
RNA chaperone assisted intramolecular annealing reaction towards oligouridylated RNA detection in cancer cells†
Kai
Zhang
ab,
Xue-Jiao
Yang
a,
Ting-Ting
Zhang
a,
Xiang-Ling
Li
*a,
Hong-Yuan
Chen
a and
Jing-Juan
Xu
 *a
*a
aState Key Laboratory of Analytical Chemistry for Life Science and Collaborative Innovation Center of Chemistry for Life Sciences, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: xlli@nju.edu.cn; xujj@nju.edu.cn
bKey Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi, Jiangsu 214063, China
First published on 22nd October 2018
Abstract
Proximity induced intramolecular nucleotide strand displacement, which can be simply performed in a single tube or in a complex cellular environment, is one of the key mechanisms for the detection of biological targets, especially for significant genetic molecules. The host factor for RNA phage Qb replication (Hfq), with two distinct single stranded RNA binding sites, has excellent properties as an affinity ligand in a proximity induced reaction. In this research, a versatile RNA chaperone-Hfq assisted RNA annealing strategy for the sensitive detection of the intermediate product, oligouridylated RNA, in a genetic regulation process was developed. Benefiting from the high binding affinity of Hfq for the probe and the target, the sensitive determination of oligouridylated RNA in cell lysis and human cervical cancer (HeLa) cells was successfully achieved. This study has also revealed that the Hfq assisted RNA annealing strategy can be further extended and applied in specific microRNA analysis, and RNA related tumorigenicity and disease diagnosis.
Introduction
Let-7, one of the first microRNAs to be discovered, acts as a cancer suppressor in many human malignant tumors, and can control the cell cycle, terminal differentiation and also cancer progression.1–4 Recent reports have revealed that RNA binding proteins – Lin28 proteins – can cause the downregulation of let-7 miRNA, which is associated with human diseases, especially with the development and progression of cancer.5 During the regulation process, the intermediate product oligouridylated pre-let-7 (OUP let-7) is resistant to dicer processing, and rapidly degraded by RNase, which inhibits the maturity of let-7 and suppresses the expression level.5–7 Obviously, the amount of intermediate product OUP let-7 can not only be associated with the expression level of Lin28 binding proteins, but also correlated with the degree of downregulation of let-7. Therefore, sensitive quantification of oligouridylated pre-let-7 transcripts is of great significance for early tumorigenesis analysis and gene regulation, and can be further used to facilitate the discovery of disease pathogenesis, evaluation of diseased states, selection of gene expression for initial therapy and monitoring of treatment progress.Lately, proximity dependent surface hybridization has enabled the ultrasensitive detection of various biomolecules.8 During this process, two oligonucleotide reagents coordinated and proximally bound to a single protein molecule, which remarkably increased the local concentrations of reagents, formed an intermolecular reaction and then significantly improved the reaction rate even at low concentrations.9–11 Recent work has revealed that the host factor for RNA phage Qb replication (Hfq), as one of the excellent RNA chaperones in RNA annealing,12–15 exhibits two distinct single stranded RNA binding sites for A/U-rich RNA sequences, which can facilitate the interaction between two RNA strands, form a transient ternary complex and increase the helix hybridization rate.6,16–20 This unique property suggests that the RNA chaperone Hfq is well-equipped for functional involvement in a proximity ligation assay.
In this research, the RNA chaperone-Hfq was used to assist intramolecular RNA annealing for highly sensitive analysis of oligouridylated pre-let-7 without any extra amplification procedure. Gold nanoparticles (AuNPs) acted as a scaffold for the assembly of capture RNAs (RNA1) and signal-output elements (RNA2). Target recognition and a strand displacement reaction were achieved by the two specific affinity bindings of capture RNA and the target to Hfq. The coupling of Hfq assisted RNA annealing with the fluorescence-quenching property of AuNPs enabled the strategy to detect genetic regulation product oligouridylated pre-let-7 RNA at a low concentration in a convenient format. This method has high sensitivity, low cost, rapidity, and simplicity, which meets the requirements for practical clinical analysis. To the best of our knowledge, such a general and versatile RNA chaperone assisted fluorescence strategy toward an oligouridylated RNA transcript assay has never been reported.
The detailed principle of the OUP let-7 assay is illustrated in Scheme 1. A thiol group terminated nucleotide (RNA1) hybridized with a fluorophore (Cy3) labeled nucleotide (RNA2) and was assembled on the AuNP for the formation of the capture probe. The fluorescence was efficiently quenched because of the proximity of the fluorophores (Cy3) to the AuNP surface. In the presence of the target OUP RNA and Hfq chaperone, target OUP RNA/Hfq chaperone complex and RNA1/Hfq, the chaperone can be formed because of the reaction assistor Hfq with two distinct RNA binding faces (the structure of an Hfq/RNA complex is shown in Fig. S1 ESI†). Subsequently, the target OUP let-7 displaced the RNA2 because of the binding induced intramolecular strand displacement reaction. As a result, fluorophores RNA2 were released and induced the recovery of the fluorescence signal thus, achieved the quantitative analysis of OUP RNA.
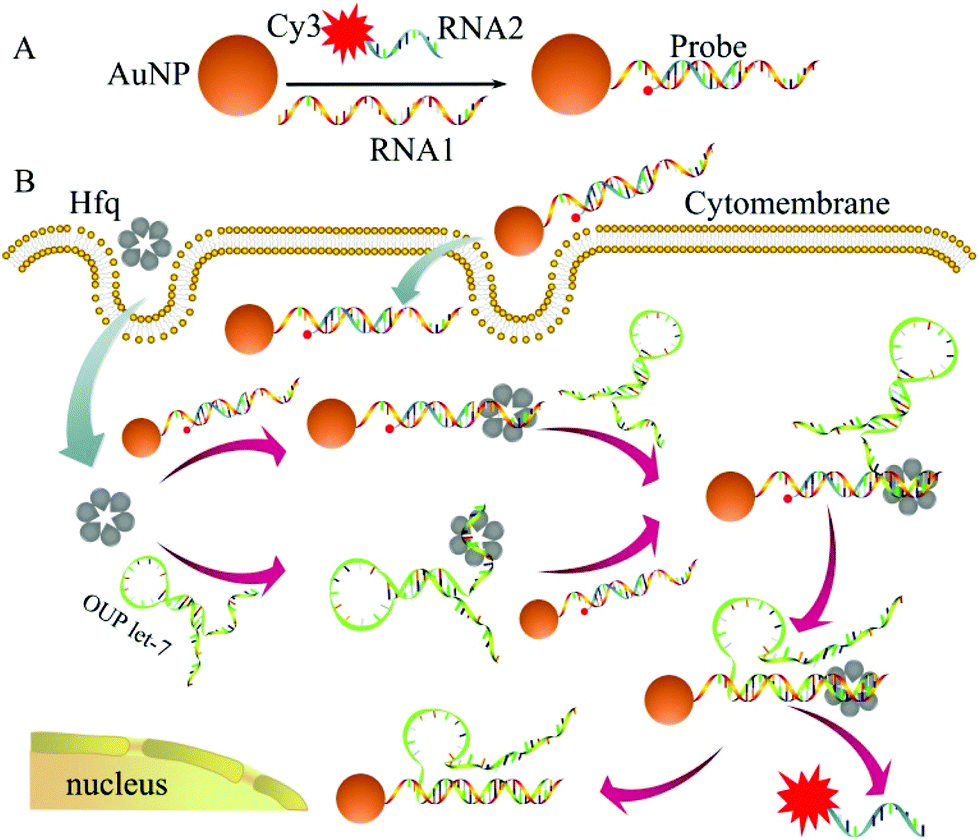 | ||
| Scheme 1 Schematic illustration of the assembly of probes (A) and the RNA chaperone, Hfq assisted oligouridylated RNA (OUP let-7) analysis (B). | ||
Results and discussion
The transmission electron microscopy (TEM) images, dynamic light scattering (DLS) measurement and the ultraviolet-visible (UV–vis) absorption were used to characterize the prepared AuNPs and the results are shown in Fig. 1. The ζ-potential of the synthesized AuNPs was determined to be −3.65 mV. When the thiol group terminated RNA1 was modified on the AuNPs, the ζ potential fell to −9.42 mV because the nucleic acids contained more negative phosphate groups (Fig. 1D).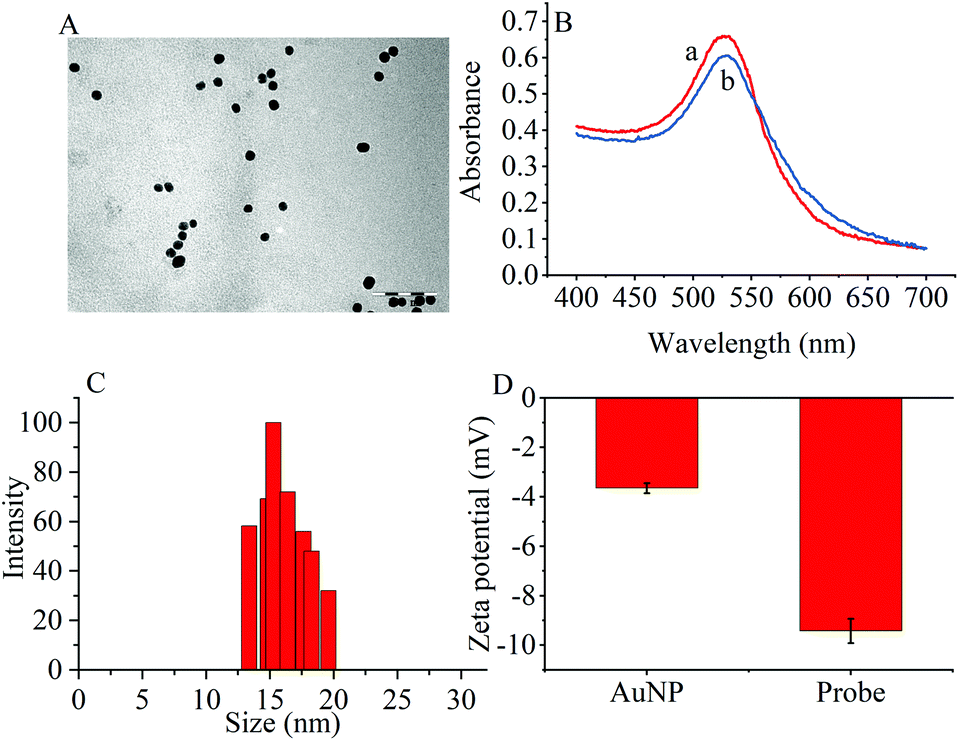 | ||
| Fig. 1 (A) TEM image of (AuNPs (scale bar: 100 nm). (B) UV-vis spectra of (a) AuNPs and (b) the probes. (C) DLS of AuNPs. (D) Zeta potentials of AuNPs and the probes. | ||
To prove the capability of the design, the recovery of the fluorescence intensities was measured in the presence of the target OUP let-7 and Hfq and the results are shown in Fig. 2A. The fluorescence intensities were greatly increased and reached a plateau at approximately 30 s. This outcome indicated that the RNA2 was efficiently displaced by the target with the help of Hfq. Meanwhile, the blank reaction without the assisting factor Hfq, showed in comparison a very marginal increase in fluorescence signal, because of the low hybridization activity of RNA 1 and the target OUP RNA.
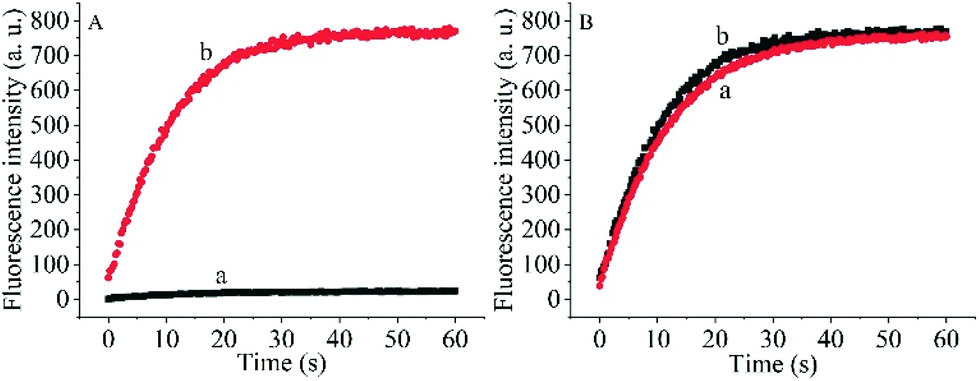 | ||
| Fig. 2 (A) The real-time fluorescence intensity of the probe and 5 μM target OUP let-7 incubated without Hfq (a) and with 5 μM Hfq (b) in PBS buffer solution. (B) The comparison of Hfq activity on the AuNPs (a) and in solution (b). | ||
Additionally, to determine whether the Hfq activity was negatively affected by the immobilization of RNA 1 on the nanoparticle surface further studies were made. A model system in solution was created by hybridizing the BHQ2 (a quencher for Cy3) modified RNA1 (Q-RNA1) with Cy3 labelled RNA2 and without assembly on the surface of the AuNPs. The target recognition activity of AuNP-immobilized substrates was compared with that of substrates in solution (Fig. 2B). The results revealed that Hfq was able to process the immobilized RNA duplexes equally well. All the results indicated that the scaffold and signal quencher-AuNPs had no negative impacts on Hfq assisted RNA detection and showed promise for the development of other related probes.
In addition to general fluorescence analysis conditions, the RNA chaperone Hfq concentration was crucial to achieve the optimal detection performance. The fluorescence intensities of different concentrations of Hfq and target OUP let-7 incubated with the probes at 30 s were measured. As shown in the results of the orthogonal experiments (Fig. 3), the fluorescence intensity was elevated with increasing concentrations of the target and the reaction assistor Hfq, and reached the highest value at a concentration of 2 μM. Then a plateau of the fluorescence intensity was reached after this concentration. Therefore, 2 μM of Hfq was taken as the optimized concentration for the RNA chaperone-Hfq in the following experiments.
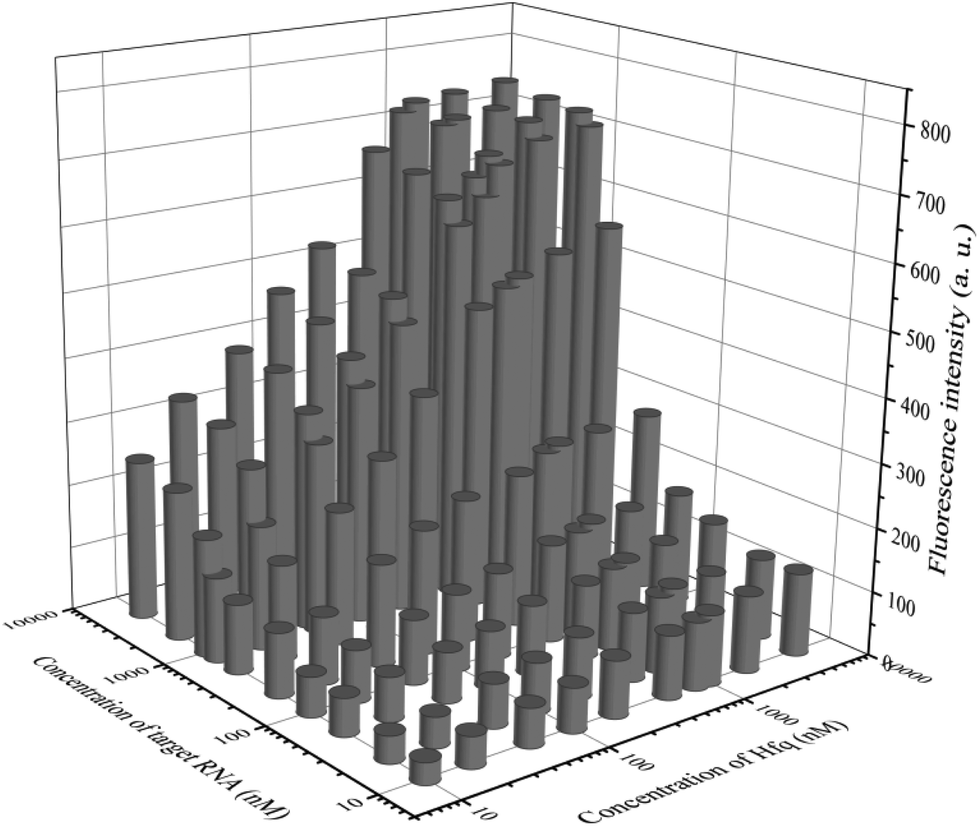 | ||
| Fig. 3 The results of orthogonal experiments of different concentrations of the target OUP RNA and Hfq. | ||
The degree of signal variation in this Hfq-assisted assay was directly related to the concentration of the target transcript. Thus, by tracking the recovery signal that monitored the amount of RNA, a new sensitive assay can be realized. As shown in Fig. 4A, the fluorescence intensity increased monotonically with the concentration of target OUP let-7, indicating that the target recognition reaction can displace and release fluorophore labelled RNA2, and then induce the recovery of the fluorescence signal and thus, quantify the oligouridylated RNA. Furthermore, a large dynamic range from 0 to 5 μM was obtained. The fluorescence intensity (at 30 s) exhibited a linear correlation to the concentration of target OUP let-7 from 0 to 800 nM with a relationship of Y = 0.788X + 27 (R2 = 0.9881), where Y is the fluorescence intensity and X is the concentration of Hfq (Fig. 4B). The detection limit of this assay, three times the standard deviation of the signal from the control test, was calculated to be 10.22 nM. This low detection limit might be attributed to the affinity binding of the target RNA and the capture RNA to one reaction assister, Hfq, which remarkably increased the local concentration of reagents and induced an intramolecular strand displacement reaction with a high ratio.
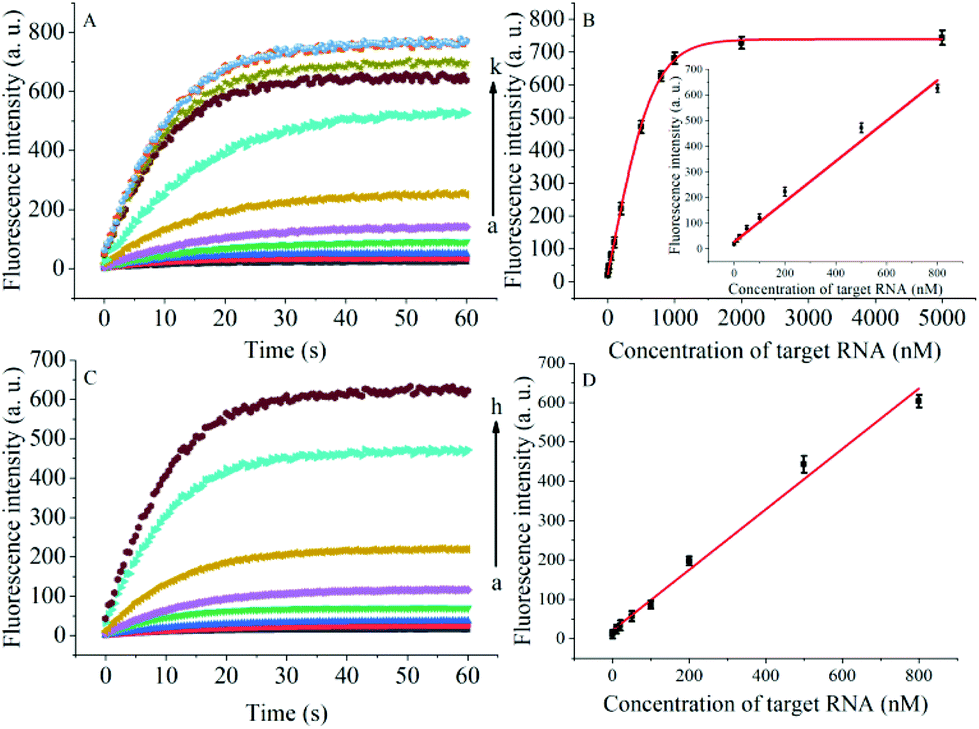 | ||
| Fig. 4 (A) RNA detection with different concentrations of target OUP let-7, from (a) to (k): 0 nM, 10 nM, 20 nM, 50 nM, 100 nM, 200 nM, 500 nM, 800 nM, 1 μM, 2 μM and 5 μM, respectively. (B) The relationship of the fluorescence intensity and the concentration of the target RNA. Inset: Linear relationship of fluorescence intensity and the concentration of target RNA over the concentration range from 0 to 800 nM. (C) Real-time assay of RNA annealing in HeLa cell lysates with different concentrations of the target OUP let-7: from (a) to (h): 0 nM, 10 nM, 20 nM, 50 nM, 100 nM, 200 nM, 500 nM, and 800 nM, respectively. (D) Linear relationship of the linear concentration range from 0 to 800 nM. | ||
To be useful in the bioassays, the developed probe should be able to tolerate the interference from the biological samples. Therefore, the feasibility of the RNA assay system in human cervical cancer (HeLa) cell lysates was investigated. As shown in Fig. 4C and D, the variance ratio of the fluorescence intensity increase was proportional to the target concentration with the linear range from 0 to 800 nM, and the detection limit was experimentally found to be 34.5 nM, which is somewhat higher than that in pure buffer. This might result from the reduction of Hfq activity or the RNA–RNA binding affinity in complex cell lysates. The box plot analysis, containing median, interquartile range, and individual data values, was performed for both the summary statistics and the distribution of the primary data acquired from a series of oligouridylated RNA detection (Fig. S2, ESI†). The result indicated the high stability and reproducibility of the Hfq assisted intramolecular reaction for the oligouridylated RNA analysis. Furthermore, other RNA analogues were used to investigate the selectivity of this analysis protocol. As shown in Fig. S3 (ESI†), only the presence of the target OUP let-7 induced a significant fluorescence signal change, which strongly suggested that the smart detection system had high selectivity in the detection of oligouridylated RNA.
Oligouridylated RNA detection in a complex cellular microenvironment was significant for practical applications. Firstly, the biocompatibility of the probe was tested using a cell viability assay. The results revealed that the cell viability was high and up to 90% even after incubation with the probes and no significant difference (P > 0.05) in cell viability was observed between the groups (Fig. S4, ESI†), which confirmed that the probe was an excellent candidate for intracellular assays. The OUP let-7 assays were then carried out in cells. HeLa cells were firstly transferred with plasmids for expressing OUP let-7 (the plasmid map is shown in Fig. S5, ESI†). Then these cells were divided into two sets, the first set was just incubated with the probe and the second set was incubated with the probe and Hfq. No obvious fluorescence signal was obtained in the first set even after leaving for 1.5 h, suggesting that the probe was stable in cell cytoplasm and no RNA recognition reaction occurred without Hfq (Fig. 5). In contrast, intense red fluorescence intensity was observed in the second set, demonstrating that this Hfq assisted RNA assay had the capability of imaging oligouridylated RNA with high sensitivity in a complex cellular microenvironment, which is beneficial for RNA annealing research, gene expression analysis and clinical diagnosis.
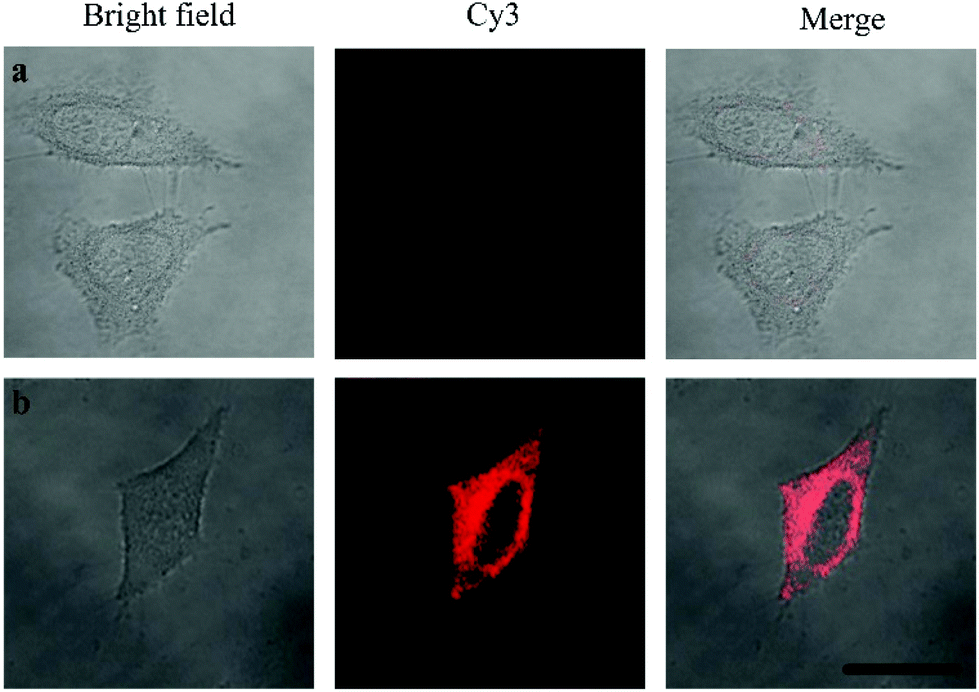 | ||
| Fig. 5 Confocal laser scanning microscopy imaging of pretreated HeLa cells incubated without Hfq (top) and with Hfq (bottom). | ||
Conclusions
In summary, we have developed a new strategy of an RNA chaperone-Hfq assisted RNA annealing reaction for sensitive detection of oligouridylated RNA. Due to the high affinity and specificity of Hfq for RNAs, this method exhibits extremely high sensitivity for the RNA assay even at low concentrations. More significantly, the Hfq assisted RNA assay also performed well in cell lysis and cancer cells, and gave a new perspective on the development of a highly sensitive detection system for genetic molecules, which holds great potential for further applications in genetic diagnostic research, cancer susceptibility and RNA related tumorigenicity.Experimental
The assembling of gold nanoparticle (AuNPs) probes
The AuNPs were prepared as described in a previous report.21 After heating 200 mL of chloroauric acid (HAuCl4) solution (0.01%) to 100 °C in round-bottomed flask, 5.0 mL of trisodium citrate (1%) was quickly added to the boiling solution under continuous stirring. The reaction mixture was stirred at 100 °C for 1 h until the color turned deep wine red and then stored at 4 °C for future use.To reduce the disulfide linkage of the thiol modified RNA1, 10 μL of 100 μM of RNA 1 was mixed with 1.5 μL of phosphate buffered saline (PBS) buffer containing 10 mM of tris(2-carboxyethyl)phosphine (TCEP, pH 7.4), 136.7 mM sodium chloride (NaCl), 2.7 mM potassium chloride (KCl), 8.72 mM disodium phosphate (Na2HPO4) 1.41 mM monopotassium phosphate (KH2PO4) 0.1% diethyl pyrocarbonate (DEPC), and 1 U μL−1 of (RNasin). After 1 h, the reduced RNA1 and 10 μL of RNA2 (100 μM) were added to 1 mL of AuNP solution in a clean glass vial with gently shaking. The vial was shaken at room temperature overnight. Afterwards, 0.1 mL of PBS solution containing 2 M of NaCl was added stepwise to the mixture (10-times) for stabilizing the probe obtained. Then the solution was centrifuged and washed with PBS buffer, and finally resuspended in 1 mL of PBS buffer.
The preparation of Hfq
Untagged Escherichia coli Hfq65, were over-expressed in E. coli BL21(DE3)Dhfq::cat-sacB cells grown in 1 L of LB Miller media (10 g L−1 tryptone, 10 g L−1 NaCl, 5 g L−1 yeast extract) supplemented with 100 mg mL−1 ampicillin until an OD600 of 0.6 at 37 °C was reached. The cells were collected using centrifugation at 5000g for 10 min. The plasmids for over-expression of Hfq proteins were created using site directed mutagenesis of plasmid pET21b-Hfq. The resuspended cell lysates of Hfq65 were clarified using ammonium sulfate precipitation after heat treatment, and the protein was purified using hydrophobic interaction chromatography. Finally, the Hfq65 were purified using cation-exchange chromatography to remove the nucleic acids. The eluate was dialyzed into a cation load buffer and concentrated using ultracentrifugation before storage at −80 °C in preparation for the next experiments.OUP let-7 in vitro analysis
For sensitive and selective detection of the target molecule, different concentrations of the target OUP Let-7 and Hfq were added into the probe reaction mixture and the final reaction volume of 100 μL in PBS buffer (pH 7.4, 136.7 mM NaCl, 2.7 mM KCl, 8.72 mM Na2HPO4, 1.41 mM KH2PO4, 0.1% DEPC, and 1 U μL−1 RNasin) was kept at 37 °C, and the fluorescence intensities of the reaction mixture were measured using spectrofluorophotometer.Cell culture and OUP RNA analysis in cancer cells
HeLa cells were cultured in a flask containing Dulbecco's modified Eagle media (DMEM, Gibco) supplemented with 10% fetal bovine serum, penicillin (100 μg mL−1), and streptomycin (100 μg mL−1) at 37 °C in a humidified atmosphere containing 5% carbon dioxide. The cell number was determined using a Petroff-Hausser cell counter (Hausser Scientific, PA, USA).The cDNA (the structure of cDNA is shown in Fig. S3, ESI†) of OUP RNA was cloned into a pcDNA 3.1(−) vector according to the manufacturer's instructions by using the endonuclease cutting sites of BmtI and HindIII. The plasmids were then transfected into cells, according to the manufacturer's instructions, using Lipofectamine 3000 (Invitrogen, Grand Island, NY, USA) in a 6-well plate for 24 h with 1 × 106 cells per well.
After washing twice with DEPC-treated PBS, the plasmid transfected cells were incubated with 25 μL of probe in 100 μL of culture medium for 1.5 h. The cells were then washed with 4% (w/v) paraformaldehyde in 10 mM PBS at room temperature for 20 min. After fixation, the cells were washed twice with DEPC treated PBS buffer and then dehydrated in a series of 70%, 85% and 99.5% solutions of ethanol for 3 min in each solution. Then, the fixed cells were washed twice with PBS at room temperature. The cells were divided into two sets: the control set was incubated without RNA chaperone-Hfq, and the experimental set was incubated with Hfq. Then the confocal fluorescence assay of each group was recorded using a TCS SP5 confocal laser scanning microscope (Leica, Germany).
For the cytotoxicity assay, HeLa cells were incubated with different concentrations of probe for various times, and then the incubation solution was replaced with fresh medium and the cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay after 48 h incubation. The statistical software, SPSS (version 24.0) and Student's t test were used for statistical analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the National Natural Science Foundation (Grants 21327902, 21605072, 21705061) of China. This work was also supported by a Project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- J. Takamizawa, H. Konishi, K. Yanagisawa, S. Tomida, H. Osada, H. Endoh, T. Harano, Y. Yatabe, M. Nagino, Y. Nimura, T. Mitsudomi and T. Takahashi, Cancer Res., 2004, 64, 3753 CrossRef CAS PubMed.
- S. M. Johnson, H. Grosshans, J. Shingara, M. Byrom, R. Jarvis, A. Cheng, E. Labourier, K. L. Reinert, D. Brown and F. J. Slack, Cell, 2005, 120, 635 CrossRef CAS PubMed.
- Y. Cheng, L. Dong, J. Zhang, Y. Zhao and Z. Li, Analyst, 2018, 143, 1758 RSC.
- L. Wang, Y. Cheng, H. Wang and Z. Li, Analyst, 2012, 137, 3667 RSC.
- I. Heo, C. Joo, J. Cho, M. Ha, J. Han and V. N. Kim, Mol. Cell, 2008, 32, 276 CrossRef CAS PubMed.
- M. J. Ellis, R. S. Trussler and D. B. Haniford, Mol. Microbiol., 2015, 96, 633 CrossRef CAS PubMed.
- H.-M. Chang, R. Triboulet, J. E. Thornton and R. I. Gregory, Nature, 2013, 497, 244 CrossRef CAS PubMed.
- H. Zhang, F. Li, B. Dever, X.-F. Li and X. C. Le, Chem. Rev., 2013, 113, 2812 CrossRef CAS PubMed.
- S. Fredriksson, M. Gullberg, J. Jarvius, C. Olsson, K. Pietras, S. M. Gustafsdottir, A. Ostman and U. Landegren, Nat. Biotechnol., 2002, 20, 473 CrossRef CAS PubMed.
- F. Li, Y. Tang, S. M. Traynor, X.-F. Li and X. C. Le, Anal. Chem., 2016, 88, 8152 CrossRef CAS PubMed.
- J. Chen, B. Deng, P. Wu, F. Li, X.-F. Li, X. C. Le, H. Zhang and X. Hou, Chem. Commun., 2016, 52, 1816 RSC.
- A. Santiago-Frangos, J. R. Jeliazkov, J. J. Gray and S. A. Woodson, eLife, 2017, 6, e27049 CrossRef PubMed.
- T. A. Geissmann and D. Touati, EMBO J., 2004, 23, 396 CrossRef CAS PubMed.
- Y. Cao, X. Li, F. Li and H. Song, ACS Synth. Biol., 2017, 6, 1679 CrossRef CAS PubMed.
- I. Moll, T. Afonyushkin, O. Vytvytska, V. R. Kaberdin and U. Blasi, RNA, 2003, 9, 1308 CrossRef CAS PubMed.
- S. Panja, R. Paul, M. M. Greenberg and S. A. Woodson, Angew. Chem., Int. Ed., 2015, 54, 7281 CrossRef CAS PubMed.
- L. Fantappiè, F. Oriente, A. Muzzi, D. Serruto, V. Scarlato and I. Delany, Mol. Microbiol., 2011, 80, 507 CrossRef PubMed.
- M. Beich-Frandsen, B. Večerek, P. V. Konarev, B. Sjöblom, K. Kloiber, H. Hämmerle, L. Rajkowitsch, A. J. Miles, G. Kontaxis, B. A. Wallace, D. I. Svergun, R. Konrat, U. Bläsi and K. Djinović-Carugo, Nucleic Acids Res., 2011, 39, 4900 CrossRef CAS PubMed.
- T. Soper, P. Mandin, N. Majdalani, S. Gottesman and S. A. Woodson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9602 CrossRef CAS PubMed.
- H. Otaka, H. Ishikawa, T. Morita and H. Aiba, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13059 CrossRef CAS PubMed.
- K. Zhang, X.-J. Yang, W. Zhao, M.-C. Xu, J.-J. Xu and H.-Y. Chen, Chem. Sci., 2017, 8, 4973 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an01662c |
| This journal is © The Royal Society of Chemistry 2019 |