re-dissolved in 4 ml of 0.2 M HF/0.5 M HCl and subjected to anion-exchange separation of molybdenum. The separation procedure was modified from that described previously.22 Disposable 5 ml pipette tips were fitted with plugs of cotton wool at their outlets and filled with ∼0.5 ml of Bio-Rad AG® 1-X8 anion exchange resin to produce chromatographic columns. The columns were cleaned with 3 M HNO3, followed by deionised water and conditioning with 0.2 M HF/0.5 M HCl. Samples prepared as described above were loaded onto the columns. Matrix elements were eluted with 5 ml of 0.2 M HF/0.5 M HCl solution, followed by 5 ml of 4 M HCl. Mo was eluted with 8 ml of 3 M HNO3, evaporated to near dryness in a Teflon beaker and re-constituted in 0.3 M HNO3. Quantitative recovery of Mo was verified on aliquots of sample solutions taken before and after the separation. Procedural blanks were run along with the samples in all manipulations. Total procedural blanks were found to contain less than 2 ng of molybdenum.Mo concentration and isotope ratio measurements
Prior to Mo isotope ratio measurement, concentrations of Mo and other elements, including Zr, Ru, and Pd, in samples were determined by single collector ICP-MS (Element-2, Thermo Scientific, Germany) with external calibration versus single element ICP standard solutions (SPEX Plasma Standards, USA). Once concentration of Mo was determined, the samples and Mo isotope standard were diluted to desired concentration ranging from 10 to 40 ng ml−1 Mo with 0.3 M HNO3 and spiked with Pd at 100 ng ml−1 Pd, followed by isotope ratio measurements by MC-ICPMS.Mo isotope ratio measurements were performed using a Neptune MC-ICPMS (Thermo Scientific, Bremen, Germany). Typical operating conditions for the instrument are shown in Table 1. Samples were introduced into the plasma through Apex-Q high sensitivity sample inlet system (Elemental Scientific Inc., USA). The analyses were conducted at low mass resolution and in static mode. Amplifier gain calibration was performed at the beginning of each measurement session. A single measurement consisted of one block, comprising 50 cycles of 4.2 second duration. The intensities of six Mo and two Pd ion beams – 94Mo+, 95Mo+, 96Mo+, 97Mo+, 98Mo+, 100Mo+, 104Pd+ and 105Pd+ – were measured simultaneously at low 4, low 3, low 2, low 1, central, high 1, high 3 and high 4 Faraday cup positions, respectively. The 105Pd/104Pd isotope ratio was used as an internal standard to correct for instrumental mass bias. The exponential model was used for correction as described in detail elsewhere.23–25 Samples were analysed in a sequence of isotope standard, two “unknown” samples, isotope standard and so on. Procedural blanks were also included in the measurements. Concentrations of samples and bracketing standards were matched to within 20%. NIST SRM 3134 Mo standard solution was used as Mo isotope standard in measurements. NIST SRM 3138 Pd standard solution was used as the internal standard. Normalisation to Pd was efficient in correcting for drift in the instrumental mass bias during measurement session and improving reproducibility of δX/95Mo values.
| RF power, W | 1250 |
| Sample and skimmer cones | Nickel, 1.1 and 0.8 mm (X type) orifice diameter, respectively |
| Argon gas flow rate, l min−1 | |
| Cool/plasma | 16 |
| Auxiliary | 1.0 |
| Nebuliser (connected to Apex unit) | 0.8 |
| Additional gas, N (connected to Apex unit) | 0.2 |
| Sample uptake rate, l min−1 | 0.1 |
| Ion lens settings | Optimised for maximum intensity of Mo+ signal |
| Detector current amplifiers, ohm | 1011 |
| Mass resolution, R(5–95%) | ∼450 |
| Typical 98Mo sensitivity, V/(mg l−1) | ∼100 |
Molybdenum ion current intensities in each measurement were corrected for procedural blank. The on-line data processing included removal of outliers using a 2σ-test and calculation of the ion beam intensity ratios and standard deviation. Further statistical treatment of the data was performed offline. For the presentation of results, the δ-notation was utilized, as defined by the relationship:
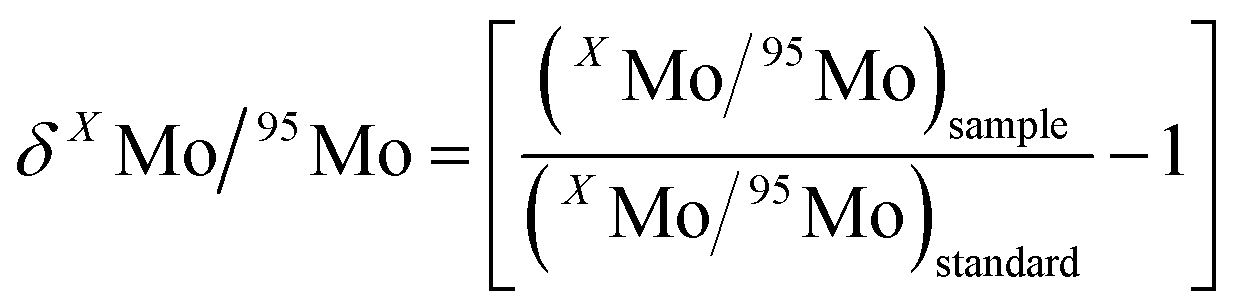 | (1) |
Results and discussion
Improved Mo isotopic analysis at Mo concentration level of 10 to 100 ng g−1
It is worth noting that attempts to determine Mo isotope ratios in samples with low concentration of molybdenum have been made in previous studies.26–30 Concentration of Mo at 30 ppb in solution measured by MC-ICPMS was reported as a minimum one sufficient for Mo isotopic analysis.28 An interesting feature of these studies was that the double spike method was used solely to correct for instrumental mass discrimination of Mo isotope ratios. The method of double spike is well known for its ability to achieve reproducible results in heavy matrices. Its only disadvantages are laborious sample preparation and inability to detect mass-independent isotope fractionation in samples, should it occur in them. In this study, we relied on normalisation to an admixed internal standard, a rival method to the double spike one, to correct for instrumental mass bias.Apex-Q desolvating sample inlet system and X type skimmer cones were means of achieving higher sensitivity for molybdenum. Sensitivity of measurement in the Apex-Q is improved by removal of solvent and enhancing transport efficiency for an analyte by heating and subsequent rapid cooling of sample aerosol. We used a 50 μl min−1 PFA nebulizer in free aspiration mode to introduce sample solution into a cyclonic spray chamber of the Apex-Q, kept heated at 140 °C, which is followed by a thermoelectric cooler operated by the Peltier effect, cooling the sample aerosol to about 2 °C.
A significant improvement in the performance of the Apex desolvating system for molybdenum has been made by adding small amount of EDTA in solution to be measured. At least two-fold enhancement of Mo ion intensities and better reproducibility of Mo isotope ratios were observed in measurements of solutions containing 0.04% EDTA in the matrix as compared to those with 0.3 M HNO3 alone. It is known that molybdenum forms stable complexes with EDTA which seem to account for better transmission of the element through the heating/cooling cycles of the desolvating unit.31 A 10- to 12-fold improvement in sensitivity of Mo relative to a conventional pneumatic nebulisation was observed in the measurements using the Apex-Q and 0.04% EDTA solution matrix. By using this approach we were able to measure Mo isotope ratios in solutions with Mo concentrations of 10 to 15 ng g−1 and within run standard deviation (1σ) ranging from 0.1‰ to 0.2‰ in terms of 98Mo/95Mo isotope ratios. No Mo isotope effect due to the interaction with EDTA was observed. It was demonstrated by the experiments in which Mo isotope ratios of NIST SRM 3134 solutions containing EDTA were measured relative to the same Mo standard in solutions without EDTA with the obtained δX/95Mo values not different from zero within the limits of measurement precision.
It should be noted that the Apex desolvating system has an option of additional removal of solvent and matrix components by attaching a membrane desolvation module. However, none of two available Apex membrane modules were used in the measurements. These were a cooled micro-porous Nafion® membrane (ACM) and a heated macro-porous Teflon® membrane (Spiro TMD). Adding a membrane module to sample flow path resulted in a decrease of sensitivity for molybdenum, as compared to that achievable by Apex-Q unit alone, and poorly reproducible results. These observations show that sample aerosol permeation and evaporation in membrane desolvation are complex processes which can be accompanied by loss of analyte and isotope fractionation.
Validation of the measurement method
Performance characteristics of the method, including Mo recovery, repeatability of Mo isotope ratios, run and matrix variation effects, were assessed through replicate measurements of NIST SRM 1547 (peach leaves) and independently processed sample aliquots of powdered and thoroughly mixed leaves of rosebay willowherb plant collected from a single sampling site. It was verified that the anion-exchange chromatography used to separate Mo was efficient in removing concomitant matrix elements and provided quantitative recovery of Mo with the yield ranging from 95% to 99% with the mean value of 97%. It is worth mentioning that plant material is characterised by lower concentration of total dissolved solids and low elemental ratio of Fe to Mo than geological samples. This noticeably alleviates the task of Mo separation from Fe, which proved to be a significant analytical challenge for many geological samples. Variations in the isotopic composition of Mo in the in-house standard after repeated independent runs through the chromatographic procedure were within the standard deviation of the measurements.Repeatability of Mo isotope ratios was assessed by repeated measurements of subsamples of rosebay willowherb leaves over period of twelve months and shown Fig. 1 in terms of δ98Mo/95Mo values. Repeatability of 98Mo/95Mo isotope ratios, expressed as combined standard deviation of independent replicate measurements, was evaluated to be 0.13‰. Combined standard uncertainties were then calculated as follows32,33
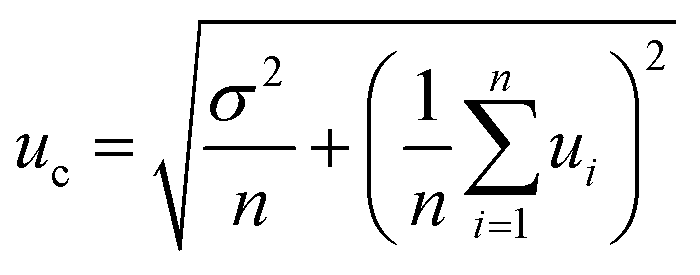 | (2) |
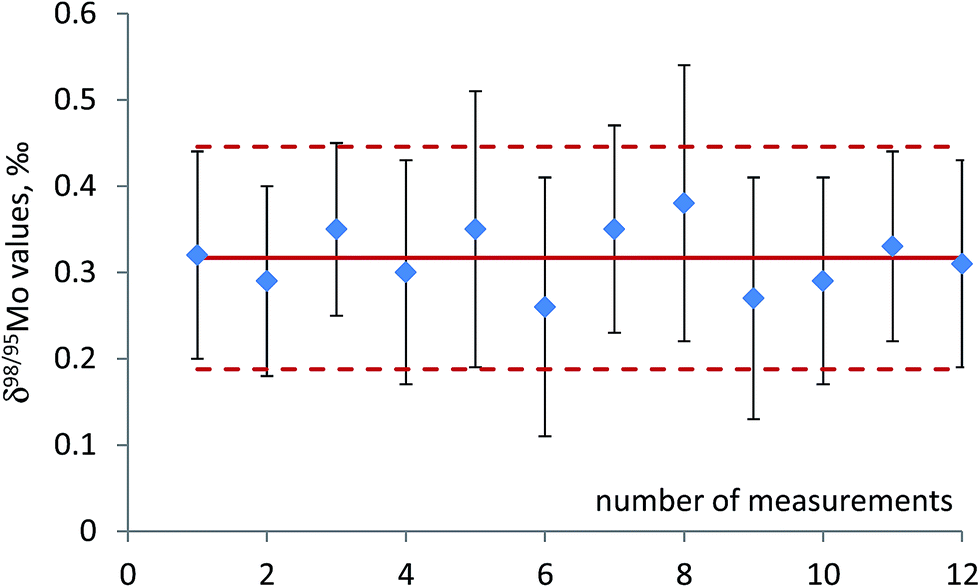 | ||
| Fig. 1 Repeatability of δ98Mo values for independent subsamples of rosebay willowherb leaves collected from a single location and thoroughly mixed (see text for details). The measured solutions have Mo concentration ranging from 10 to 15 ng g−1. Uncertainty bars are standard deviation of individual measurement (1σ). Solid line is the mean of 12 replicates; dotted lines show expanded uncertainty with k factor = 1. | ||
The quality of Mo isotope ratio measurements can also be assessed by using three isotope plot. In this approach, the slope of regression through experimental data points is compared with the exponent of isotope fractionation calculated by using known atomic masses of the isotopes involved in the isotope ratios. The exponent of isotope fractionation, β, for each triad of Mo isotopes can be calculated. The rationale for calculating values of the isotope fractionation exponent is described in detail elsewhere.34Fig. 2 shows linearised δ′97Mo vs. δ′98Mo plot for the samples measured in this study. As can be seen from Fig. 2, regression through the experimental data points yields the slope which agrees well with theoretical ones (kinetic and equilibrium) within the limits of measurement uncertainties. This serves as an indication of the absence of spectral interferences and stability of instrumental mass discrimination during the measurement session. Similar calculations were made, and the agreement of the experimentally obtained β values with theoretical ones was observed for other triads of Mo isotopes.
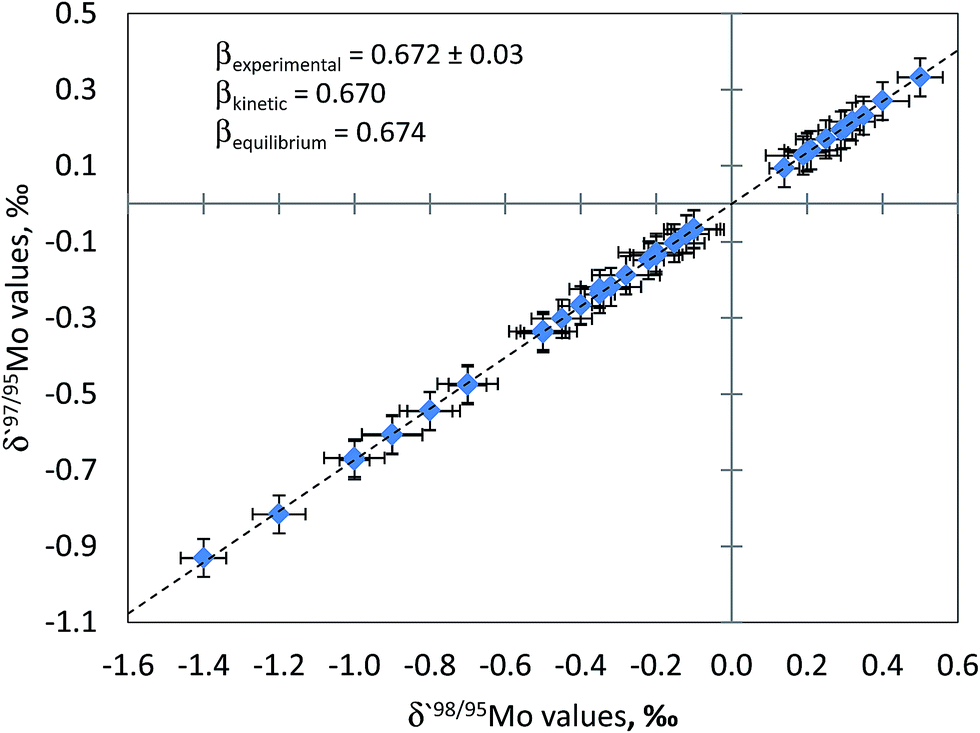 | ||
| Fig. 2 Three isotope plot constructed using the approach of Young et al. (2002) and Mo isotope ratio data for plant material. Uncertainty bars are one standard deviation. The fractionation exponent derived from the measured data set (β = 0.672 ± 0.003, R2 = 0.9997, n = 48) is indistinguishable from that expected for both kinetic and equilibrium isotope fractionation within the limits of uncertainty. The agreement of the experimentally determined fractionation exponent with the theoretical value and linearity of the array serve as an indication of insignificant spectral interferences on the measured Mo isotopes and stability of mass bias over MC-ICPMS sessions. | ||
It should also be noted that there is currently no isotopic reference material of Mo in biological samples which can be used for purposes of quality control. In the light of this fact, results we obtained demonstrate that the method is fit for purpose.
Mo isotope fractionation in plants
Results of Mo isotopic analyses of plants collected in this study are shown in Fig. 3. As can be seen from this figure, Mo isotope fractionation between different anatomical parts was observed in three out of four plant species studied. Although direction of isotope fractionation was the same in all studied species with lighter Mo isotopes progressively enriched in stems and leaves relative to roots, the extent of the fractionation was different. Most significant shift in δ98Mo values between leaves and roots of ∼1.2‰ was observed in common juniper. Blue and lingon berries were characterised by smaller magnitude of Mo isotope fractionation between leaves and roots of ∼0.6‰ and ∼0.5‰ in terms of δ98Mo values, respectively. What is particularly interesting is that although δ98Mo values of the samples vary depending on sampling sites the differences in δ98Mo values between roots, stems and leaves in the same plant species remain very similar. It suggests that the same process or a set of processes are responsible for isotope fractionation during translocation of molybdenum in plants of the same genus. Site-to-site differences in the isotopic composition of Mo in roots are most likely caused by geochemical features of bedrocks. These differences are then translated into the corresponding variations in Mo isotope abundances in stems and leaves. | ||
| Fig. 3 Mo isotope fractionation in plants: (A) common juniper; (B) rosebay willowherb; (C) blue berry; (D) lingon berry. Three out of four plant species are characterised by Mo isotope fractionation concomitant Mo translocation. An interesting observation is that although δ98Mo values of the samples vary depending on sampling sites the differences in δ98Mo values between roots, stems and leaves in the same plant species remain very similar. | ||
No Mo isotope fractionation, within the limits of measurement precision, was observed between different anatomical parts of rosebay willowherb. Mo concentrations in roots of rosebay willowherb were slightly higher than those in stems and leaves of the plant, being 0.7 ± 0.1 μg g−1 and 0.5 ± 0.1 μg g−1, respectively. The concentration data have not provided insight into the understanding of the absence of Mo isotope fractionation in rosebay willowherb. Although we are unable to explain it with rather limited data we have so far, the absence of Mo isotope fractionation can potentially be a very useful indicator of specific processes involved in the uptake and transport of Mo in plants.
It was suggested previously that chemical processes occurring during transport of metals in plants are likely to induce isotope fractionation.18 As molybdate oxyanion (MoO42−) is the predominant chemical form of molybdenum in soil solutions at pH higher than 4, it requires active transport across the plasma membrane of plant root cells for uptake. However, as pointed out recently, despite the fact that Mo transport and homeostasis have been well characterised in bacteria and some eukaryotes, relatively little is known about mechanisms of Mo uptake, transport and distribution in plants.35 It has long been assumed that uptake of molybdate could be mediated unspecifically by phosphate (HPO4−) and sulfate (SO42−) anions. The latter can be particularly important as molybdate and sulfate anions are similar in chemical structure and size, and both possess a double negative charge. Recent studies also identified and characterised Mo specific transporters, referred to as MOT1 and MOT2 families of proteins.17,36 Once uptaken, Mo is transported into different parts of plant via phloem and xylem. As molybdate anion was observed in both phloem and xylem it was assumed that MoO42− is a major chemical form of molybdenum in these two long-distance transport systems in plants.37
Although the data obtained in this study does not allow us discriminating between many processes capable of inducing Mo isotope fractionation in plants it is worth noting that diffusion can play an important role. It was shown previously that diffusion can isotopically fractionate transition metals, including molybdenum, resulting in enrichment of lighter isotopes at the diffusion front.38,39 It has also been shown that the magnitude of Mo isotope fractionation during diffusion is dependent on chemical form of molybdenum.39 The isotopic pattern observed in blueberry, lingonberry and the common juniper in this study is consistent with the hypothesis that differences in Mo isotope ratios between roots, stems and leaves are diffusion-driven. Other important processes can include oxidation–reduction and ion exchange.
Conclusions
We have developed a new method for determination of Mo isotope ratios in plants by MC-ICPMS. The method features a single step anion-exchange chromatographic procedure for separation of Mo from sample matrix and the use of a desolvating nebulizer as a means of Mo signal enhancement. Addition of a complexing agent, EDTA, to the sample solutions at the concentration level of 0.04% was used in the measurements and found very effective in improving the performance of the desolvating nebulizer. The presence of EDTA in the solutions allowed for better transmission of Mo through the desolvating unit and results in higher sensitivity for Mo and more reproducible Mo isotope ratios. By using this approach Mo isotope ratios were measured in sample solutions with Mo concentrations as low as 10 to 15 ng g−1 and within run precision of 0.1 to 0.2‰.Mo isotope fractionation was shown to occur during long-distance transport of Mo in plants. An important observation was that the difference in the isotopic composition of Mo between roots, stems and leaves of the same plant species remained very reproducible from one sampling site to another, suggesting that the same set of processes can drive the observed isotope fractionation. These data may prove to be useful for developing a better understanding of Mo isotope budget in the biogeochemical cycling of the element. It is anticipated that the developed measurement method will offer a wide potential in biogeochemical and other fields of Mo isotope research.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful to the staff of Kola Science Center and Division of Applied Geology, Luleå University of Technology for useful discussion and technical assistance.References
- K. H. Wedepohl, Geochim. Cosmochim. Acta, 1995, 59, 1217–1232 CrossRef CAS.
- J. R. De Laeter, J. K. Böhlke, P. De Bièvre, H. Hidaka, H. S. Peiser, K. J. R. Rosman and P. D. P. Taylor, Pure Appl. Chem., 2003, 75, 683–800 CrossRef CAS.
- C. Siebert, T. F. Nägler, F. von Blanckenburg and J. D. Kramers, Earth Planet. Sci. Lett., 2003, 211(3–4), 159–171 CrossRef.
- A. D. Anbar and O. Rouxel, Annu. Rev. Earth Planet. Sci., 2007, 35, 717–746 CrossRef CAS.
- C. R. Pearce, A. S. Cohen, A. L. Coe and K. W. Burton, Geology, 2008, 36, 231–234 CrossRef CAS.
- R. Mathur, S. Brantley, A. Anbar, F. Munizaga, V. Maksaev, R. Newberry, J. Vervoort and G. Hart, Miner. Deposita, 2010, 45, 43–50 CrossRef CAS.
- N. D. Greber, B. A. Hofmann, A. R. Voegelin, I. M. Villa and T. F. Nägler, Geochim. Cosmochim. Acta, 2011, 75, 6600–6609 CrossRef CAS.
- S. Song, K. Hu, H. Wen, Y. Zhang, K. Li and H. Fan, Chin. Sci. Bull., 2011, 56, 2221–2228 CrossRef CAS.
- F. L. H. Tissot, N. Dauphas, C. T. Reinhard, T. W. Lyons, D. Asael and O. Rouxel, Mo and U geochemistry and isotopes, in Reading the archive of Earth's oxygenation: Global Events and the Fennoscandian Arctic Russia – Drilling Early Earth Project, ed. Melezhik V. A., Kump L. R., Fallick A. E., Strauss H., Hanski E., Prave A. R. and Lepland A., Springer-Verlag, Berlin, 2013, Vol. 3, pp. 1500–1506 Search PubMed.
- N. Breillat, C. Guerrot, E. Marcoux and P. Négrel, J. Geochem. Explor., 2016, 161, 1–15 CrossRef CAS.
- Y. Wang, L. Zhou, S. Gao, J.-W. Li, Z.-F. Hu, L. Yang and Z.-C. Hu, Miner. Deposita, 2016, 51, 201–210 CrossRef CAS.
- R. R. Mendel and R. Hänsch, J. Exp. Bot., 2002, 53, 1689–1698 CrossRef CAS PubMed.
- B. N. Kaiser, K. L. Gridley, J. Brady, T. Phillips and S. D. Tyerman, Ann. Bot., 2005, 96, 745–754 CrossRef CAS PubMed.
- R. Hille, The Mononuclear Molybdenum Enzymes, Chem. Rev., 1996, 96, 2757–2816 CrossRef CAS PubMed.
- G. Schwarz and R. R. Mendel, Annu. Rev. Plant Biol., 2006, 57, 623–647 CrossRef CAS PubMed.
- I. Baxter, B. Muthukumar, H. C. Park, P. Buchner, B. Lahner, J. Danku, K. Zhao, J. Lee, M. J. Hawkesford, M. L. Guerinot and D. E. Salt, PLoS Genet., 2008, 4(2), e1000004 Search PubMed.
- F. Bittner, Frontiers in plant science, 2014, 5, 1–6 CrossRef PubMed.
- F. Von Blanckenburg, N. von Wirén, M. Guelke, D. J. Weiss and T. D. Bullen, Elements, 2009, 5, 375–380 CrossRef CAS.
- R. Hänsch and R. R. Mendel, Curr. Opin. Plant Biol., 2009, 12, 259–266 CrossRef PubMed.
- Eisler's encyclopedia of environmentally hazardous priority chemicals, Elsevier, Amsterdam, 2007 Search PubMed.
- D. J. Reuter, J. B. Robinson, K. I. Perevill, G. H. Price and M. J. Lambert, Guidelines for collecting, handling and analysing plant material, in Plant Analysis: An Interpretation Manual, ed. D. J. Reuter and J. B. Robinson, CSIRO Publishing, Collingwood, Australia, 2nd edn, 1997, pp. 53–70 Search PubMed.
- C. R. Pearce, A. S. Cohen and I. J. Parkinson, Geostand. Geoanal. Res., 2009, 33, 219–229 CrossRef CAS.
- D. C. Baxter, I. Rodushkin, E. Engström and D. Malinovsky, J. Anal. At. Spectrom., 2006, 21, 427–430 RSC.
- D. Malinovsky, P. J. H. Dunn and H. Goenaga-Infante, J. Anal. At. Spectrom., 2016, 31, 1978–1988 RSC.
- D. Malinovsky, I. Rodushkin, D. C. Baxter, J. Ingri and B. Öhlander, Int. J. Mass Spectrom., 2005, 245, 94–107 CrossRef CAS.
- C. Archer and D. Vance, Nat. Geosci., 2008, 1, 597–600 CrossRef CAS.
- C. R. Pearce, K. W. Burton, P. A. E. Pogge von Strandmann, R. H. James and S. R. Gíslason, Earth Planet. Sci. Lett., 2010, 295, 104–114 CrossRef CAS.
- A. R. Voegelin, T. F. Nägler, T. Pettke, N. Neubert, M. Steinmann, O. Pourret and I. M. Villa, Geochim. Cosmochim. Acta, 2012, 86, 150–165 CrossRef CAS.
- J. Liu, H. Wen, Y. Zhang, H. Fan and C. Zhu, J. Anal. At. Spectrom., 2016, 31, 1287–1297 RSC.
- M. Willbond, K. Hibbert, Y.-J. Lai, H. Freymuth, R. C. Hin, C. Coath, F. Vils and T. Elliott, Geostand. Geoanal. Res., 2016, 40, 389–403 Search PubMed.
- K. Ogura and Y. Enaka, Electrochim. Acta, 1978, 23(8), 767–772 CrossRef CAS.
- Joint Committee for Guides in Metrology (JCGM/WG 1), “Evaluation of measurement data – Guide to the expression of uncertainty in measurement”, JCGM, 2008.
- D. Malinovsky and N. Kashulin, Anal. Methods, 2016, 8, 5921–5929 RSC.
- E. D. Young, A. Galy and H. Nagahara, Geochim. Cosmochim. Acta, 2002, 66, 1095–1104 CrossRef CAS.
- M. Schiavon, M. Pittarello, E. A. H. Pilon-Smits, M. Wirtz, R. Hell and M. Malagoli, Environ. Exp. Bot., 2012, 75, 41–51 CrossRef CAS.
- M. Tejada-Jiménez, A. Chamizo-Ampudia, A. Galván, E. Fernández and Á. Llamas, Metallomics, 2013, 5, 1191–1203 RSC.
- W. Zimmer and R. Mendel, Plant Biol., 1999, 1, 160–168 CrossRef CAS.
- I. Rodushkin, A. Stenberg, H. Andrén, D. Malinovsky and D. C. Baxter, Anal. Chem., 2004, 76, 2148–2151 CrossRef CAS PubMed.
- D. Malinovsky, D. C. Baxter and I. Rodushkin, Environ. Sci. Technol., 2007, 41, 1596–1600 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |