
Exploring the efficacy and cellular uptake of sorafenib in colon cancer cells by Raman micro-spectroscopy†
H. K.
Yosef
 a,
T.
Frick
a,
M. K.
Hammoud
a,
A.
Maghnouj
b,
S.
Hahn
b,
K.
Gerwert
*a and
S. F.
El-Mashtoly
a
a,
T.
Frick
a,
M. K.
Hammoud
a,
A.
Maghnouj
b,
S.
Hahn
b,
K.
Gerwert
*a and
S. F.
El-Mashtoly
a
aDepartment of Biophysics, Ruhr-University Bochum, Germany. E-mail: gerwert@bph.rub.de; Fax: +49 234 3214238; Tel: +49 234 3224461
bDepartment of Molecular GI-Oncology, Clinical Research Center, Ruhr-University Bochum, 44780 Bochum, Germany
First published on 7th November 2018
Abstract
In recent years, many subcellular proteins have emerged as promising therapeutic targets in oncology. One crucial target is the epidermal growth factor receptor. Inhibition of this receptor has significantly improved the survival rate of patients for many cancers. However, oncogenic mutations such as B-RAFV600E have rendered tumours resistant to this therapeutic approach. Therefore, this mutation has emerged as a potential target for cancer therapy. Sorafenib is developed to overcome the B-RAFV600E mutation and restore the response of the mutated tumour to therapy. Here, we explore the efficacy and distribution of sorafenib at a cellular level using colon cancer cell lines with B-RAFV600E or K-RASG12V mutations. The Raman results detected significant sorafenib-induced spectral differences in both cell lines. In addition, the western blot and real-time cell analysis in vitro assays revealed that the ERK phosphorylation and the cellular proliferation of cells are inhibited, respectively, in the sorafenib-treated cells. Thus, the observed Raman spectral changes illustrate the potent effect of sorafenib on cells despite the presence of the B-RAFV600E or K-RASG12V mutations. These results are in agreement with the clinical studies, where patients with the B-RAFV600E mutation respond to sorafenib. Furthermore, the Raman spectral imaging results have shown the uptake and the distribution of sorafenib in colon cancer cells with the B-RAFV600E mutation through its label-free marker bands in the fingerprint region. The present results of sorafenib efficacy and distribution in cells demonstrate the potential of Raman micro-spectroscopy as the in vitro assay for the assessment of drugs, which is important in drug discovery.
Introduction
Despite the extensive efforts and huge investments made on accelerating the development of drugs in oncology, cancer therapy remains one of the highest demanding medical needs.1–3 The development of the molecular targeting strategy has offered a great improvement in cancer therapy.4 In this approach, a specific protein that contributes to cancer progression is targeted and inhibited by a therapeutic agent. Epidermal growth factor receptor (EGFR) is one of the most prominent targets in cancer therapy.5,6 This is due to its major role in the regulation of cell proliferation and tumour growth.7 It is well established that EGFR activation by growth factors initiates a cascade of downstream signalling through an RAS/mitogen-activated protein kinase (MAPK) pathway, which leads to genetic transcription engaging cell proliferation and survival (Fig. 1).8,9 EGFR activation can be blocked using monoclonal antibodies or tyrosine kinase inhibitors (TKIs). The blocking of EGFR inhibits MAPK that ceases cell proliferation.10 The MAPK pathway consists of RAF, MEK, and ERK proteins.9,11 RAF exists in three isoforms: A-RAF, B-RAF, and C-RAF.12 B-RAF received more attention than the other types after the detection of the B-RAF mutation in 66% of melanomas and in various types of human solid cancers.13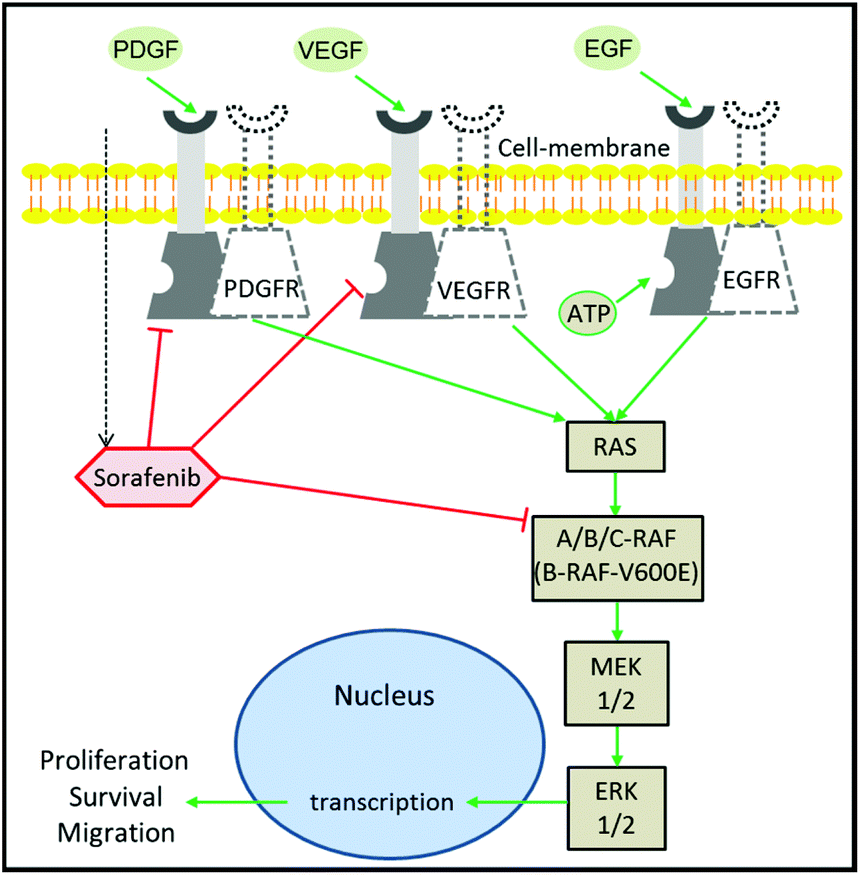 | ||
| Fig. 1 Mechanism of sorafenib action. Sorafenib is a multi-kinase inhibitor that targets RAF, PDGFR, and VEGFR, inhibiting cellular proliferation. The dotted and non-dotted receptors undergo homodimerization upon ligand binding to the receptor triggering autophosphorylation. This leads to the activation of the tyrosine kinase signal transduction pathways, which modulate cellular proliferation through the MAPK pathway. | ||
Among the 100 mutations that have been identified in B-RAF, V600E is the most occurring one and has been reported in several aggressive cancers including metastatic colon cancer,14 thyroid cancer,15 non-small-cell lung cancer,16 and melanoma.17 Furthermore, clinical studies indicated that metastatic colorectal and lung cancer patients with the B-RAFV600E oncogenic mutation were non-responders to EGFR inhibitors such as cetuximab and erlotinib.18,19 This is because the RAFV600E mutation causes a prolonged activation of the MAPK pathway regardless of EGFR activation or inhibition, leading to an activation of cell proliferation that confers a refractory character to the EGFR-targeting therapy.13,19 As a consequence, the mutated B-RAFV600E protein has emerged as one of the new targets in cancer therapy.17,20
Sorafenib (Nexavar™) is a multi-kinase inhibitor that is developed to block the phosphorylation of RAF and its isoforms including the B-RAFV600E mutation, preventing cell proliferation and tumour progression.21,22 It also targets the vascular endothelial growth factor receptors (VEGFR), platelet-derived growth factor receptor-β (PDGFR), and fibroblast growth factor receptor 1.22–24 In addition, sorafenib inhibits the MAPK pathway and ERK phosphorylation in a panel of human breast, pancreatic, and colon cancer and melanoma cell lines expressing either mutant B-RAF, mutant K-RAS, or wildtype RAF or RAS.25,26 Sorafenib has been approved by the U.S. Food and Drug Administration and European Medicines Agency for the treatment of thyroid cancer, and hepatocellular and renal cell carcinomas.27–29
Sorafenib has also demonstrated promising results concerning the treatment of colorectal cancer when it is used alone or in combination with other anticancer drugs and it is still in clinical trials for metastatic colon cancer.30,31 Here, we examine the sorafenib efficacy and distribution in colon cancer cells with and without B-RAF and K-RAS mutations using Raman micro-spectroscopy.
Raman micro-spectroscopy has been implemented extensively in several biomedical and clinical applications as well as in the drug discovery process.31–39 It offers great potential in discriminating between different cells and eventually can screen cancer cells,40–44 and also for the assessment of drugs at the cellular level.45–57 For instance, Raman micro-spectroscopy has been used to monitor the efficacy of several anti-cancer agents or drugs such as cisplatin, vincristine, doxorubicin, panitumumab, and erlotinib on cancer cells.44,46,47,56,58,59 Furthermore, the label-free Raman imaging of the distribution of small molecule inhibitors or drug carriers containing functional groups such as an alkyne, nitrile, or isotopic label (deuterium) in cells has been reported in the literature.45,52,54,60–64 The label-free drug distribution approach is based on the fact that these functional groups have Raman bands in the silent region of the cell spectrum (1800–2800 cm−1) and they can be used as label-free markers. However, small molecule inhibitors such as TKIs, which are promising candidates used also as label-free Raman markers, exhibit strong and sharp Raman lines in the fingerprint region.50,64 For instance, Fu et al. used the Raman bands of imatinib and nilotinib at 1300 cm−1, of chloroquine at 1370 cm−1, and of GNF-2 and GNF-5 at 1600 cm−1 to monitor their distribution in the cells.50 Aljakouch et al. also used a Raman band near 1386 cm−1 to monitor the distribution of neratinib in different cancer cells.64
Here, we utilized Raman micro-spectroscopy to explore the potency and efficacy of sorafenib on three colon cancer cell lines with B-RAF (HT29 cells) and K-RAS (SW480 cells) mutations and without these mutations (SW48 cells). The Raman results displayed large sorafenib-induced differences in the cells irrespective of whether they harbour B-RAF or K-RAS mutations. The common in vitro assays such as western blot and real-time cell analysis (RTCA) indicated that sorafenib inhibited ERK phosphorylation and the proliferation of colon cancer cells, respectively, in the case of HT29 and SW480 cells. These results suggest that the observed Raman spectral changes in the cells upon treatment with sorafenib are produced as a result of a cellular response. Furthermore, the label-free distribution of sorafenib in the cells was monitored through its Raman bands in the fingerprint region. The present results demonstrate the potential of Raman micro-spectroscopy as a companion diagnostics for screening anti-cancer agents and could be of importance in drug discovery, especially for preclinical in vitro screening.
Experimental section
Cell culture
HT29 (HTB-38), SW48 (CCL-231), and SW480 (CCL-228) human colon cancer cell lines were obtained from the American Type Culture Collection. HT29 cells have the B-RAFV600E mutation,65,66 whereas SW480 cells harbour the K-RASG12V mutation.67 The cell culture is conducted in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, USA) that is supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, USA), 2 mM L-glutamine, and 5% penicillin/streptomycin. The cells were incubated under a 10% CO2 atmosphere at 37 °C. CaF2 slides (Korth Kristalle, Kiel, Germany) were used as the adherent surface for cell culture and as the substrate for the Raman measurements, in order to avoid background signals from conventional glass slides. The cells were incubated with sorafenib (50 μM, 4 hours) for the drug uptake experiment and with sorafenib (15–25 μM, 16 hours) for the drug effect experiment under a 10% CO2 atmosphere. Afterwards, the cells were fixed in 4% paraformaldehyde (VWR International, Darmstadt, Germany) and submerged in phosphate-buffered saline (PBS; Invitrogen, Carlsbad, USA) until further use.Confocal Raman microscopy
Raman micro-spectroscopy imaging was conducted using a WITec alpha300 AR confocal Raman microscope (Ulm, Germany), which is described previously.40,45–47,68,69 Two laser sources, a frequency-doubled Nd:YAG laser source of 532 nm (Crystal Laser, Reno, USA) and a single-frequency diode laser of 785 nm (Toptica Photonics AG, Munich, Germany), were used for monitoring the sorafenib (Fig. S1†) effect on the cells and its localization within the cells. The Raman intensity of the sorafenib bands is stronger with 785 nm excitation compared to that with 532 nm excitation. Therefore, a 532 nm laser was used in the case of the drug effect experiment with an output power of 12 mW approximately at the sample. In the case of drug uptake measurements in which the cells were treated with a high sorafenib concentration (50 μM), a 785 nm excitation was used with an output power of 120 mW at the sample. A Nikon NIR APO (60×/1.00 NA) water-immersion objective is used for all measurements. The spot size of the laser beam is ∼1 μm. The Raman imaging measurements were performed by raster-scanning the laser beam over the cells and obtaining a full Raman spectrum at a speed of 0.5 seconds per pixel and the pixel resolution is 500 nm.Multivariate analysis
Hierarchical cluster analysis (HCA) and principal component analysis (PCA) were utilized to analyse the Raman spectra of the cells. The Raman hyperspectral results were exported to MATLAB 8.2 (MathWorks Inc., MA), and the data pre-processing and cluster analyses were conducted using in-house built scripts. For measurements with 532 nm excitation, the Raman spectra that had no C–H band at 2850–3000 cm−1 were treated as the background and deleted. The cosmic spikes were removed using an impulse noise filter, and the Raman spectra were interpolated to a reference wavenumber scale. All spectra were then baseline-corrected by a third-order polynomial and were vector normalized. We performed the HCA in the regions of 700–1800 and 2800–3050 cm−1 using Ward's clustering in combination with the Pearson correlation distance for the datasets obtained with 532 nm excitation. The PCA was performed on the average spectra of the control and the sorafenib-treated cells in the region of 700–3050 cm−1 to obtain the principal component (PC) scores and loadings. The first three PCs generated the maximum variances within the Raman results, highlighting the different spectral features between the control and the sorafenib-treated cells. In the case of 785 nm excitation, the HCA was performed in the region of 700–1800 cm−1.Results and discussion
Cellular response to sorafenib by RTCA
To examine whether SW48, HT29, and SW480 colon cancer cells respond to sorafenib or not, a standard RTCA bioassay was used. RTCA evaluates cellular proliferation, migration and invasion based on the impedance detection of the cell viability in a label-free manner.70 This technique monitors non-invasively the growth, shape, and damage of the cell over an extended period of time. In an RTCA instrument, gold electrodes located at the bottom of each cell culture well act as electronic sensors that measure changes in the impedance induced by the adherent cells. When the cells are seeded into the well, the impedance signal expressed in the cell index value increases with cells attaching to the bottom of the cell culture well. The division of the cells causes an increase in impedance over time reflecting cell proliferation, whereas it decreases as the cells detach when they die. RTCA (see the ESI†) was conducted on SW48, HT29 (B-RAF mutation), and SW480 (K-RAS mutation) cells treated with 15, 25, and 50 μM concentrations of sorafenib, as well as on cells without sorafenib treatment (control), and the results are shown in Fig. 2. These concentrations are similar to the detected sorafenib concentrations in the plasma (5–31 μM) of patients diagnosed with hepatocellular carcinoma, after treatment with a single oral dose of 200 or 400 mg per day.71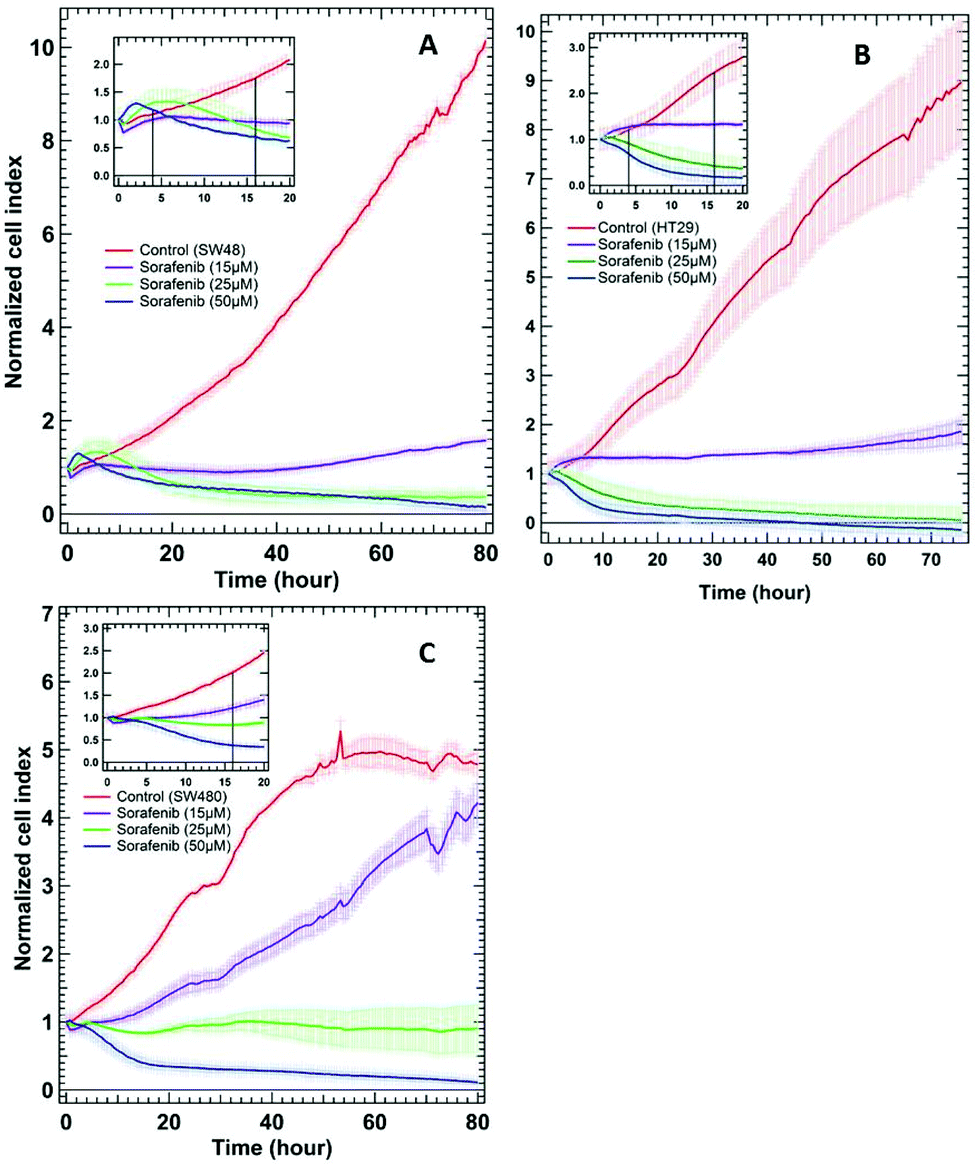 | ||
| Fig. 2 RTCA of SW48 (A), HT29 (B), and SW480 (C) cells of the control and the cells treated with different sorafenib concentrations. The insets show only the first 20 hours after drug treatment. | ||
In the case of the control, the results revealed an increase in the cell index value over time, indicating that the cells still proliferate. In contrast, the cell index values decreased to ∼0.5–0 after ∼30 hours when the cells were treated with higher sorafenib concentrations (25 μM in the case of SW48 and HT29 cells and 50 μM in the case of all cell lines). However, the cell index values of the cells treated with lower sorafenib concentrations (15 μM in the case of SW48 and HT29 cells and 25 μM in the case of SW480 cells) remain almost constant at ∼1 but did not increase significantly as in the control nor dropped to ∼0.5–0 as in the case of the cells treated with higher sorafenib concentrations. These results suggest that lower sorafenib concentrations induce a cytostatic effect by interfering with the cell-cycle machinery leading to growth arrest. On the other hand, higher concentrations of sorafenib may induce a cytotoxic effect on the cells after ∼30 hours of incubation leading to necrosis, apoptosis and/or cell lysis.72 These results indicate that the SW48, HT29, and SW480 cells responded to sorafenib irrespective of whether they harbour B-RAF or K-RAS mutations. Therefore, we used the SW48 and HT29 cells treated with 15 μM sorafenib and the SW480 cells treated with 25 μM sorafenib for 16 hours to monitor its efficacy by Raman micro-spectroscopy. At these concentrations, sorafenib itself is not detectable. On the other hand, the cells treated with 50 μM sorafenib for 4 hours were used to localize sorafenib in the cells. At these concentrations and incubation times, most of the cells are viable and adherent to the cell culture well.
Inhibition of ERK phosphorylation by sorafenib
In order to confirm that sorafenib inhibits the activity (phosphorylation) of RAF to prevent cellular proliferation, we targeted the ERK proteins which can be used as an indicator of RAF protein activity. It is well established that RAF activation increases the phosphorylation of ERK, whereas inhibiting RAF can block ERK phosphorylation.73 We used the western blot assay to evaluate the inhibitory effect of sorafenib on ERK phosphorylation in SW48 (A), HT29 (B), and SW480 (C) cells as shown in Fig. 3. The results indicate an increase in the activation level of phospho-ERK (P-ERK) in the cells treated with epidermal growth factor (EGF; a positive control) compared to the control cells that exhibit ERK phosphorylation. This can be attributed to the activation of EGFR by EGF binding that in turn activates the RAS-RAF-ERK pathway as shown in Fig. 1.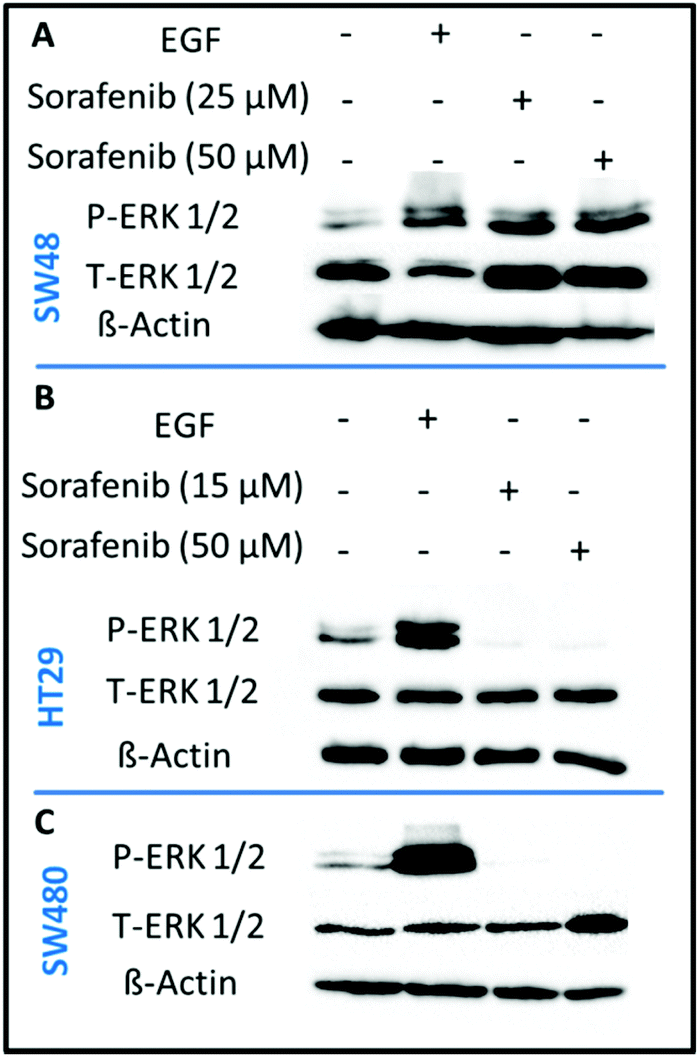 | ||
| Fig. 3 Effect of sorafenib on ERK phosphorylation in SW48 (A), HT29 (B), and SW480 (C) cells. Cell lysates were resolved by SDS-PAGE and western blot analysis using antibodies that recognize phosphospecific ERK1/ERK2 (P-ERK1/2) and total ERK1/ERK2 (T-ERK1/2). β-Actin was used as a loading control. | ||
In the case of 15–50 μM sorafenib-treated HT29 (B) and SW480 (C) cells, the signal of P-ERK is clearly inhibited as a result of an inhibition of an upstream RAF protein, which is expressed in these cells (Fig. S2†). In contrast, the P-ERK signal was not inhibited in the sorafenib-treated SW48 cells (A) but it is increased. The amount of T-ERK was also increased upon sorafenib treatment. The reproducibility of the P-ERK results for the sorafenib-treated SW48 cells was confirmed using three different cell batches. However, the reason for these results is not clear. Therefore, the used sorafenib concentrations clearly inhibited the cell growth and proliferation as revealed by the RTCA results (Fig. 2) for all cell lines. In addition, western blot (Fig. 3) showed that sorafenib inhibited the P-ERK expression in both HT29 and SW480 cells.
Cellular response to sorafenib by Raman micro-spectroscopy
Raman spectral imaging was conducted on the SW48, HT29, and SW480 cells treated with sorafenib for 16 hours as well as on the cells without treatment. The average Raman spectra of the control and the sorafenib-treated cells (approximately 50–60 cells per cell line) are displayed in Fig. S3.† As indicated by RTCA, the adherent cells still remain viable after drug treatment for 16 hours (Fig. 2). The white dots in the images (Fig. 4A and B) reflect the presence of lipid droplets that have a strong Raman intensity and a characteristic Raman spectrum.46,47 The average Raman spectrum of the control (Fig. 4C(a)) contains bands at 1008 cm−1 (phenylalanine ring-breathing mode), 1098 cm−1 (phospholipid C–C stretching), 1130 cm−1 (C–N stretching), 1260–1350 cm−1 (amide III vibration of peptide linkages), 1451 cm−1 (C–H deformation or CH2 bending modes), 1660 cm−1 (amide I), and 2800–3020 cm−1 (C–H stretching of CH3, CH2, and CH).35,46,47 The tentative assignment of the main Raman bands is also indicated in Table 1.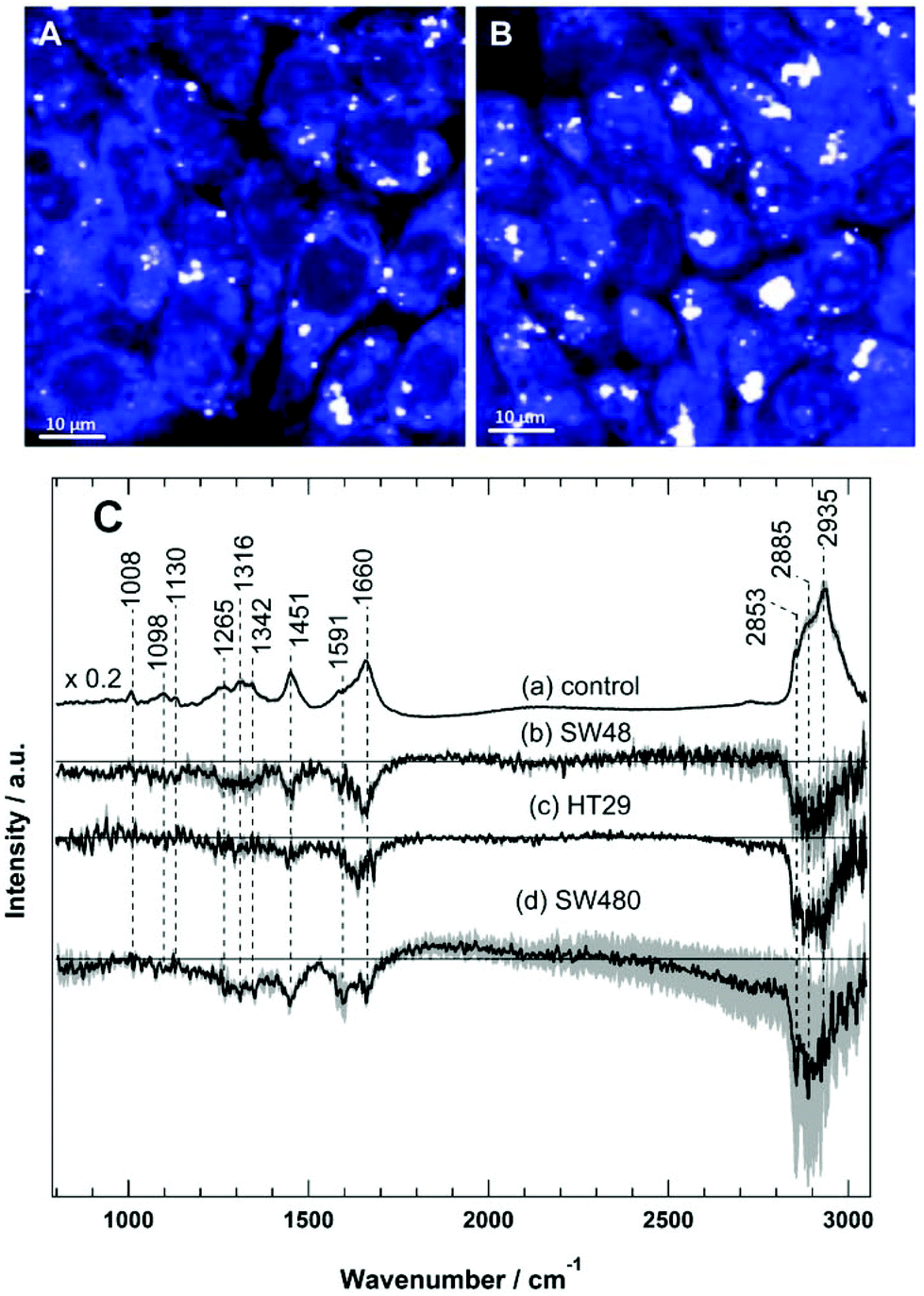 | ||
| Fig. 4 Raman spectral imaging of HT29 cells with 532 nm excitation. The integrated Raman intensity images of the C–H stretching region (2800–3050 cm−1) of the control (A) and the sorafenib-treated cells (B). (C) Average Raman spectra of HT29 cells of the control (a) and the difference spectra (control – sorafenib-treated cells) of SW48 (b), HT29 (c), and SW480 (d) cells. | ||
| Wavenumber (cm−1) | Assignment |
|---|---|
| 1008 | Ring-breathing mode in phenylalanine |
| 1098 | Symmetric PO2 stretching vibration of the DNA backbone or phospholipid C–C stretching |
| 1130 | C–N stretching |
| 1169 | C–H in plane-bending mode of tyrosine and phenylalanine; cytosine, guanine |
| 1205 | Polysaccharides |
| 1250–1340 | Amide III (C–N stretching, N–H bending, proteins), and/or PO2 asymmetric stretching |
| 1308 | CH3 and/or CH2 twisting or bending mode of lipids or collagen |
| 1451 | CH (CH2) bending mode in proteins and lipids |
| 1588 | Adenine, guanine and/or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bending mode of phenylalanine C bending mode of phenylalanine |
| 1660 | Amide I (CO stretching, C–N stretching, and N–H bending, proteins) |
| 2850–2945 | C–H symmetric stretching (lipids and proteins) |
The difference spectra of the untreated cells versus the sorafenib-treated cells are determined and presented in Fig. 4C(b–d). These difference spectra revealed the molecular changes in cancer cells upon treatment with sorafenib. They display clear negative bands in the fingerprint region around 1451 cm−1 (lipids) and 1660 cm−1 (proteins), as well as the bands in the C–H stretching region near 2840–2960 cm−1 (lipids and proteins).46,47 These results may suggest that the lipid and protein contents are increased upon sorafenib treatment. This is perhaps due to drug-induced cell stress and subsequent apoptosis.74,75
The PC scatter plots for the Raman results displayed in Fig. 5 show a distinctive separation between the control and the sorafenib-treated SW48 (A), HT29 (B), and SW480 (C) cells. In addition, the PC loadings are presented in Fig. 5 for the SW48 (D), HT29 (E), and SW480 (F) cells. All the three PCs show variation in the lipid, protein, nucleic acid, amino acid, and polysaccharide contents of the cells. For instance, the PC1 of the SW48 cells is dominated by the lipid (1446 cm−1) and protein (amide I: 1664 cm−1) contribution. The PC2 shows variations in proteins (Tyr: 1169 cm−1, Phe: 1588 cm−1, and amide I: 1630 cm−1), lipids (1308 and 1445 cm−1), nucleic acids (1308 cm−1), and polysaccharides (1205 cm−1). The PC3 also depicts variations in proteins (amide III: 1250–1340 cm−1 and amide II: 1552 cm−1), lipids (1453 cm−1), and nucleic acids (1525 cm−1). The lipids and proteins in all the three PCs also contribute to the 2850–2945 cm−1 region. In addition, the three PCs of the HT29 cells (E) display variations in lipids (1453 cm−1) and proteins (amide III: 1250–1320 cm−1 and amide I: 1630 and 1657 cm−1), and both of them contribute also to the 2850–2945 cm−1 region. Furthermore, the three PCs of the SW480 cells (F) show variations in lipids (1453 cm−1), proteins (amide III: 1250–1320 cm−1 and amide I: 1630 and 1657 cm−1), and nucleic acids (1090 cm−1 and 1190 cm−1), and PC1 and PC3 have contributions also to the lipids and proteins in the 2850–2945 cm−1 region. It is noted that H–O–H bending of water may contribute to the PC2 of both HT29 (E) and SW480 (F) cells, where a broad band around 1640 cm−1 is present in PC2 loading.76
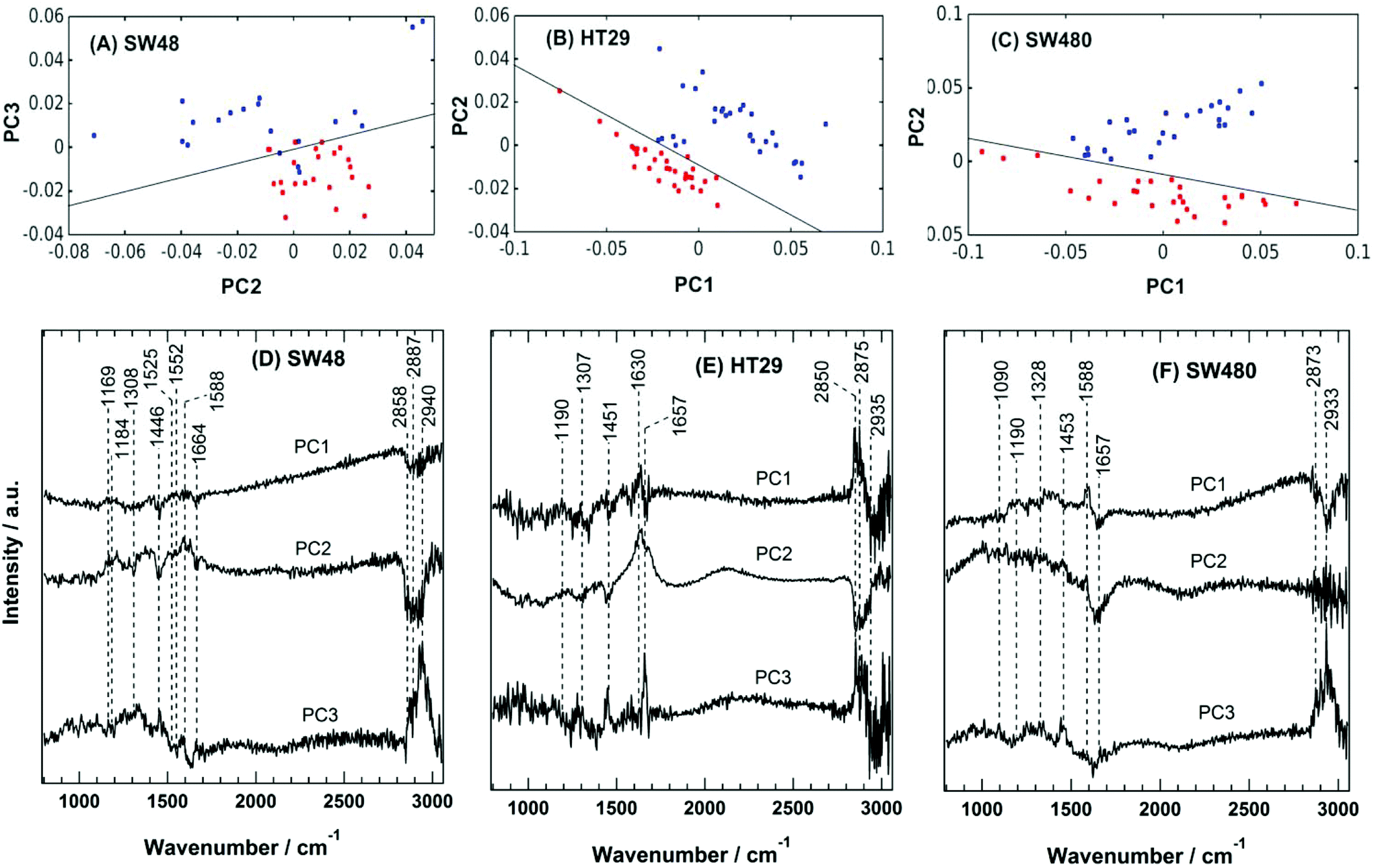 | ||
| Fig. 5 Principal component analysis (PCA) of the Raman results for colon cancer cells. The PCA scatter plots for (A) SW48, (B) HT29, and (C) SW480 cells. The control and sorafenib-treated cells are shown in red and blue, respectively. Principal component loadings of the Raman results for (D) SW48, (E) HT29, and (F) SW480 cells. | ||
These variations indicate a clear alteration in the protein, lipid, and nucleic acid contents of the cells upon sorafenib treatment. They also refer to a major metabolic shift within the cell, which can be attributed to the apoptotic changes induced by sorafenib treatment. The observed variations in the loadings (mainly in lipids and proteins) are in good agreement with those spectral changes detected in the Raman difference spectra (Fig. 4C).
To monitor the effect of sorafenib on the subcellular components, the average cluster spectra of the plasma membrane region, cytoplasm, nucleus, and lipid droplets for the sorafenib-treated cells and the control of, for example, the HT29 cells were recorded as described previously,47 and the results are displayed in Fig. S4 and S5.† Only the average spectra of the nucleus contain the DNA marker band near 790 cm−1 (O–P–O backbone stretching). The average spectra of the lipid droplets are similar to those of high lipid or phospholipid contents.46,47 The difference spectra (untreated cells – sorafenib-treated cells) of the plasma membrane region, cytoplasm, nucleus and lipid droplets were also determined and are shown in Fig. S6.† These difference spectra revealed spectral changes implying variations in the subcellular organelles upon treatment with sorafenib.
Taking together the Raman results, RTCA, and the western blot analysis, the detected Raman spectral changes can be considered as a cellular response to sorafenib.46,47 However, there is a discrepancy in the case of SW48 cells between the RTCA and Raman results on the one hand and those of ERK phosphorylation on the other hand. Both the RTCA and Raman results suggest that the SW48 cells responded to sorafenib, while ERK phosphorylation was not inhibited by sorafenib under similar experimental conditions. Such discrepancy has been observed previously for the SW480 cells treated with panitumumab. The Raman results suggested that the SW480 cells did not respond to panitumumab due to the K-RAS mutation similar to the clinical observations, while ERK phosphorylation was inhibited by panitumumab.47
In our previous study,46 the Raman difference spectra of the untreated cells versus the erlotinib-treated cells displayed major spectral changes in the SW48 cells harbouring wild-type B-RAF, whereas minor/limited changes were detected in the case of the HT29 cells containing the B-RAFV600E mutation. These minor/limited changes were observed because the HT29 cells harbouring the B-RAFV600E mutation have a limited response to EGFR inhibitors.13 In the present study, sorafenib overcomes the B-RAFV600E mutation, and larger spectral changes especially in the C–H stretching region and consequently a clear effect on the HT29 cells were observed. These results are in agreement with the clinical results that indicate a positive response of B-RAFV600E-mutated tumours to sorafenib treatment.27–30
The SW480 cells harbouring the K-RAS mutation showed no response to EGFR inhibitors such as panitumumab and erlotinib.46,47 These results are in agreement with the observed response of colorectal cancer patients in clinics. In contrast, the SW480 cells showed a response to sorafenib, which is a multi-kinase inhibitor. These results are in the same line with the clinical trial Phase II (NEXIRI) results, conducted on patients with K-RAS-mutated metastatic colorectal cancer. The results showed that using sorafenib in combination with irinotecan enhances the tumour response of patients who are initially resistant to irinotecan monotherapy.26
Label-free distribution of sorafenib in cells by Raman spectral imaging
One of the major evaluation criteria of drug pharmacokinetics is the detection of the subcellular uptake of the therapeutic agent. However, imaging the uptake of small molecule drugs or drug carriers is highly challenging using conventional Raman micro-spectroscopy. This is because drugs or their carriers often accumulate at lower concentrations within cells. There is a general consensus to use functional groups such as an alkyne, nitrile, or isotopic label (deuterium or 13C) as a label-free marker to improve the Raman sensitivity of the molecule of interest since these groups have Raman bands in the silent region of the cell spectrum (1800–2800 cm−1).45,52,54,60–64 These label-free Raman tags are in some cases inherent to the chemical structure of the molecule under examination such as the presence of an alkyne group in erlotinib and EdU, and a nitrile group in neratinib,64 or alternatively, they may be integrated by the chemical modification of the parent structure.45,51 On the other hand, the sorafenib chemical structure (Fig. 6) does not contain any of these label-free Raman tags that can provide vibrational bands in the silent region of the Raman cell spectrum. Consequently, it is more challenging to detect the intracellular uptake of sorafenib using only Raman bands in the fingerprint region of the Raman spectrum.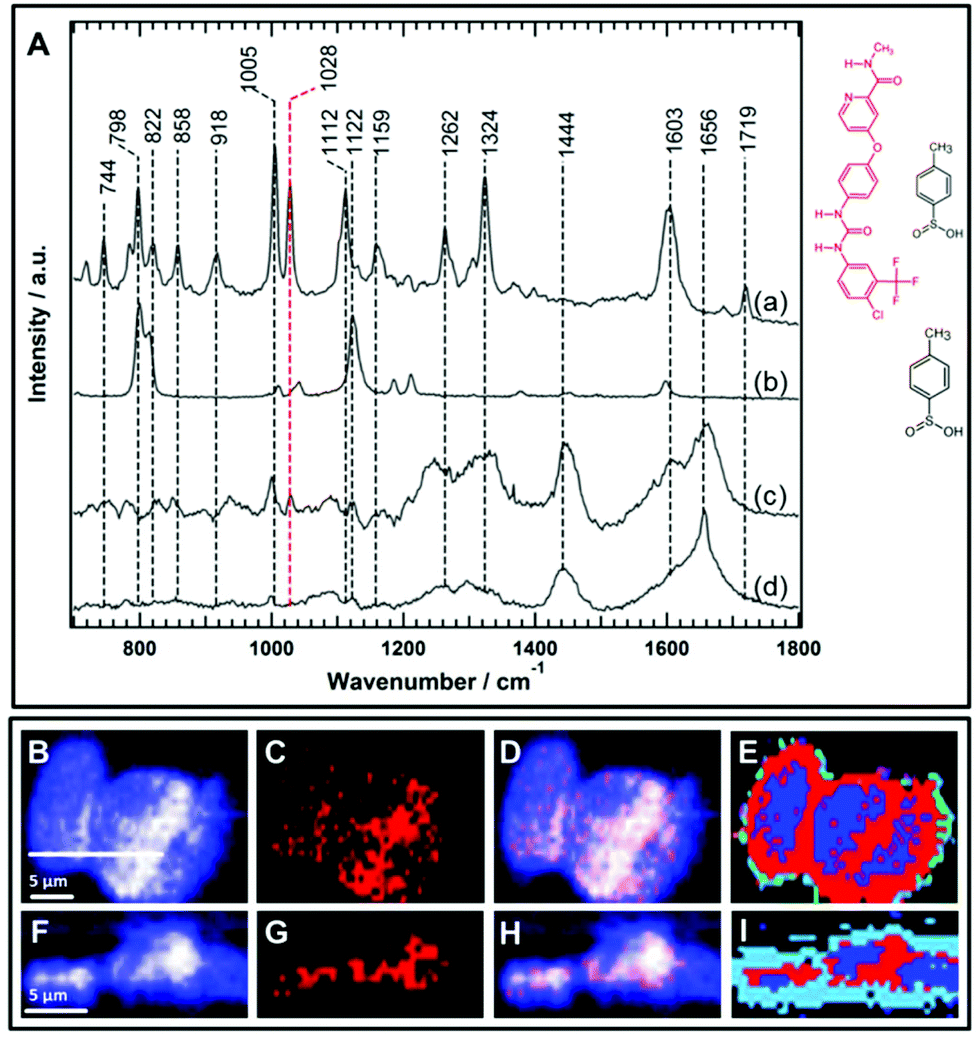 | ||
| Fig. 6 (A) The Raman spectra of free sorafenib tosylate (a) and p-toluenesulfonic acid (b), the average Raman spectrum of the sorafenib-containing cluster in HT-29 cells (c), and the average Raman spectrum of the control cells (d) excited at a wavelength of 785 nm. The chemical structures of sorafenib tosylate and p-toluenesulfonic acid are also shown. (B–I) Raman imaging of the HT-29 cells treated with 50 μM sorafenib for 4 h. Raman images reconstructed from the band's intensities at 1420–1470 cm−1 (B) and 1015–1040 cm−1 (C). Panel C represents sorafenib (red structure only) without its tosylate. (D) Overlay of Panels B and C. (F–H) Cross-sectional Raman images along the x–z axis of the same cells. The scanning positions are indicated by the white line in Panel B. (D and H) represent the distribution of sorafenib (red structure) in the cells. (E and I) The HCA results based on the Raman data shown in Panels B and F and the red clusters. (E and I) represent sorafenib. | ||
Sorafenib-p-toluenesulfonate (sorafenib tosylate) is a commonly used form of sorafenib in clinics.77,78 Introducing the tosylate moiety to sorafenib can enhance its water solubility due to salt formation, which facilitates oral drug applications. The Raman spectrum (a) of pure sorafenib tosylate reveals bands at around 744, 798, 822, 858, 918, 1005, 1028, 1112, 1159, 1262, 1324, 1603, and 1719 cm−1 as depicted in Fig. 6A. The strong sorafenib bands at 798, 1005, 1028, 1112, 1324, and 1603 cm−1 are potential candidates to monitor the localization of sorafenib within the HT29 cells. However, the Raman bands near 798 and 1112 cm−1 may originate from the p-toluenesulfonate moiety as indicated by the Raman spectrum of p-toluenesulfonic acid (spectrum b). It is noted that the band at 1122 cm−1 in p-toluenesulfonic acid is downshifted in sorafinib tosylate (1112 cm−1) most likely due to salt formation.79 Since sulfonic acid salts offer a relatively higher dissolution rate compared to other salts moieties,80 the p-toluenesulfonate moiety is expected to dissociate before sorafenib interacts with the RAF protein. It is noted that the dissociation of the p-toluenesulfonate moiety was not detected in the present study. Therefore, the 798 and 1112 cm−1 bands cannot be used to monitor the distribution of sorafenib within the cells.
Furthermore, the Raman spectrum (d) of the HT29 cells displays Raman bands near 1005 cm−1 (ring-breathing mode of phenylalanine), ∼1324 cm−1 (amide III), and ∼1656 cm−1 (amide I) that overlap with sorafenib bands at 1005, 1324, and 1603 cm−1, respectively. Thus, it is safer to conclude that only the sorafenib Raman band at 1028 cm−1 (spectrum a), which has no corresponding band in the cell spectrum (d), can be used as a label-free marker candidate to monitor sorafenib localization within the HT29 cells. The Raman spectral imaging was conducted on the HT29 cells treated with 50 μM sorafenib for 4 hours. Most of the cells are viable at that time as shown by the RTCA results (Fig. 2). By constructing the Raman intensity image of HT-29 cells at 1420–1470 cm−1 (C–H deformation region), the major cellular architecture can be visualized as depicted in Fig. 6B, whereas the Raman intensity image constructed around the band of 1028 cm−1 reveals the distribution of sorafenib within the cells (Panel C). By overlaying images (B) and (C), the subcellular distribution of sorafenib (red structure in Fig. 6) without its salt can be visualized in image (D), which indicates a clear spatial distribution of sorafenib in the HT29 cells.
Raman spectral data were analysed using the HCA, conducted on the spectral range of 700–1800 cm−1. In the HCA index-colour image, the subcellular distribution of a sorafenib-containing cluster is indicated in red (E). The average Raman spectra of the HCA are shown in Fig. S7.† It is noticed that the HCA image displays more sorafenib-containing clusters than the corresponding Raman intensity image (Fig. S8†). This is because the HCA clustering assigns each pixel exclusively to a cluster, producing a better image than the univariate Raman intensity image. The average spectrum of the blue cluster (Fig. S7†) contains a DNA marker band (784 cm−1). Thus, the blue cluster is tentatively assigned to the nucleus. It also contains sorafenib since the average Raman spectrum of the nucleus displays a band at 1028 cm−1. An average Raman spectrum of the sorafenib-containing cluster within the HT29 cells is depicted in Fig. 6A(c). Although the average Raman spectrum of the sorafenib-containing cluster in the cells contains strong cellular contribution, the sorafenib marker bands at 1028 cm−1 are clearly visible. In addition, most likely the sorafenib bands at 1603 and 1005 cm−1 (spectrum a) contribute to the spectrum of the sorafenib-containing cluster (spectrum c).
Furthermore, cross-sectional Raman imaging in the x–z axis was also conducted to confirm that the detected sorafenib is confined within the cell and not precipitated extracellularly, as shown in Fig. 6(F–I). In order to validate the reproducibility of these findings, Raman measurements of three more replicates of HT29 cells were performed and shown in Fig. S9–S14.† Similar results were obtained using SW48 cells (Fig. S15 and S16†). Thus, Raman spectral imaging demonstrates the sorafenib uptake, utilizing Raman marker bands in the fingerprint region. The concentration of sorafenib within the cancer cells is presumably in the mM range as quantitatively estimated for other tyrosine kinase inhibitors using Raman microscopy.50,64
Sorafenib is a multi-kinase inhibitor blocking the phosphorylation of RAF and its isoforms including B-RAFV600E. A combination of Raman spectral imaging followed by fluorescence imaging was used to monitor the distribution of not only sorafenib but also the B-RAF in the same cell as shown in Fig. 7 and S11–13.† The HCA of the Raman results and fluorescence imaging are shown in Panels B and C, respectively. The sorafenib-containing cluster (red) from (B) is overlaid with the B-RAF (green) from (C) and the results are shown in (D). The overlaid region (yellow) suggests that a large amount of sorafenib is colocalized with B-RAF, which is one of the targets of sorafenib. These results suggest that some of the sorafenib binds to B-RAF, blocking its phosphorylation and then inhibiting the MAPK pathway and subsequently the cellular proliferation.21,22
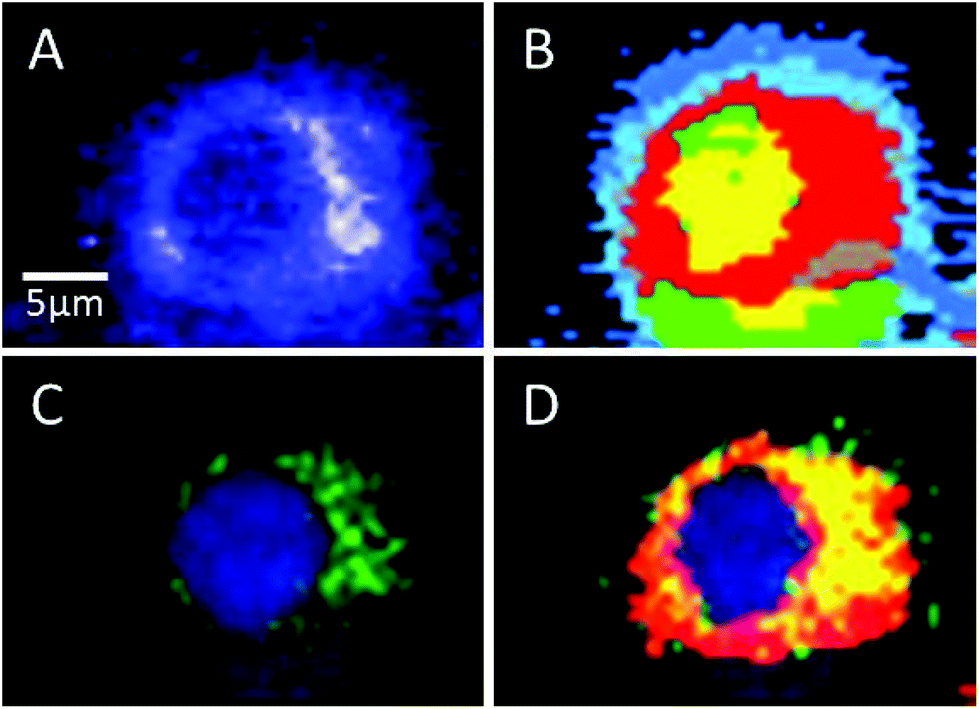 | ||
| Fig. 7 (A) Integrated Raman image of the HT29 cell treated with sorafenib. (B) The HCA of the Raman data shown in (A) containing sorafenib (red). (C) Fluorescence image of the same cell that shows B-RAF (green) and the nucleus (blue). (D) Overlay of the sorafenib-containing cluster (red) from (B) and B-RAF (green) from (C); the overlaid regions are shown in yellow. | ||
Conclusions
The present Raman results detected large sorafenib-induced differences in HT29 colon cancer cells harbouring the B-RAFV600E mutation. Under the same experimental conditions, cellular proliferation and ERK phosphorylation are inhibited in the sorafenib-treated HT29 cells as illustrated by the in vitro RTCA and western blot assays, respectively. Thus, the Raman micro-spectroscopic results illustrate the potent effect of sorafenib on HT29 cells despite the presence of the B-RAFV600E-resistant mutation. These results are also in agreement with the clinical studies, where patients with the B-RAFV600E mutation respond to sorafenib. Similar results were also observed for SW480 cells harbouring the K-RAS mutation. On the other hand, SW48 cells showed a discrepancy in the results of different conventional in vitro assays. Furthermore, the Raman spectral imaging results have provided a proof of the uptake and the label-free distribution of sorafenib in cancer cells and its colocalization with B-RAF. The results demonstrate that the capability of Raman micro-spectroscopy in drug uptake detection is not only limited to the Raman marker bands in the silent region of the cell spectra (1800–2800 cm−1), but it can also be extended to bands in the fingerprint region as shown here for sorafenib. Taking together the results of sorafenib efficacy and distribution in cells, the capacity of Raman micro-spectroscopy for monitoring both drug efficacy and distribution can be further extended for drug evaluation in the in vivo environment such as mouse models, which is the next step in pre-clinical evaluation and drug discovery.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Prof. Dr Axel Mosig for useful discussion. This research was supported by the Protein Research Unit Ruhr within Europe (PURE), the Ministry of Innovation, Science, and Research (MIWF) of North-Rhine Westphalia, Germany and the European Regional Development Fund, European Union and North-Rhine Westphalia, Germany.References
- K. Dhingra, Ann. Oncol., 2015, 26, 2347–2350 CrossRef CAS PubMed.
- J. C. Yao, F. Meric-Bernstam, J. J. Lee and S. G. Eckhardt, Clin. Cancer Res., 2013, 19, 4305–4308 CrossRef PubMed.
- S. Turajlic and C. Swanton, Nat. Rev. Drug Discovery, 2017, 16, 441–442 CrossRef CAS PubMed.
- C. Brown, Nature, 2016, 537, S106–S108 CrossRef CAS PubMed.
- L. Vecchione, B. Jacobs, N. Normanno, F. Ciardiello and S. Tejpar, Exp. Cell Res., 2011, 317, 2765–2771 CrossRef CAS PubMed.
- J. R. Grandis and J. C. Sok, Pharmacol. Ther., 2004, 102, 37–46 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- P. J. Roberts and C. J. Der, Oncogene, 2007, 26, 3291–3310 CrossRef CAS PubMed.
- G. L. Johnson, Science, 2002, 298, 1911–1912 CrossRef CAS PubMed.
- C. M. Rocha-Lima, H. P. Soares, L. E. Raez and R. Singal, Cancer Control J. Moffitt Cancer Cent., 2007, 14, 295–304 CrossRef PubMed.
- H. Rajagopalan, A. Bardelli, C. Lengauer, K. W. Kinzler, B. Vogelstein and V. E. Velculescu, Nature, 2002, 418, 934–934 CrossRef CAS PubMed.
- C. Wellbrock, M. Karasarides and R. Marais, Nat. Rev. Mol. Cell Biol., 2004, 5, 875–885 CrossRef CAS PubMed.
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett, W. Bottomley, N. Davis, E. Dicks, R. Ewing, Y. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. C. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Seigler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton and P. A. Futreal, Nature, 2002, 417, 949–954 CrossRef CAS PubMed.
- K. Korphaisarn and S. Kopetz, Cancer J., 2016, 22, 175–178 CrossRef CAS PubMed.
- Y. Cohen, M. Xing, E. Mambo, Z. Guo, G. Wu, B. Trink, U. Beller, W. H. Westra, P. W. Ladenson and D. Sidransky, J. Natl. Cancer Inst., 2003, 95, 625–627 CrossRef CAS PubMed.
- A. Marchetti, L. Felicioni, S. Malatesta, M. Grazia Sciarrotta, L. Guetti, A. Chella, P. Viola, C. Pullara, F. Mucilli and F. Buttitta, J. Clin. Oncol., 2011, 29, 3574–3579 CrossRef CAS PubMed.
- M. Holderfield, M. M. Deuker, F. McCormick and M. McMahon, Nat. Rev. Cancer, 2014, 14, 455–467 CrossRef CAS PubMed.
- H.-C. Hsu, T. K. Thiam, Y.-J. Lu, C. Y. Yeh, W.-S. Tsai, J. F. You, H. Y. Hung, C.-N. Tsai, A. Hsu, H.-C. Chen, S.-J. Chen and T.-S. Yang, Oncotarget, 2016, 7, 22257–22270 Search PubMed.
- K. Ohashi, L. V. Sequist, M. E. Arcila, T. Moran, J. Chmielecki, Y.-L. Lin, Y. Pan, L. Wang, E. de Stanchina, K. Shien, K. Aoe, S. Toyooka, K. Kiura, L. Fernandez-Cuesta, P. Fidias, J. C.-H. Yang, V. A. Miller, G. J. Riely, M. G. Kris, J. A. Engelman, C. L. Vnencak-Jones, D. Dias-Santagata, M. Ladanyi and W. Pao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E2127–E2133 CrossRef CAS PubMed.
- J. H. Strickler, C. Wu and T. Bekaii-Saab, Cancer Treat. Rev., 2017, 60, 109–119 CrossRef CAS PubMed.
- T. B. Lowinger, B. Riedl, J. Dumas and R. A. Smith, Curr. Pharm. Des., 2002, 8, 2269–2278 CrossRef CAS PubMed.
- S. Wilhelm, C. Carter, M. Lynch, T. Lowinger, J. Dumas, R. A. Smith, B. Schwartz, R. Simantov and S. Kelley, Nat. Rev. Drug Discovery, 2006, 5, 835–844 CrossRef CAS PubMed.
- G. M. Keating and A. Santoro, Drugs, 2009, 69, 223–240 CrossRef CAS PubMed.
- S. M. Wilhelm, L. Adnane, P. Newell, A. Villanueva, J. M. Llovet and M. Lynch, Mol. Cancer Ther., 2008, 7, 3129–3140 CrossRef CAS PubMed.
- M. Beeram, A. Patnaik and E. K. Rowinsky, J. Clin. Oncol., 2005, 23, 6771–6790 CrossRef CAS PubMed.
- E. Samalin, O. Bouché, S. Thézenas, E. Francois, A. Adenis, J. Bennouna, J. Taieb, F. Desseigne, J. F. Seitz, T. Conroy, M. P. Galais, E. Assenat, E. Crapez, S. Poujol, F. Bibeau, F. Boissière, P. Laurent-Puig, M. Ychou and T. Mazard, Br. J. Cancer, 2014, 110, 1148–1154 CrossRef CAS PubMed.
- L. Thomas, S. Y. Lai, W. Dong, L. Feng, R. Dadu, R. M. Regone and M. E. Cabanillas, Oncologist, 2014, 19, 251–258 CrossRef CAS PubMed.
- R. C. Kane, A. T. Farrell, R. Madabushi, B. Booth, S. Chattopadhyay, R. Sridhara, R. Justice and R. Pazdur, Oncologist, 2009, 14, 95–100 CrossRef CAS PubMed.
- R. R. Shah, S. A. Roberts and D. R. Shah, Br. J. Clin. Pharmacol., 2013, 76, 396–411 CrossRef CAS PubMed.
- R. Brandão Moreira, R. D'Alpino Peixoto and M. Rocha de Sousa Cruz, Case Rep. Oncol., 2015, 8, 83–87 CrossRef PubMed.
- K. Martchenko, I. Schmidtmann, T. Thomaidis, V. Thole, P. R. Galle, M. Becker, M. Möhler, T. C. Wehler and C. C. Schimanski, World J. Gastroenterol., 2016, 22, 5400 CrossRef CAS PubMed.
- C. Krafft, M. Schmitt, I. W. Schie, D. Cialla-May, C. Matthäus, T. Bocklitz and J. Popp, Angew. Chem., Int. Ed., 2017, 56, 4392–4430 CrossRef CAS PubMed.
- H. J. Byrne, M. Baranska, G. J. Puppels, N. Stone, B. Wood, K. M. Gough, P. Lasch, P. Heraud, J. Sulé-Suso and G. D. Sockalingum, Analyst, 2015, 140, 2066–2073 RSC.
- K. Kong, C. Kendall, N. Stone and I. Notingher, Adv. Drug Delivery Rev., 2015, 89, 121–134 CrossRef CAS PubMed.
- M. Diem, J. M. Chalmers and P. R. Griffiths, Vibrational spectroscopy for medical diagnosis, John Wiley & Sons, Chichester, England; Hoboken, NJ, 2008 Search PubMed.
- A. F. Palonpon, M. Sodeoka and K. Fujita, Curr. Opin. Chem. Biol., 2013, 17, 708–715 CrossRef CAS PubMed.
- L. Sun, B. A. Simmons and S. Singh, Biotechnol. Bioeng., 2011, 108, 286–295 CrossRef CAS PubMed.
- I. Notingher and L. L. Hench, Expert Rev. Med. Devices, 2006, 3, 215–234 CrossRef CAS PubMed.
- I. Pence and A. Mahadevan-Jansen, Chem. Soc. Rev., 2016, 45, 1958–1979 RSC.
- H. K. Yosef, S. D. Krauß, T. Lechtonen, H. Jütte, A. Tannapfel, H. U. Käfferlein, T. Brüning, F. Roghmann, J. Noldus, A. Mosig, S. F. El-Mashtoly and K. Gerwert, Anal. Chem., 2017, 89, 6893–6899 CrossRef CAS PubMed.
- S. D. Krauß, H. K. Yosef, T. Lechtonen, H. Jütte, A. Tannapfel, H. U. Käfferlein, T. Brüning, F. Roghmann, J. Noldus, S. F. El-Mashtoly, K. Gerwert and A. Mosig, J. Chemom., 2018, 32, e2973 CrossRef.
- L. T. Kerr, K. Domijan, I. Cullen and B. M. Hennelly, Photonics Lasers Med., 2014, 3, 193–224 Search PubMed.
- I. W. Schie, J. Rüger, A. S. Mondol, A. Ramoji, U. Neugebauer, C. Krafft and J. Popp, Anal. Chem., 2018, 90, 2023–2030 CrossRef CAS PubMed.
- M. K. Hammoud, H. K. Yosef, T. Lechtonen, K. Aljakouch, M. Schuler, W. Alsaidi, I. Daho, A. Maghnouj, S. Hahn, S. F. El-Mashtoly and K. Gerwert, Sci. Rep., 2018, 8, 15278 CrossRef PubMed.
- S. F. El-Mashtoly, D. Petersen, H. K. Yosef, A. Mosig, A. Reinacher-Schick, C. Kötting and K. Gerwert, Analyst, 2014, 139, 1155–1161 RSC.
- H. K. Yosef, L. Mavarani, A. Maghnouj, S. Hahn, S. F. El-Mashtoly and K. Gerwert, Anal. Bioanal. Chem., 2015, 407, 8321–8331 CrossRef CAS PubMed.
- S. F. El-Mashtoly, H. K. Yosef, D. Petersen, L. Mavarani, A. Maghnouj, S. Hahn, C. Kötting and K. Gerwert, Anal. Chem., 2015, 87, 7297–7304 CrossRef CAS PubMed.
- K. Meister, J. Niesel, U. Schatzschneider, N. Metzler-Nolte, D. A. Schmidt and M. Havenith, Angew. Chem., Int. Ed., 2010, 49, 3310–3312 CrossRef CAS PubMed.
- H. Nawaz, F. Bonnier, P. Knief, O. Howe, F. M. Lyng, A. D. Meade and H. J. Byrne, Analyst, 2010, 135, 3070–3076 RSC.
- D. Fu, J. Zhou, W. S. Zhu, P. W. Manley, Y. K. Wang, T. Hood, A. Wylie and X. S. Xie, Nat. Chem., 2014, 6, 614–622 CrossRef CAS PubMed.
- H. Yamakoshi, K. Dodo, A. Palonpon, J. Ando, K. Fujita, S. Kawata and M. Sodeoka, J. Am. Chem. Soc., 2012, 134, 20681–20689 CrossRef CAS PubMed.
- B. Kann, H. L. Offerhaus, M. Windbergs and C. Otto, Adv. Drug Delivery Rev., 2015, 89, 71–90 CrossRef CAS PubMed.
- G. P. S. Smith, C. M. McGoverin, S. J. Fraser and K. C. Gordon, Adv. Drug Delivery Rev., 2015, 89, 21–41 CrossRef CAS PubMed.
- L. Wei, F. Hu, Y. Shen, Z. Chen, Y. Yu, C.-C. Lin, M. C. Wang and W. Min, Nat. Methods, 2014, 11, 410–412 CrossRef CAS PubMed.
- A. F. Palonpon, J. Ando, H. Yamakoshi, K. Dodo, M. Sodeoka, S. Kawata and K. Fujita, Nat. Protoc., 2013, 8, 677–692 CrossRef CAS PubMed.
- H. Nawaz, A. Garcia, A. D. Meade, F. M. Lyng and H. J. Byrne, Analyst, 2013, 138, 6177 RSC.
- Z. Farhane, F. Bonnier, O. Howe, A. Casey and H. J. Byrne, J. Biophotonics, 2018, 11, e201700060 CrossRef PubMed.
- H. Nawaz, F. Bonnier, A. D. Meade, F. M. Lyng and H. J. Byrne, Analyst, 2011, 136, 2450–2463 RSC.
- Z. Farhane, F. Bonnier and H. J. Byrne, Anal. Bioanal. Chem., 2017, 409, 1333–1346 CrossRef CAS PubMed.
- S. Hong, L. Lin, M. Xiao and X. Chen, Curr. Opin. Chem. Biol., 2015, 24, 91–96 CrossRef CAS PubMed.
- W. J. Tipping, M. Lee, A. Serrels, V. G. Brunton and A. N. Hulme, Chem. Soc. Rev., 2016, 45, 2075–2089 RSC.
- F. Hu, L. Wei, C. Zheng, Y. Shen and W. Min, Analyst, 2014, 139, 2312–2317 RSC.
- C. Matthäus, A. Kale, T. Chernenko, V. Torchilin and M. Diem, Mol. Pharm., 2008, 5, 287–293 CrossRef PubMed.
- K. Aljakouch, T. Lechtonen, H. K. Yosef, M. K. Hammoud, W. Alsaidi, C. Kötting, C. Mügge, R. Kourist, S. F. El-Mashtoly and K. Gerwert, Angew. Chem., Int. Ed., 2018, 57, 7250–7254 CrossRef CAS PubMed.
- C. Roma, A. M. Rachiglio, R. Pasquale, F. Fenizia, A. Iannaccone, F. Tatangelo, G. Antinolfi, P. Parrella, P. Graziano, L. Sabatino, V. Colantuoni, G. Botti, E. Maiello and N. Normanno, Cancer Biol. Ther., 2016, 17, 840–848 CrossRef CAS PubMed.
- Y. H. Tan, Y. Liu, K. W. Eu, P. W. Ang, W. Q. Li, M. Salto-Tellez, B. Iacopetta and R. Soong, Pathology, 2008, 40, 295–298 CrossRef CAS PubMed.
- K. Shigeta, T. Hayashida, Y. Hoshino, K. Okabayashi, T. Endo, Y. Ishii, H. Hasegawa and Y. Kitagawa, PLoS One, 2013, 8, e66302 CrossRef CAS PubMed.
- L. Mavarani, D. Petersen, S. F. El-Mashtoly, A. Mosig, A. Tannapfel, C. Kötting and K. Gerwert, Analyst, 2013, 138, 4035–4039 RSC.
- D. Petersen, L. Mavarani, D. Niedieker, E. Freier, A. Tannapfel, C. Kötting, K. Gerwert and S. F. El-Mashtoly, Analyst, 2017, 142, 1207–1215 RSC.
- N. Ke, X. Wang, X. Xu and Y. A. Abassi, in Mammalian Cell Viability, ed. M. J. Stoddart, Humana Press, Totowa, NJ, 2011, vol. 740, pp. 33–43 Search PubMed.
- C. Fucile, S. Marenco, M. Bazzica, M. L. Zuccoli, F. Lantieri, L. Robbiano, V. Marini, P. Di Gion, G. Pieri, P. Stura, A. Martelli, V. Savarino, F. Mattioli and A. Picciotto, Med. Oncol., 2015, 32, 335 CrossRef PubMed.
- S. Kustermann, F. Boess, A. Buness, M. Schmitz, M. Watzele, T. Weiser, T. Singer, L. Suter and A. Roth, Toxicol. In Vitro, 2013, 27, 1589–1595 CrossRef CAS PubMed.
- D. T. Leicht, V. Balan, A. Kaplun, V. Singh-Gupta, L. Kaplun, M. Dobson and G. Tzivion, Biochim. Biophys. Acta, Mol. Cell Res., 2007, 1773, 1196–1212 CrossRef CAS PubMed.
- H. J. Hülsmann, J. Rolff, C. Bender, M. Jarahian, U. Korf, R. Herwig, H. Fröhlich, M. Thomas, J. Merk, I. Fichtner, H. Sültmann and R. Kuner, Lung Cancer, 2014, 86, 151–157 CrossRef PubMed.
- J. Boren and K. M. Brindle, Cell Death Differ., 2012, 19, 1561–1570 CrossRef CAS PubMed.
- J.-J. Max and C. Chapados, J. Chem. Phys., 2009, 131, 184505 CrossRef PubMed.
- C. Lathia, J. Lettieri, F. Cihon, M. Gallentine, M. Radtke and P. Sundaresan, Cancer Chemother. Pharmacol., 2006, 57, 685–692 CrossRef CAS PubMed.
- S. Joshita, K. Yoshizawa, K. Sano, S. Kobayashi, T. Sekiguchi, S. Morita, A. Kamijo, M. Komatsu, T. Umemura, T. Ichijo, A. Matsumoto and E. Tanaka, Intern. Med., 2010, 49, 991–994 CrossRef PubMed.
- H. G. Edwards, D. Brown, J. Dale and S. Plant, J. Mol. Struct., 2001, 595, 111–125 CrossRef CAS.
- D. P. Elder, E. Delaney, A. Teasdale, S. Eyley, V. D. Reif, K. Jacq, K. L. Facchine, R. S. Oestrich, P. Sandra and F. David, J. Pharm. Sci., 2010, 99, 2948–2961 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an02029a |
| This journal is © The Royal Society of Chemistry 2018 |