
Selectivity enhancement of amperometric nitric oxide detection via shape-controlled electrodeposition of platinum nanostructures†
Sohee
Kim‡
,
Yejin
Ha‡
,
Su-jin
Kim
,
Chongmok
Lee
 * and
Youngmi
Lee
*
* and
Youngmi
Lee
*
Department of Chemistry and Nano Science, Ewha Womans University, Seoul, 03670, Republic of Korea. E-mail: youngmilee@ewha.ac.kr; cmlee@ewha.ac.kr
First published on 19th October 2018
Abstract
Nitric oxide (NO) is a biologically multifunctional gaseous signaling molecule. For electrochemical NO detections, complex membranes are commonly adopted to acquire the selectivity for NO over other oxidizable biological species. In this study, we demonstrate the improved selectivity in amperometric NO measurements at nanostructured Pt. The Pt layers were electrodeposited on Au substrate electrodes at a constant potential (−0.2 V vs. Ag/AgCl) with a constant deposition charge (0.08 C). The various distinctive nanostructures of Pt deposits were obtained via either changing the precursor concentrations (from 5 to 75 mM K2PtCl4) or using a different precursor (75 mM H2PtCl6). With a higher K2PtCl4 concentration, the Pt deposition became less sharp and the smoothest Pt was deposited with 75 mM H2PtCl6. The most greatly sharp-pointed nanostructures were generated with the lowest precursor concentration (5 mM K2PtCl4) and exhibited the highest sensitivity, which was attributed to the hydrophobic property of sharply nanostructured Pt. A hydrophobic neutral gas molecule, NO, possibly has a more favorable access to the inner surface of more hydrophobic Pt deposition and eventually increases the oxidation current. NO current sensitivity was enhanced at the more hydrophobic Pt surface, whereas the oxidation currents of acetaminophen, L-ascorbic acid, nitrite and hydrogen peroxide, four oxidizable biological interfering species, were independent of the Pt nanostructure. Conclusively, the enhanced amperometric selectivity to NO was achieved by the simple electrodeposition method without any additional membranes.
1. Introduction
Nitric oxide (NO) plays various biological/physiological functions as a vital gaseous signaling molecule in biological systems involving neurotransmission,1 vasodilation,2 inflammation3 and immune response in both mammalian and non-mammalian species. The accurate detection and quantification of NO are required to understand its biological roles more thoroughly. Since NO, which is a free radical, reacts very quickly4 and easily with other molecules such as oxygen,5 peroxides, and metals,6 real-time analytical monitoring of NO in vivo and in vitro is critical. NO measurements have been performed mainly via spectroscopic (e.g., chemiluminescence,7,8 fluorescence,9,10 and electron paramagnetic resonance (EPR) spectroscopy11,12) and electrochemical methods.13,14 While direct and sensitive measurements can be achieved with chemiluminescence or EPR, these techniques have disadvantages such as they cannot be easily used for the analysis of complex samples such as blood or wound fluid.15 In contrast, electrochemical sensors allow the continuous, precise, and real-time in situ analysis of NO with a high spatio-temporal resolution.15In an electrochemical (particularly amperometric) NO sensor, the sensor working electrode is applied with an appropriate constant potential at which NO is electrochemically oxidized, and the induced anodic current proportional to the NO concentration is measured as a responsive signal. Therefore, any species in biological samples that are oxidizable at the NO sensor applied potential can interfere with the selective amperometric sensing of NO. The most commonly encountered interfering species are nitrite, ascorbic acid (AA), uric acid, acetaminophen (AP), dopamine (DA), norepinephrine, and serotonin. To attain the selectivity for NO, the sensors generally employ one or more membranes on the sensor working electrodes. For example, selective membranes such as chloroprene, PTFE (polytetrafluoroethylene),16 fluorinated xerogel17,18 and Nafion19,20 were used for fabricating NO sensors in recent years. The sensors obtain the selectivity to NO with these permselective membranes through which the passage of general interferents are restricted via electrostatic repulsion, hydrophobicity or size exclusion. However, the adoption of these selective membranes not only makes the sensor fabrication process lengthy and more complicated, but it also causes a longer sensor response time by providing additional diffusional barrier, even to NO.
Metal nanostructures effectively modify the surface characteristics depending on the kinds of metals, structures, and dimensions.21 Although plenty of methods have been reported to synthesize metal nanostructures including wet synthesis, vapor deposition methods, template assisted methods, etc.,22,23 electrodeposition is one of the simplest and the most convenient methods for synthesizing nanostructures directly on the electrode surface.21 Many researches have been devoted to fabricate various shapes of nanostructures such as dendrites,24 flowers,25 and needles26 using the electrodeposition method by controlling the type of precursors, precursor concentrations, deposition charges, etc.
The regulation of surface wettability is of great interest in many fields such as the development of self-cleaning materials and microfluidic systems. The introduction of solid surface roughness is known to alter its wettability drastically.27 Accordingly, an electrodeposited metal with a rough surface nanostructure is rationally thought to exhibit a different wettability from that of the flat bulk metal. In fact, tree-like Pt nanostructures28 and nanoflake Pd structures21 were fabricated via electrodeposition with proper precursor concentration and deposition charge; and these nanostructures were reported to show superhydrophobicity. Taking note of these electroplated superhydrophobic metal nanostructures, in our current study, we demonstrate the feasibility of selective amperometric NO measurements without any additional membranes. The hydrophobicity of highly roughened electrode surfaces is conceived to simply enhance the selectivity to hydrophobic neutral NO over general biological interferents that are polar or charged. In fact, various nanostructured Pt layers are electrodeposited, and the currents induced by NO oxidation over other oxidizable interferents are investigated depending on the actual nanostructures along with the corresponding hydrophobicity.
2. Experimental
2.1. Chemicals and apparatus
Potassium tetrachloroplatinate(II) (K2PtCl4), chloroplatinic acid solution (H2PtCl6), sulfuric acid (H2SO4), acetaminophene (AP), L-ascorbic acid (AA), sodium nitrite (NaNO2), hydrogen peroxide (H2O2), human serum albumin (HSA) and γ-globulin were supplied by Sigma-Aldrich (St Louis, MO). Phosphate buffered-saline (PBS, pH 7.4 at 25 °C) was purchased from Fisher Scientific (Pittsburgh, PA, USA). Nitric oxide and argon (Ar) gases were obtained from Dong-A Gas Co. (Seoul, Korea). All aqueous solutions were prepared with deionized water (resistivity ≥18 MΩ cm), and chemicals were analytical reagent grade and used without additional purification.Electrochemical measurements were performed using CHI 1000A multipotentiostat (CH Instruments, Inc., TX, USA). An Au disk electrode (Bioanalytical Systems, Inc., 2 mm in diameter) was used as a substrate for electrodeposition. As the reference and counter electrodes, an Ag/AgCl electrode and a Pt wire were used, respectively. Pt structures electroplated on Au electrodes were characterized by field emission scanning electron microscopy (FE-SEM, JEOL, Japan). The water contact angles on the Pt layers were measured with ImageJ (Image Processing and Analysis in Java).
2.2. Electrodeposition of Pt structures
An Au disk working electrode was mechanically polished on a microcloth with alumina powder and used as a flat Au substrate electrode. Pt electrodeposition on the Au disk electrode was carried out in aqueous solutions containing either a Pt precursor at various concentrations (5, 15, 40 and 75 mM K2PtCl4) with 0.1 M H2SO4 or only 75 mM H2PtCl6. The solutions were purged with Ar gas for 5 min before electrodeposition. Pt nanostructure was electrochemically deposited on the Au disk electrode at −0.2 V (vs. Ag/AgCl) up to deposition charge of 0.08 C except for H2PtCl6 (−0.1 V up to 0.16 C). Water contact angles (CAs) were measured using ImageJ with a drop of 1 μL deionized water placed on the electrodeposited Pt layers.2.3. Electrochemical measurements
The electrodes were calibrated using saturated NO (1.91 mM) solutions prepared by bubbling NO gas to deaerated PBS solutions (pH 7.4).29 The electrode current responses to common biological interfering species AP, AA, NO2− and H2O2 were also measured with the addition of 5 μM each species in a deaerated PBS solution in a gas-tight container. These currents were recorded at +0.75 V (vs. Ag/AgCl), which was enough to induce NO oxidation. To measure electrochemical surface areas (ESAs) of electroplated Pt electrodes, cyclic voltammetry (sweep rate, 20 mV s−1) was performed in the potential range of −0.2–+1.1 V (vs. Ag/AgCl) in 1 M H2SO4 solution. The integrated area for a hydrogen desorption region in an obtained cyclic voltammogram was divided by a conversion factor for Pt (210 μC cm−2) to estimate the ESA.303. Results and discussion
3.1. Electrodeposited Pt nanostructures depending on the precursor concentrations
For the electrodeposition of Pt, a constant potential of −0.2 V (vs. Ag/AgCl) was applied to an Au disk electrode until a deposition charge of 0.08 V was achieved. Pt nanostructures were dependent on the concentration of K2PtCl4 in the deposition solution. It was clearly observed that as the K2PtCl4 concentration decreased from 75 mM to 5 mM, a much sharper morphology was obtained (Fig. 1A–D). Although both of the Pt deposits with 5 and 15 mM K2PtCl4 showed nano-tree structures,28 the one with 5 mM precursor had a more distinct hierarchical nanostructure with two levels: relatively larger nano-tree bodies covered with many sharp leaf-spikes than that with 15 mM precursor. The deposition with the 40 mM K2PtCl4 appeared like a mixture of nano-tree and nano-flake structures, while that with the 75 mM precursor showed a simple flake-type nanostructure. In contrast, the Pt layer deposited using a different precursor, H2PtCl6 at 75 mM, produced a round-shaped smooth structure. As seen in Fig. 1E, 10 to 200 nm round Pt particles were deposited, and sharp shapes such as tree- or flake-like structures were barely observed. A doubled charge (0.16 C) was set for the deposition with 75 mM H2PtCl6 solution, since twice the number of electrons were required for the reduction to Pt metal compared to K2PtCl4. Instead of −0.2 V, an optimized potential of −0.1 V was employed for the deposition with K2PtCl4, according to a previous report.28 | ||
| Fig. 1 SEM images of electrodeposited Pt surfaces with deposition solutions containing (A) 5 mM K2PtCl4, (B) 15 mM K2PtCl4, (C) 40 mM K2PtCl4, (D) 75 mM K2PtCl4, and (E) 75 mM H2PtCl6. The applied potentials and deposition charges are (A–D) −0.2 V and 0.08 C; (E) −0.1 V and 0.16 C, respectively. | ||
The reason for different nanostructures of Pt based on the precursor concentration is unclear but can be possibly understood as in Scheme S1 (in ESI†). At Step 1, the early stage of deposition, PtCl42− is reduced to Pt particles on the Au surface. Although the reduction rate at a higher concentration is likely to be faster than at a lower one, in the cases of all samples, Pt particles fill the surface of the Au electrode primarily. At the second stage, Step 2, Pt particles are formed on the previously deposited Pt layer. With the low concentration of PtCl42− (Scheme S1A†), the pre-deposited Pt layer has less chance to meet PtCl42− to deposit Pt. The reduction rate becomes controlled by the precursor diffusion, and slow coalescence occurs with a rather faster electron transfer during the deposition.31 In this situation, the precursor reduction occurs at the outermost edge-like sharp parts of the electrode where the mass transport of the precursor is the most greatly facilitated, and it becomes harder for PtCl42− to go inside the narrow porous Pt deposits previously formed. Thus, Pt deposition made the hierarchical tree-like structure with a low concentration PtCl42− solution. With a high concentration of PtCl42− (Scheme S1B†), the reduction can occur at most parts of the surface. They fill the gaps among Pt particles and generate a relatively even Pt surface.
3.2. Tendency of water contact angle of deposited Pt nanostructures
Static water contact angles (CAs) were measured for all the deposited Pt structures (Fig. S1, in ESI†) and the CA values as a function of the used precursors are shown in Fig. 2 (n = 10). The CA was unmeasurable for the surface deposited with 5 mM PtCl42− because a drop of water never stayed, but rolled off the surface (Fig. S1A†). This clearly indicates a great hydrophobicity of the surface. Thus, in Fig. 2, a CA of 150° was assigned to this sample based on the definition of superhydrophobicity (CA > 150°).32 The CAs of the Pt deposits gradually decreased as the concentration of PtCl42− increased from 5 mM to 75 mM, and dropped further to 99° on the surface deposited with 75 mM PtCl62−. It demonstrates a tendency of hydrophobicity of the electrode surfaces, i.e., the surface with the sharper structures shows the more hydrophobic nature.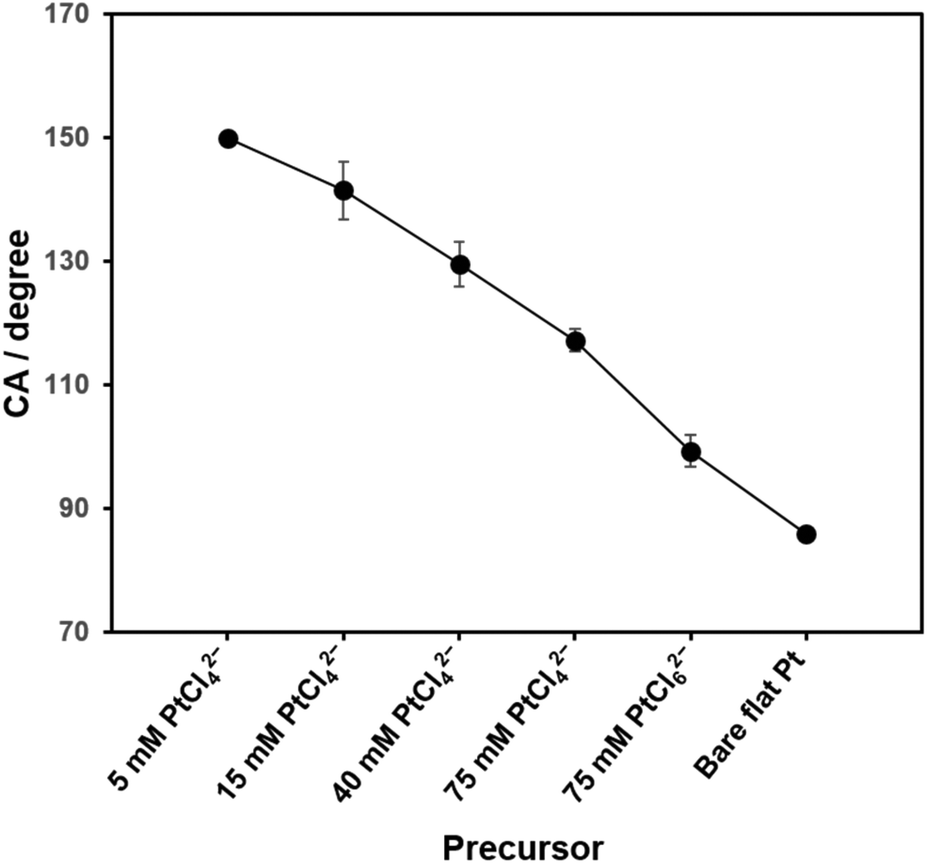 | ||
| Fig. 2 Static water contact angles of nanostructured Pt surfaces as a function of the precursor concentration. Data are represented as the means (with the error bars, standard deviations) (n = 10). | ||
In general, to obtain hydrophobicity of a metal, not only the surface is roughened but also further surface modification with a self-assembled monolayer of low-surface-energy organic molecules is applied. In the current study, electrodeposited Pt nanostructures themselves present the hydrophobicity even without additional surface modification. This is possibly attributed to the effect of airborne hydrocarbons, which reduces the surface free energy.33
There are two general hypothetic models to account for the hydrophobicity depending on the surface roughness: Winzel model describes that the increased surface area with roughening simply increases hydrophobicity and a water droplet interacting with the surface is in wet-contact mode; and Cassie model states that air can be trapped within the rough surface below a water droplet and they are in non-wet-contact mode.27,32 The Cassie situation is generally accepted for very hydrophobic or very rough surfaces where water CA is related to fractional interfacial area as in eqn (1).
cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ* = ϕwcosθ − ϕa θ* = ϕwcosθ − ϕa | (1) |
With CA of 86° on a bare flat Pt, the calculated ϕa decreases from 0.74 to 0.36 as the concentration of PtCl42− increases from 5 mM to 75 mM. Even smaller ϕa (0.24) was estimated for the Pt deposit with 75 mM PtCl62−. Especially for the Pt sample deposited with the lowest precursor concentration, the flawless hierarchical tree-like nanostructures, which had clear micro-trees surrounded by nano-leaves formed the two-level lotus-like structures and showed the superhydrophobic property.32
3.3. Selective electrochemical measurements of NO over interfering species
The Pt nanostructures deposited at various Pt precursor concentrations were used as the electrodes for the anodic measurements of NO. Fig. 3A shows the representative dynamic response curves of the electrodes to NO oxidation at an electrode potential of +0.75 V (vs. Ag/AgCl). Current densities of NO oxidation measured with various Pt electrodeposited electrodes increased along with five consecutive additions of a NO stock solution (1.91 mM) to increase the NO concentration as 0.382, 1.15, 2.67, 4.95, and 8.75 μM in a test solution. Note that the increased NO concentration (Δ[NO]) with each injection was not identical through the dynamic response curve in Fig. 3A. To eliminate the areal difference effect among the electrodes, the currents were normalized to the electrochemical surface areas (ESAs) of the electrodes, which were estimated from hydrogen adsorption/desorption.28 The measured ESAs (mean (±SD) for n = 5) were 4.8 (±0.6), 5.0 (±0.4), 4.5 (±0.5) and 4.7 (±0.6) cm2 for the Pt deposits with 5, 15, 40 and 75 mM PtCl42−, respectively. Very similar ESAs were obtained for the Pt electrodes deposited using the PtCl42− precursor, regardless of the concentration. In contrast, the one deposited with 75 mM PtCl62− showed a much greater ESA of 8.4 (±0.4) cm2. The reason for this ESA discrepancy is unclear, but it is likely to be caused by the much smoother surface morphology of Pt deposited with 75 mM PtCl62− linked to the relatively hydrophilic surface property as seen in Fig. 1E and 2.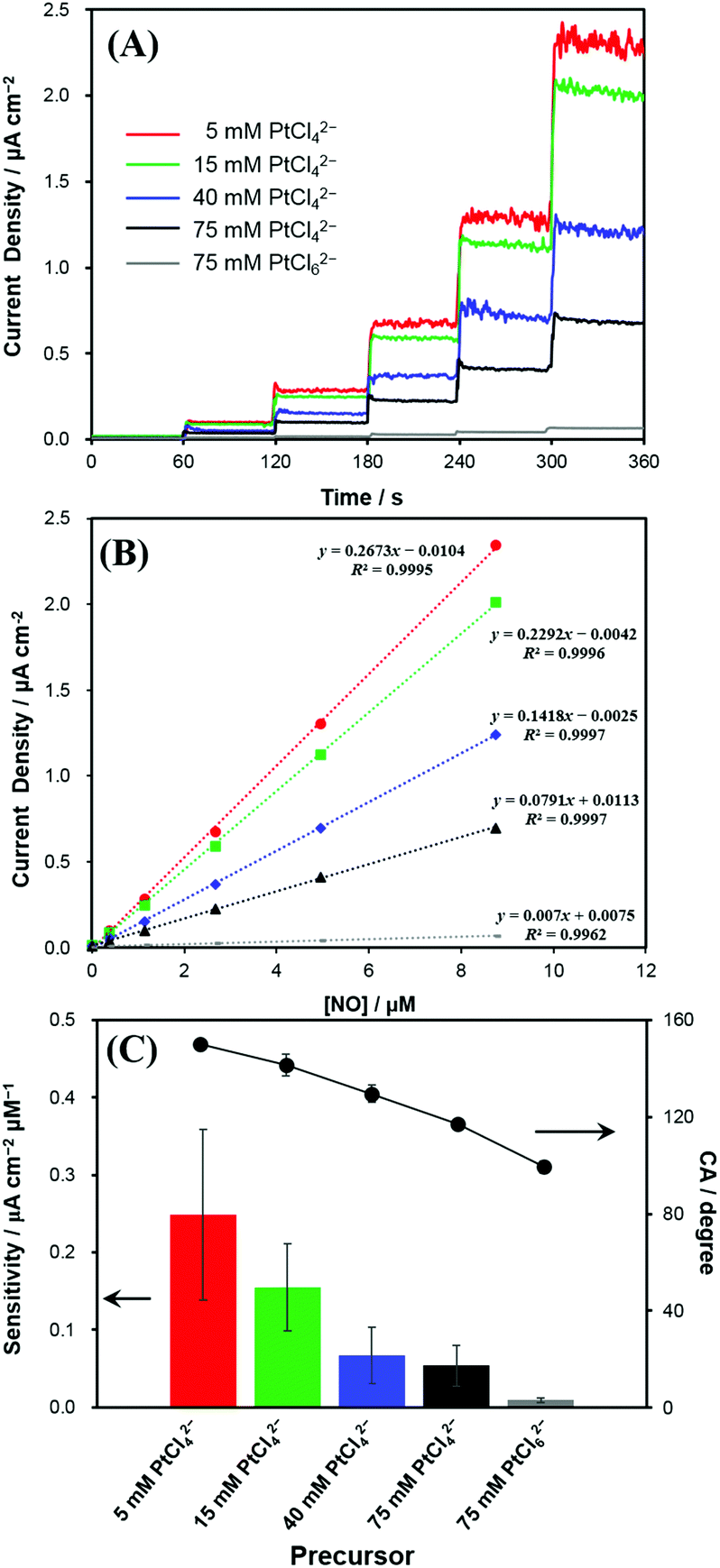 | ||
| Fig. 3 (A) Typical dynamic response curves to successive NO concentration changes (0 to 8.75 μM) obtained for variously platinized Pt electrodes. (B) Calibration curves corresponding to (A). The applied potential to the electrodes was +0.75 V vs. Ag/AgCl. (C) NO sensitivities and corresponding water contact angles for the electrodes (n = 5). | ||
In Fig. 3A, the Pt electrode deposited with 5 mM PtCl42−, the lowest concentration, exhibited the highest current-density increase, responding to the successive stepwise NO concentration increment. As the precursor concentration increases, the produced Pt electrode exhibited a smaller enhancement in the current density responding to the identical concentration increase of NO. The calibration plots showed good linearities (R2 > 0.99) between the current densities and NO concentrations in the tested concentration range (Fig. 3B). Accordingly, the NO sensitivity was the greatest for the Pt deposit with 5 mM PtCl42−, which became lower with increasing PtCl42− concentration, and then exhibited the lowest value for the Pt electrode deposited with 75 mM PtCl62− (Fig. 3C).
The sensitivity was calculated as follows:
| SNO = J/CNO | (2) |
As presented in Table S1,† the NO sensitivity differences among the electrodes were statistically significant (paired t-test). This clearly indicates that the anodic sensitivity of the electrode to NO is dependent of the electrodeposited Pt nanostructures. Interestingly, the greater sensitivity the electrode has, the sharper the morphology of the electrode becomes. This tendency of the NO sensitivity is well matched with the tendency of the electrode hydrophobicity (Fig. 3C). As the hydrophobicity of electrodes increased, current density changes to NO oxidations were significantly increased. The limit of detection (LOD) also showed a similar tendency to the sensitivity: the lowest LOD at the most hydrophobic Pt. The LOD values were 5, 6, 10, 12 and 103 nM for the Pt deposits with 5, 15, 40, 75 mM PtCl42− and 75 mM PtCl62−, respectively (S/N = 3).
For comparison, anodic current responses of the electrodes to four common biological interferents were investigated. Two charged species, AA (pKa1 = 4.25, pKa2 = 11.8)34 and NO2−; and two neutral species, AP (pKa = 9.5)35 and H2O2 (pKa = 11.7)36 present at physiological pH were tested as representative interferents. When AP, AA, NO2− and H2O2 standard solutions were injected to a PBS solution individually to achieve the bulk concentration of 5 μM, the anodic currents increased by the oxidation of interferents (insets of Fig. 4). In contrast to the case of NO (Fig. 3), the current responses of the electrodes were close to one another, regardless of the surface structures of the electrodes (Fig. 4). This observation suggests that the oxidations of interferents are not related to the Pt nanostructures, but more likely dependent on the apparent geometric surface area (GSA) of the electrode. The electrodes had very similar GSAs within a range of 0.032–0.035 cm2, slightly larger than the GSA of an Au disk substrate electrode (1 mm in radius).
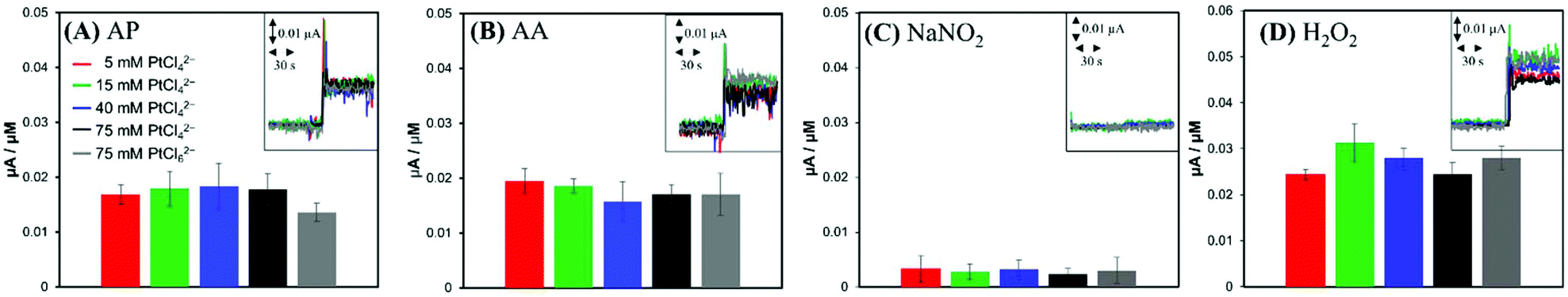 | ||
| Fig. 4 Current sensitivities (n = 5) of variously electrodeposited Pt electrodes to (A) acetaminophen (AP, 5 μM), (B) L-ascorbic acid (AA, 5 μM), (C) sodium nitrite (NaNO2, 5 μM) and (D) hydrogen peroxide (H2O2, 5 μM). Insets are current responses to AA, AP, NO2− and H2O2 at the various electrodes. | ||
Park et al. reported that a faradaic current of a reactant proceeding with a fast heterogeneous electron transfer reaction is proportional to the GSA of the electrode, irrespective of the electrode roughness, while a faradaic current for a sluggish reaction increases with the electrode ESA.37 Based on this effect, non-enzymatic amperometric measurements of glucose were proposed.37,38 Similarly, much higher anodic current induced by NO compared to that of tested interferents can be ascribed to the different rates of the electrochemical oxidations: the electrochemical oxidation of NO, known to be a kinetically controlled reaction, can utilize a larger Pt surface area including deeper layers of nanostructured Pt deposition, but AP, AA, NO2− and H2O2 are oxidized mostly at the outermost projected area of Pt deposits via the relatively faster electron transfer.
A notable finding in our current study is that the current induced by NO oxidation depends on the electrode surface nanostructures in addition to its roughness. In fact, a higher oxidation current was generated at a sharper Pt nanostructure morphology even with the similar ESAs (i.e., similar roughness). This is conceived to be related to the Pt surface hydrophobicity level. Hydrophobic neutral NO molecule possibly has a more favourable access to the inner Pt surface of the more hydrophobic Pt layer. Therefore, the selectivity for hydrophobic NO over interfering species could be further improved by enhancing the electrode surface hydrophobicity, which was achieved simply by controlling the precursor concentrations without any additional modifications.
The selectivity coefficient was determined according to eqn (3).
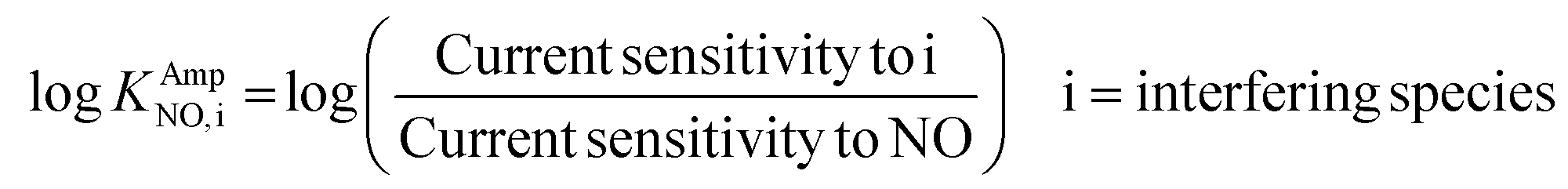 | (3) |
The obtained selectivity coefficients of various Pt electrodes, including the flat bare Pt for NO against interferents (AP, AA, NO2− and H2O2), are summarized in Table S2.† Compared to the least hydrophobic (deposited with 75 mM PtCl62−) Pt, the most hydrophobic Pt (deposited with 5 mM PtCl42−) exhibited ca. three-fold enhanced selectivity coefficients to NO over AP, AA and H2O2. To the best of our knowledge, there is no previous report on the selectivity coefficients to NO over various interfering species depending on the electrode morphology without any selective membrane. Only the selectivity coefficient of a flat bare Pt to NO over NO2− (logKAmpNO, nitrite) was estimated to be −1.51 in a previous study, which was done in a similar experimental condition to ours.39 This value is well matched with the one we measured in the current study (−1.406, Table S2†), supporting the validity of our experimental scheme.
The reasonable resistivity to protein adsorption was also confirmed. In a PBS solution (pH 7.4) containing human serum albumin (HSA, 10 mg mL−1) and γ-globulin (1 mg mL−1), the Pt deposit with 5 mM PtCl42− maintained 88% and 85% of the NO sensitivity without HSA and γ-globulin, respectively (Fig. S2†).
4. Conclusions
Variously nanostructured Pt layers were electrodeposited by changing the precursor concentration (from 5 to 75 mM K2PtCl4) or using a different precursor (75 mM H2PtCl6). From 75 mM to 5 mM of PtCl42−, the nanostructures of Pt varied from nano-flake to hierarchical nano-tree structures and 75 mM H2PtCl6 produced a relatively smooth deposition. Depending on the actual morphology, the Pt deposits showed different hydrophobicity: the sharper the structure, the more hydrophobic it became. Current sensitivity for NO oxidation was more greatly increased as the Pt nanostructure became sharper and then more hydrophobic, even with the similar ESA. This proposes that the NO transport is facilitated through more hydrophobic Pt nanostructure layer, utilizing larger Pt surfaces for NO oxidation. Since the oxidations of interferents AP, AA, NO2− and H2O2 were independent of the Pt nanostructures, a decent selectivity to NO was obtained with hydrophobic Pt deposition. The greatest hydrophobic Pt layer plated with 5 mM K2PtCl4 showed the highest NO sensitivity and selectivity against AP, AA, NO2− and H2O2: the selectivity coefficient for AP, AA and H2O2 was three-fold greater than the Pt deposition with 75 mM H2PtCl6. Our current study demonstrates the feasibility of amperometric NO detection without any selective membranes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science, ICT and Future Planning (NRF-2017R1A2A2A14001137 for YL and NRF-2017R1A2B4002159 for CL) and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1A6A1A03025340).References
- Y. Cury, G. Picolo, V. P. Gutierrez and S. H. Ferreira, Nitric Oxide, 2011, 25, 243–254 CrossRef CAS PubMed.
- L. Jia, C. Bonaventura, J. Bonaventura and J. S. Stamler, Nature, 1996, 380, 221–226 CrossRef CAS PubMed.
- T. Trepels, A. M. Zeiher and S. Fichtlscherer, Endothelium, 2006, 13, 423–429 CrossRef PubMed.
- S. H. Snyder and D. S. Bredt, Sci. Am., 1992, 266, 68–77 CrossRef CAS PubMed.
- M. Kelm, M. Feelisch, R. Spahr, H.-M. Piper, E. Noack and J. Schrader, Biochem. Biophys. Res. Commun., 1988, 154, 236–244 CrossRef CAS PubMed.
- J. S. Beckman, J. Chen, H. Ischiropoulos and J. P. Crow, in Methods Enzymol, Academic Press, 1994, vol. 233, pp. 229–240 Search PubMed.
- J. Wang, M. Lu, F. Yang, X. Zhang, W. R. G. Baeyens and A. M. García Campaña, Anal. Chim. Acta, 2001, 428, 173–181 CrossRef CAS.
- A. Menou, D. Babeanu, H. N. Paruit, A. Ordureau, S. Guillard and A. Chambellan, J. Breath Res., 2017, 11, 036008 CrossRef CAS PubMed.
- R. Miao, L. Mu, H. Zhang, H. Xu, G. She, P. Wang and W. Shi, J. Mater. Chem., 2012, 22, 3348–3353 RSC.
- H. Thomsen, N. Marino, S. Conoci, S. Sortino and M. B. Ericson, Sci. Rep., 2018, 8, 9753 CrossRef PubMed.
- B. Fink, S. Dikalov and N. Fink, Pharmacol. Rep., 2006, 58, 8–15 Search PubMed.
- S. D. Dumitrescu, A. T. Meszaros, S. Puchner, A. Weidinger, M. Boros, H. Redl and A. V. Kozlov, Magn. Reson. Med., 2017, 77, 2372–2380 CrossRef CAS PubMed.
- B. J. Privett, J. H. Shin and M. H. Schoenfisch, Chem. Soc. Rev., 2010, 39, 1925–1935 RSC.
- E. Dumitrescu, K. N. Wallace and S. Andreescu, Nitric Oxide, 2018, 74, 32–38 CrossRef CAS PubMed.
- M. D. Brown and M. H. Schoenfisch, ACS Sens., 2016, 1, 1453–1461 CrossRef CAS.
- Y. Lee and J. Kim, Anal. Chem., 2007, 79, 7669–7675 CrossRef CAS PubMed.
- J. H. Shin, B. J. Privett, J. M. Kita, R. M. Wightman and M. H. Schoenfisch, Anal. Chem., 2008, 80, 6850–6859 CrossRef CAS PubMed.
- R. A. Hunter, B. J. Privett, W. H. Henley, E. R. Breed, Z. Liang, R. Mittal, B. P. Yoseph, J. E. McDunn, E. M. Burd, C. M. Coopersmith, J. M. Ramsey and M. H. Schoenfisch, Anal. Chem., 2013, 85, 6066–6072 CrossRef CAS PubMed.
- N. J. Finnerty, S. L. O'Riordan, E. Palsson and J. P. Lowry, J. Neurosci. Methods, 2012, 209, 13–21 CrossRef CAS PubMed.
- J. Zajda, N. J. Schmidt, Z. Zheng, X. Wang and M. E. Meyerhoff, Electroanalysis, 2018, 30, 1602–1607 CrossRef CAS.
- H. Jeong and J. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 7129–7135 CrossRef CAS PubMed.
- S. Guo and E. Wang, Acc. Chem. Res., 2011, 44, 491–500 CrossRef CAS PubMed.
- A. J. Mieszawska, R. Jalilian, G. U. Sumanasekera and F. P. Zamborini, Small, 2007, 3, 722–756 CrossRef CAS PubMed.
- J. K. Kawasaki and C. B. Arnold, Nano Lett., 2011, 11, 781–785 CrossRef CAS PubMed.
- M. Q. Guo, H. S. Hong, X. N. Tang, H. D. Fang and X. H. Xu, Electrochim. Acta, 2012, 63, 1–8 CrossRef CAS.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, L. Cui, B. Ren and Z.-Q. Tian, Chem. Commun., 2006, 4090–4092 RSC.
- A. Lafuma and D. Quéré, Nat. Mater., 2003, 2, 457–460 CrossRef CAS PubMed.
- S. Choi, S. Kweon and J. Kim, Phys. Chem. Chem. Phys., 2015, 17, 23547–23553 RSC.
- Y. Ha, J. Sim, Y. Lee and M. Suh, Anal. Chem., 2016, 88, 2563–2569 CrossRef CAS PubMed.
- S. Trasatti and O. A. Petrii, J. Electroanal. Chem., 1992, 327, 353–376 CrossRef CAS.
- B. Lim and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 76–85 CrossRef CAS PubMed.
- S. Wang and L. Jiang, Adv. Mater., 2007, 19, 3423–3424 CrossRef CAS.
- P. Liu, L. Cao, W. Zhao, Y. Xia, W. Huang and Z. Li, Appl. Surf. Sci., 2015, 324, 576–583 CrossRef CAS.
- M. Valko, M. Izakovic, M. Mazur, C. J. Rhodes and J. Telser, Mol. Cell. Biochem., 2004, 266, 37–56 CrossRef CAS PubMed.
- M. Shalaeva, J. Kenseth, F. Lombardo and A. Bastin, J. Pharm. Sci., 2008, 97, 2581–2606 CrossRef CAS PubMed.
- M. G. Evans and N. Uri, Trans. Faraday Soc., 1949, 45, 224–230 RSC.
- S. Park, T. D. Chung and H. C. Kim, Anal. Chem., 2003, 75, 3046–3049 CrossRef CAS PubMed.
- J. H. Shim, K. Jang, Y. Lee and C. Lee, Electroanalysis, 2011, 23, 2063–2069 CrossRef CAS.
- J. H. Shin, S. W. Weinman and M. H. Schoenfisch, Anal. Chem., 2005, 77, 3494–3501 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an01518j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |