
Conductometric immunoassay of alpha-fetoprotein in sera of liver cancer patients using bienzyme-functionalized nanometer-sized silica beads
Jiaming
Liang
 ,
Jing
Wang
,
Luwei
Zhang
,
Sijia
Wang
,
Cuiping
Yao
and
Zhenxi
Zhang
*
,
Jing
Wang
,
Luwei
Zhang
,
Sijia
Wang
,
Cuiping
Yao
and
Zhenxi
Zhang
*
Institute of Biomedical Analytical Technology and Instrumentation, Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology, Xi'an Jiaotong University, Xi'an 710049, Shaanxi, China. E-mail: zxzhang@mail.xjtu.edu.cn
First published on 13th October 2018
Abstract
A conductometric immunoassay protocol was designed for the sensitive detection of a liver cancer biomarker, alpha-fetoprotein (AFP), in biological fluids by using enzyme-conjugated nanometer-sized enzyme-doped silica beads. Initially, urease molecules were doped into nanosilica particles by using the reverse micelle method. Thereafter, arginase-labeled anti-AFP antibodies were covalently conjugated onto the surface of the synthesized nanoparticles. The immunoreaction was carried out in a monoclonal anti-AFP capture antibody-coated microplate with a sandwich-type assay format by using bienzyme-functionalized silica nanobeads as the recognition elements. Upon L-arginine introduction, the substrate was cleaved into urea and L-ornithine on the basis of the arginase enzymatic reaction, and the as-produced urea was then decomposed into ammonia (NH4+) and bicarbonate (HCO3−) ions by the doped urease, thus causing the variation in the local conductivity of the detection solution on an interdigitated conductometric transducer. Under optimal conditions, the developed immunosensing system exhibited good conductometric responses toward target AFP within a dynamic linear range of 0.01–100 ng mL−1 at a relatively low detection limit of 4.8 pg mL−1 based on the 3sB criterion. Importantly, good reproducibility, high specificity and acceptable method accuracy were acquired for the analysis of human serum specimens in liver cancer patients.
Introduction
Liver cancer, also known as hepatic cancer, is very common that starts in the liver. The most common types are hepatocellular carcinoma (HCC), which makes up 80% of the cases, and cholangiocarcinoma.1 The less common types include mucinous cystic neoplasm and intraductal papillary biliary neoplasm.2 The diagnosis may be supported by blood tests and medical imaging with confirmation by tissue biopsy. Alpha-fetoprotein (AFP; a major plasma glycoprotein with a molecular weight of ∼70 kDa) is involved in the regulation of fatty acids in both the fetal (where it functions to block the transport of estradiol across the placenta) and proliferating adult liver cells, and its expression is found to be increased in acute liver injuries.3 Usually, the AFP levels decrease by the second year of birth and its ectopic expression is linked with tumors. In patients with chronic hepatitis C, high levels of this protein are related to the increased risk of developing HCC.4 Therefore, the AFP level in the amniotic fluid serves as a tumor biomarker for hepatocellular carcinoma.Recently, various methods and strategies have been reported for the quantitative monitoring of the AFP levels in biological fluids, on the basis of different signal-generation principles, e.g., impedimetric immunoassay,5 nano-optical sensor,6 electrochemical aptasensor,7 enzyme immunoassay,8 potentiometric immunoassay9 and fluorescent immunosensor.10 Inspiringly, the rapidly emerging research field of conductometric sensing strategies provides exciting new possibilities for the advanced development of new analytical tools for bioanalytical and biotechnological applications, thanks to their unique advantages, e.g., being suitable for miniaturization and large-scale production, not requiring a reference electrode and requiring a low driving voltage.11 Typically, conductometric sensors usually consist of a planar glass support with interdigitated gold electrode pairs on one surface in a planar configuration, and the signal derives from the electrical resistance between two parallel electrodes by many biochemical reactions to change the ionic compositions in solution.12 Tang et al. designed a conductometric immunosensor for the detection of AFP on an anti-AFP capture antibody-modified interdigitated gold transducer using horseradish peroxidase (HRP)-labeled carbon nanoparticles to induce a H2O2–KI system.13 Korotcenkov and Cho discussed the methods of the formation of metal oxide composites and the advantages of their usage in the development of conductometric gas sensors.14 Jaffrezic-Renault and Dzyadevych presented principles of conductometric measurements in ionic media and equivalent electrical circuits of different designs for the measurement of biocatalytic reactions.15 Interestingly, Dewan and Roy proposed a conductometric study on ion-pair and triple-ion formation in a low permittivity solvent.16 Despite many advances in this field, there is still a requirement to explore innovative schemes for the development of conductometric sensing strategies while preserving the essential benefits of sensitivity and simplification.
To achieve low limits of detection and quantification, there are two basic aspects for the development of high-efficiency conductometric sensing protocols. The first key issue is to exploit an advanced signal-generation pathway. Routine approaches are usually carried out by quantitatively monitoring the consumption or production of charged species by the enzymatic reactions.17 Kucherenko et al. used hexokinase to construct a conductometric biosensor for the detection of adenosine triphosphate, and the signal was generated exclusively due to the enzymatic reaction accompanying the changes in the charged species (D-glucose + ATP + hexokinase → D-glucose-6-phosphate + ADP).18 Liang et al. utilized HRP to consume the iodide ions in the presence of hydrogen peroxide for the conductometric detection of interleukin-6.19 Recently, Soldatkina et al. developed a conductometric arginine biosensor based on arginase and urease for the generation of ammonia and bicarbonate ions to induce a change in the solution conductivity.20 To this end, our motivation herein is designing a conductometric immunosensor focusing on bienzyme-based generation of the charged species for the detection of AFP.
Another important concern is to study the signal-amplification method. Generally, the catalytic efficiency of enzymes alone is limited toward the corresponding substrates, thanks to the limitation of the enzymatic activity.21,22 An alternative strategy that combines enzyme labels with nano labels would be advantageous because of the high surface-to-volume ratio and quantum-size effect.23,24 In this regard, nanometer-sized silica beads are expected to enhance the loading amount of enzyme molecules due to their overwhelming merits: (i) molecular tags such as fluorophores and indicators are easily doped into silica beads during the synthesis process by using the reverse micelle method (also known as water-in-oil microemulsion systems); and (ii) the silica shells can facilitate a wide variety of surface reactions and allow conjugation with biomolecules such as proteins and DNA.25,26 In this contribution, arginase-coated nanometer-sized urease-doped silica beads are utilized for the construction of a conductometric immunosensing system (Scheme 1). Upon reaction with silica beads, arginase-labeled anti-AFP secondary antibodies are simultaneously conjugated onto urease-doped silica beads. The assay is performed in anti-AFP capture antibody-coated microplates in sandwiched reaction mode. In the presence of target AFP, bienzyme-functionalized silica beads along with the immunocomplexes can catalyze the conversion of L-arginine into ammonia (NH4+) and bicarbonate (HCO3−) ions, thereby resulting in the change in the conductivity of the solution, which can be quantitatively monitored on an interdigitated gold conductometric transducer. This work aims to devise a new conductometric immunoassay for the high-sensitivity detection of low-abundance proteins for disease diagnoses.
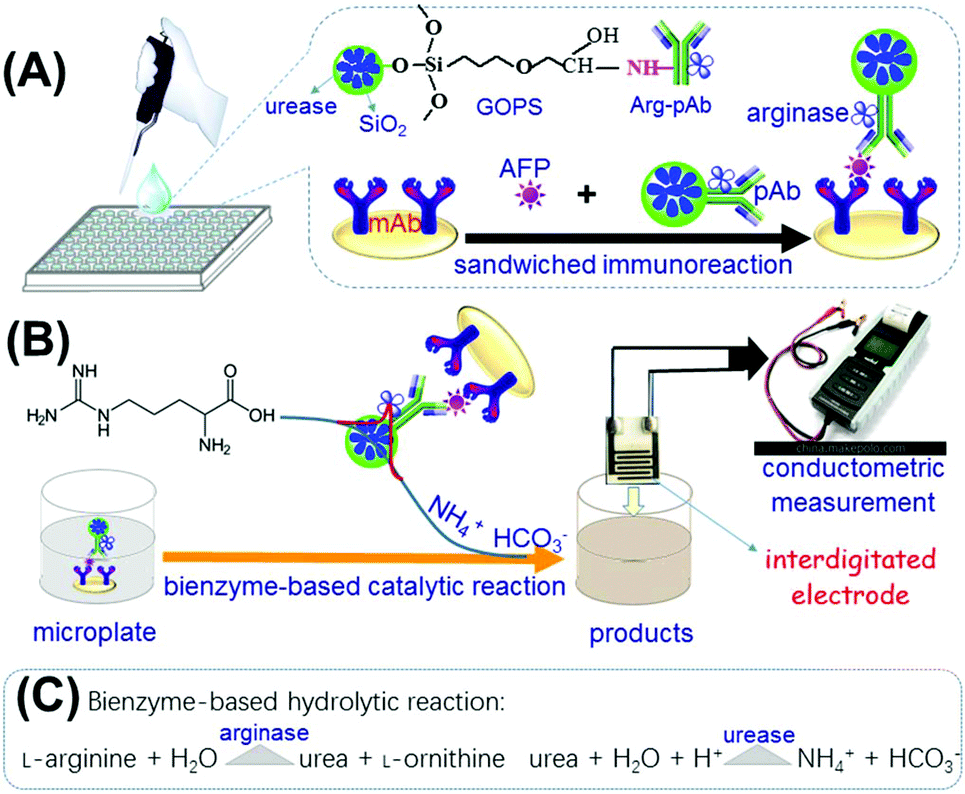 | ||
| Scheme 1 Schematic illustration of the bienzyme-based conductometric immunoassay for target alpha-fetoprotein (AFP) on monoclonal mouse anti-human AFP antibody (mAb)-coated microplate by using L-arginase-coated urease-encapsulated nanometer-sized silica beads: (A) immunoreaction protocol for target AFP between mAb and polyclonal rabbit anti-human AFP antibody–arginase conjugate (Arg–pAb)-labeled urease-functionalized silica nanoparticle; (B) conductometric measurement; and (C) bienzyme-based hydrolytic reaction. | ||
Experimental section
Materials and chemicals
Monoclonal mouse anti-human AFP capture antibody (mAb; clone C3, purified from hybridoma cell culture, MW: ∼74 kDa, 1.0 mg mL−1), polyclonal rabbit anti-human AFP secondary antibody (pAb; 0.4 μg mL−1), human AFP ELISA kits including the different-concentration standards, bovine serum albumin (BSA, 96%–99%), L-arginase from bovine liver (protein ≥70% by biuret, powder; ≥100 units per mg; one unit causes the hydrolysis of 1.0 μmol of L-arginine per minute at pH 9.5 and 37 °C), (3-glycidyloxypropyl) trimethoxysilane (C9H20O5Si, GOPS), bis-(2-ethylhexyl) sodium sulfosuccinate (AOT), tetramethoxysilane (TEOS), urease from Canavalia ensiformis (Jack bean, Type III, powder, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000 units per g solid; one micromolar unit liberates 10 μmol of NH3 from urea per min at pH 7.0 and 25 °C) and glutaraldehyde solution (Grade II, 25 wt% in H2O) were purchased from Sigma-Aldrich (St Louis, MO, USA). Cyclohexane, n-hexanol, and ammonium hydroxide (25 wt%) were purchased from Merck (Darmstadt, Germany). All the high-binding polystyrene 96-well microtiter plates (Ref. 655061) were obtained from Greiner (Frickenhausen, Germany). All buffers including 5.0 mM Tris-HCl and phosphate-buffered saline (PBS) solutions were the products of Sigma-Aldrich. A pH 9.6 coating buffer (1.59 g of Na2CO3, 2.93 g of NaHCO3 and 0.2 g of NaN3) was prepared by adding the chemicals into 1000 mL of distilled water. The blocking buffer and washing buffer were obtained by adding 1.0% BSA (w/v) and 0.05% Tween 20 (v/v) in PBS, respectively. Ultrapure water obtained from a Millipore water purification system (18.2 MΩ cm, Milli-Q, Millipore) was used in all runs.
000–50000 units per g solid; one micromolar unit liberates 10 μmol of NH3 from urea per min at pH 7.0 and 25 °C) and glutaraldehyde solution (Grade II, 25 wt% in H2O) were purchased from Sigma-Aldrich (St Louis, MO, USA). Cyclohexane, n-hexanol, and ammonium hydroxide (25 wt%) were purchased from Merck (Darmstadt, Germany). All the high-binding polystyrene 96-well microtiter plates (Ref. 655061) were obtained from Greiner (Frickenhausen, Germany). All buffers including 5.0 mM Tris-HCl and phosphate-buffered saline (PBS) solutions were the products of Sigma-Aldrich. A pH 9.6 coating buffer (1.59 g of Na2CO3, 2.93 g of NaHCO3 and 0.2 g of NaN3) was prepared by adding the chemicals into 1000 mL of distilled water. The blocking buffer and washing buffer were obtained by adding 1.0% BSA (w/v) and 0.05% Tween 20 (v/v) in PBS, respectively. Ultrapure water obtained from a Millipore water purification system (18.2 MΩ cm, Milli-Q, Millipore) was used in all runs.
Preparation of arginase-labeled anti-AFP secondary antibody (Arg-pAb)
Arginase molecules were conjugated to polyclonal rabbit anti-human AFP secondary antibody by using a one-step glutaraldehyde protocol referring to ref. 27. Prior to the experiment, 2.0 mL of a mixture containing 2.0 mg mL−1 pAb antibody and 5.0 mg mL−1 arginase was prepared in a 20 mM sodium phosphate solution containing 0.15 M NaCl, pH 7.4. Thereafter, 20 μL of 25 wt% glutaraldehyde was injected into the antibody/enzyme solution. After the reaction for 2 h at 4 °C under gentle shaking on a shaker, sodium borohydride was added to reduce the resultant Schiff bases and any excess aldehydes at a final concentration of 10 mg mL−1 (60 min at 4 °C). To remove any insoluble polymers that may have formed, the resulting mixture was filtered through a 0.45 μm filter (Whatman). The Arg–mAb conjugates were purified by dialysis in PBS (10 mM, pH 7.4), and stored at 4 °C for use.Synthesis and bioconjugation of urease-doped silica nanoparticles [(UA)SiNP–Arg–pAb]
Urease-doped silica nanoparticles [denoted as (UA)SiNPs] were synthesized similar to previous literature28 by using the reverse micelle method. Initially, 1.0 mL of urease (1.0 mg mL−1) in PBS (10 mM, pH 7.4) was added to 20 mL of cyclohexane containing 5.3 g of Triton X-100 and 5.0 mL of n-hexanol. After vigorous stirring, 2.5 mL of TEOS and 1.8 mL of NH3·H2O (25 wt%) were injected into the resulting mixture, and reacted for 12 h at 4 °C under the same conditions. Following that, 5.0 mL of acetone was added, and the resultant mixture was centrifuged (10 min, 10000g, 4 °C) and washed alternately with ethanol and water three times. Finally, the collected precipitate (i.e., urease-doped silica beads) was used for the labeling of Arg–pAb conjugates as follows by (i) adding the doped silica bead into 5.0 mL of 5% GOPS (v/v) in dry toluene and reacting for 6 h at 4 °C on a shaker; (ii) injecting the above-prepared Arg-pAb conjugates (100 μL) into 2.0 mL of the functional beads (dispersed into 10 mM PBS, pH 7.4) after centrifugation (10 min, 10000g, 4 °C) and washing three times with toluene and ethanol alternately; and (iii) incubating the precipitates with 10 mg mL−1 BSA–PBS for 60 min at 37 °C to block the unreacted and nonspecific sites. Finally, the Arg–pAb-conjugated urease-doped silica nanoparticles, denoted as (UA)SiNP–Arg–pAb, were dispersed into 10 mM PBS, pH 7.4, and stored at 4 °C at a final concentration of 25 mg mL−1 when not in use.
Immunoreaction protocol and conductometric measurement
Scheme 1 shows a schematic illustration of conductometric immunoassay for target AFP by using Arg–pAb-coated urease-doped silica beads as the detection antibodies in sandwich-type reaction mode. Prior to the measurement, monoclonal mouse anti-AFP capture antibody-coated microplates were prepared in a high-binding polystyrene 96-well microtiter plate by adding the mAb antibody (50 μL per well, 1.0 mg mL−1 in 0.05 M sodium carbonate buffer, pH 9.6) into the well. The microplates were covered with an adhesive plastic plate sealing film to prevent evaporation. On the following day, the plates were washed three times with the washing buffer, and then incubated with 300 μL per well of blocking buffer for 60 min at 37 °C with gentle shaking. The plates were then washed as before. Following that, 50 μL of AFP standards or samples with various concentrations were added into the well, and incubated for 45 min at room temperature under shaking. After washing, 50 μL of the prepared (UA)SiNP–Arg–pAb suspension (25 mg mL−1) was added into the well and incubated for 45 min under the same conditions. The plates were washed again, and 100 μL of Tris-HCl buffer (pH 8.0, 5.0 mM) containing arginine (0.5 mM) was added into each well. The plates were shaken for 20 min on a plate shaker for the bienzyme reactions. Finally, the conductivity of the resulting solution was determined and registered by using an interdigitated micro-comb gold electrode in an open circuit. To avoid possible errors during the readout, the response for each assay was recorded until equilibrium was reached (shift less than 1.0 μS min−1). The control tests with normal samples and the evaluations of clinical specimens were performed accordingly. The change of conductivity relative to the target AFP standard was calculated using the following equation: ΔS = Sn − S0 (where S0 and Sn represent the steady-state conductivity (μS) of the immunosensing platform in the absence and presence of target AFP, respectively). The signals in all experiments referred to the average responses of reactions with corresponding standard deviations in triplicate, unless otherwise indicated. The sigmoidal curves were calculated by mathematically fitting experimental points using Rodbard's four parameter function with the Origin 6.0 software. Graphs were plotted in the form of conductivity (μS) against the logarithm of the AFP concentration (ng mL−1). All measurements were performed at room temperature (25 ± 1.0 °C).Statistical analyses
A statistical data analysis was performed using Statistical Analysis System (SAS) ver. 9.0 and Statistical Program for Social Sciences (SPSS) ver. 9.0 software packages. Comparisons between dependent variables were performed using analysis of variance (ANOVA), Duncan's multiple range test, correlation analysis and multiple regression analysis. Results are expressed as mean value ± standard deviation (SD) of three determinations and the statistical significance was defined as P ≤ 0.05.All experiments were performed in accordance with the guidelines of Xi'an Jiaotong University, and approved by the ethics committee at Xi'an Jiaotong University, China. Informed consents were obtained from the human participants of this study.
Results and discussion
Design and characterization of bienzyme-based conductometric immunoassay
For the development of a new conductometric immunoassay, signal amplification is very crucial. In this work, enzyme-coated nanometer sized enzyme-doped silica beads are expected to enhance the sensitivity of the conductometric immunoassay by coupling with enzyme labels and nano labels due to high chemical/thermal stability, final suspendability in aqueous solutions, and good compatibility with the environment. Initially, soluble urease molecules are doped into silica beads during the synthesis on the basis of a water-in-oil microemulsion method. In this system, a hydrophilic head region is formed which contains numerous nanometer-sized water cores, each with a hydrophobic tail that extends into an apolar continuous phase. The nanodroplets of water in the bulk-oil phase act as nanoreactors for discrete particle formation. Thereafter, L-arginase-labeled anti-AFP antibodies are covalently conjugated onto the surface of urease-doped silica beads through the epoxy-amino reaction. In this case, numerous enzyme molecules can be doped and labeled on one silica bead. When one antibody on the bead reacts with the antigen, all the doped/coated enzyme molecules can participate in the catalytic reaction toward the substrate. In this configuration, the as-synthesized (UA)SiNP–Arg–pAb is employed to detect AFP on the anti-AFP capture antibody-coated microplate in a sandwiched immunoassay format since bienzyme reactions can be accompanied by changes in the solution conductivity as follows:Immunoreaction:
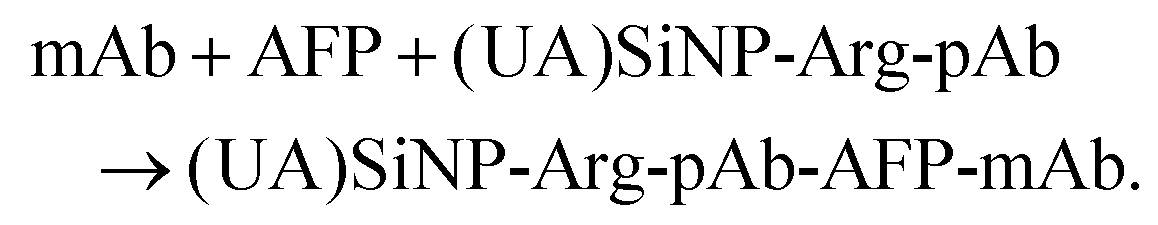
Enzyme reactions:
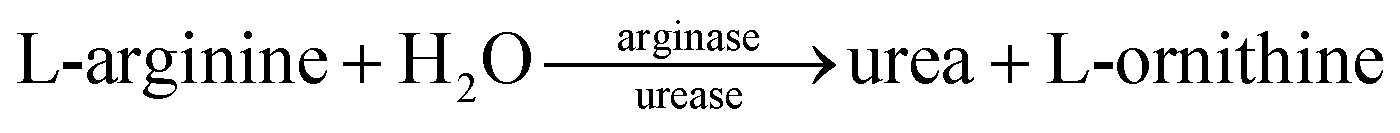
| Urea + H2O + H+ → NH4+ + HCO3− |
The coated arginase on the nanobeads initially decomposes L-arginine into urea and L-ornithine, and then the as-produced urea is hydrolyzed into ammonia (NH4+) and bicarbonate (HCO3−) ions by the doped urease in silica nanoparticles, thus resulting in the local change in the solution conductivity.20 By monitoring the change in the conductivity on a conductometric transducer, we can quantitatively evaluate the concentration of target AFP in the sample.
To construct such an immunosensing platform, the preparation of bienzyme-functionalized silica nanoparticles would be very important. First, we investigated the surface micro-architecture and morphological feature of nanometer-sized silica beads by using scanning electron microscopy (SEM; Hitachi 4800, Japan). Fig. 1A shows a typical SEM image of the urease-doped silica nanoparticles [(UA)SiNPs], and the average size is ∼105 nm diameter. Moreover, almost all the nanoparticles had the same sizes, and the surface was relatively smooth. Logically, one puzzling question arises as to whether urease molecules were successfully doped into the nanobeads. To clarify this issue, the as-prepared (UA)SiNPs were thoroughly ground, and the obtained powder was characterized by using Fourier transform infrared (FTIR) spectroscopy (Vector 22, Bruker, Germany) (Fig. 1B). For comparison, silica nanoparticles without the doped urease molecules were prepared by using a similar synthesis method. As seen from curve ‘a’, a silica bead alone exhibited the typical Si–O–Si bands associated with the formed network at 1125, 938, 805 and 465 cm−1.29 The high intensity and broad absorption band at 1125 cm−1 was ascribed to the asymmetric stretching bonds of Si–O–Si associated with the motion of oxygen, and the shoulder at 938 cm−1 was attributed to the vibrations of Si–OH around silica beads.30 Significantly, another two characteristic bands at 1682 and 1578 cm−1 appeared when urease molecules were present in the water-in-oil microemulsion system (curve ‘b’). As is well known, the shapes of the infrared absorption bands of amide I groups at 1610–1690 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of peptide linkages and amide II groups around 1500–1600 cm−1 from a combination of N–H bending and C–N stretching can provide detailed information on the secondary structure of proteins.28 The comparative study indicated that urease could be doped into silica nanoparticles by using the reverse micelle method.
O stretching vibrations of peptide linkages and amide II groups around 1500–1600 cm−1 from a combination of N–H bending and C–N stretching can provide detailed information on the secondary structure of proteins.28 The comparative study indicated that urease could be doped into silica nanoparticles by using the reverse micelle method.
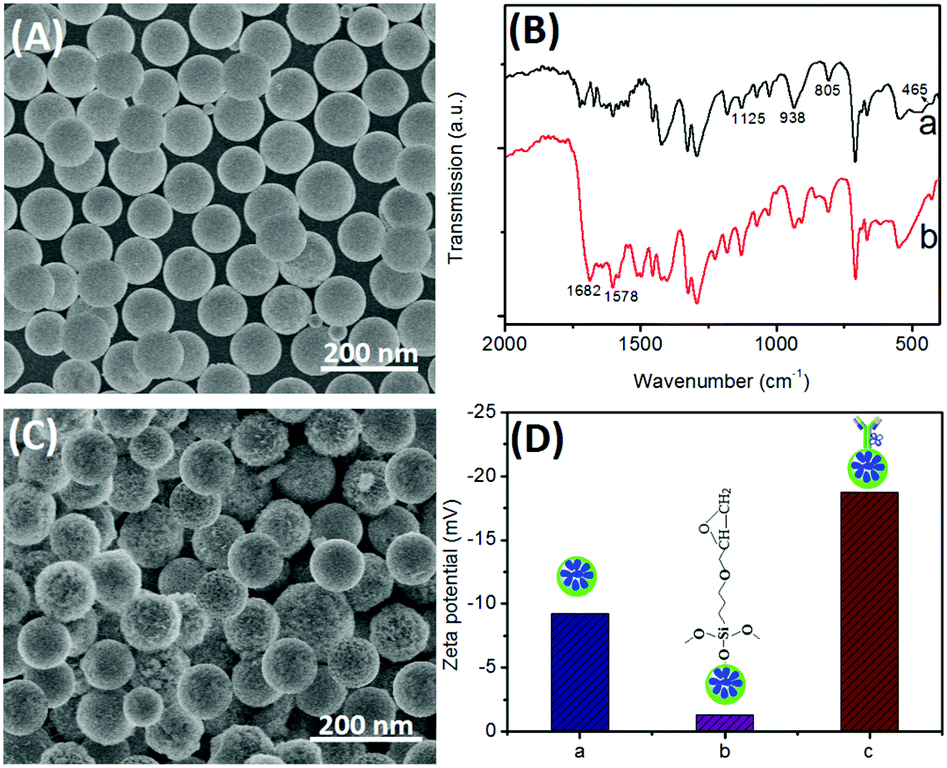 | ||
| Fig. 1 (A,C) SEM images of (A) urease-doped silica nanoparticles and (C) (UA)SiNP–Arg–pAb; (B) FTIR spectra of (a) silica nanoparticles and (b) urease-doped silica nanoparticles; and (D) zeta potentials of (a) (UA)SiNPs, (b) GOPS-functionalized (UA)SiNPs and (c) (UA)SiNP–Arg–pAb. | ||
Next, the as-prepared (UA)SiNPs were used for the labeling of Arg–pAb conjugates by using the epoxy-amino reaction. As seen from the SEM image in Fig. 1C, the surface became rougher than that of (UA)SiNPs (Fig. 1A) after bioconjugation with arginase-labeled anti-AFP antibody using GOPS. Vaguely, some branched topological structures on the surface of (UA)SiNPs were observed. To further investigate the conjugation process of Arg–pAb on (UA)SiNPs, we also employed dynamic light scattering (DLS; Zetasizer Nano S90, Malvern, UK) to monitor the biofunctionalized silica beads after each step (Fig. 1D). The zeta potential (ζ) of (UA)SiNPs was −9.2 mV (column ‘a’), which was attributed to the –OH groups on the surface of silica beads in aqueous solution. Upon introduction of GOPS on the surface of (UA)SiNPs, the zeta potential decreased to −1.3 mV (column ‘b’), which was ascribed to the reaction of the –OH groups with GOPS followed by the formation of the uncharged epoxy groups. After GOPS-functionalized (UA)SiNPs reacted with Arg–pAb conjugates, the surface potential of the formed (UA)SiNP–Arg–pAb became more negative (−18.7 mV) (column ‘c’). The reason might be the fact that the isoelectric point of the Arg–pAb conjugate was lower than pH 7.4. In this case, the labeled Arg–pAb conjugate had a negatively charged species. Furthermore, the size of (UA)SiNP–Arg–pAb (128 ± 9.4 nm) was bigger than that of (UA)SiNP (112 ± 6.8 nm). The size of (UA)SiNPs obtained from DLS data was slightly bigger than that from the SEM result, which was due to the fact that DLS data were derived from the hydration radius of the nanoparticles in aqueous solution. These results also revealed that (UA)SiNP–Arg–pAb could be formed by using the epoxy-amino reaction.
Evaluation of bienzyme activity and feasibility analysis
As mentioned above, the detectable signal of the conductometric immunoassay originated from the change in the conductivity of the detection solution via the doped/coated urease/arginase toward the hydrolysis of arginine. The bienzyme activity in the silica nanobeads (2.5 mg used in this case) was investigated in Tris-HCl buffer (100 μL, pH 8.0, 5.0 mM) containing the arginine substrate with different concentrations on a conductometric transducer. As seen from plot ‘a’ in Fig. 2A, the as-synthesized (UA)SiNP–Arg–pAb exhibited a classical Michaelis–Menten behavior, indicating that doped/coated bienzymes showed significant biocatalytic responses toward arginine. The steady-state conductivity was achieved when the balance between the rate of ion production due to an enzymatic reaction and the influx of ions through a mediated transport mechanism with buffer species acting as a carrier was reached. As the control tests, (UA)SiNP and SiNP–Arg–pAb were prepared by using the same synthesis route in the absence of urease or arginase, respectively. These two functionalized silica nanobeads were monitored in the presence of different-concentration arginine substrates (plots ‘b and c’). Obviously, all the conductivity values were relatively low. Moreover, the values did not increase with the increase of the arginine concentration, suggesting that the change in the conductivity stemmed from the combined action of urease and arginase.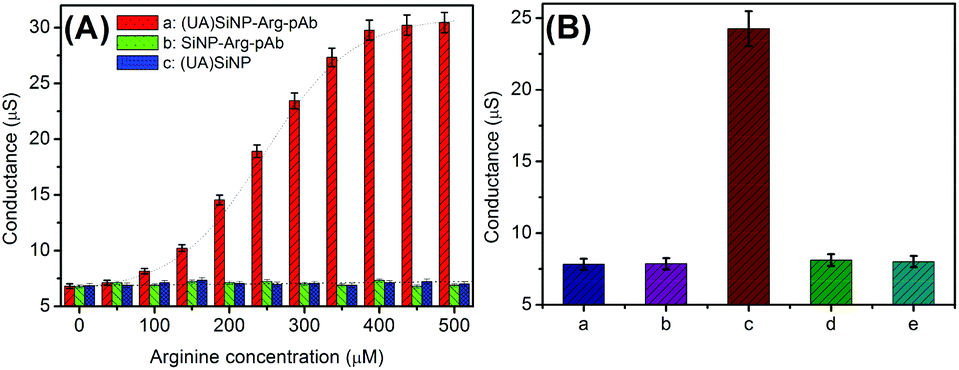 | ||
| Fig. 2 (A) Enzymatic activities of (a) (UA)SiNP–Arg–pAb, (b) SiNP–Arg–pAb and (c) (UA)SiNP in Tris-HCl buffer (100 μL, pH 8.0, 5.0 mM) containing arginine at different levels on an interdigitated conductometric transducer; and (B) conductometric responses of (a) mAb-coated microplate + arginine, (b) mAb-coated microplate + 10 ng mL−1 AFP + arginine, (c) mAb-coated microplate + 10 ng mL−1 AFP + (UA)SiNP–Arg–pAb + arginine, (d) mAb-coated microplate + 10 ng mL−1 AFP + (UA)SiNP–Arg–pAb, and (e) mAb-coated microplate + (UA)SiNP–Arg–pAb + arginine in Tris-HCl buffer (100 μL, pH 8.0, 5.0 mM). | ||
Maybe, one question should be answered whether the as-prepared (UA)SiNP–Arg–pAb could be used for the detection of target AFP in the conductometric immunoassay. To demonstrate this point, mAb-coated microplates and (UA)SiNP–Arg–pAb were utilized for the determination of 10 ng mL−1 AFP (as an example) in sandwiched reaction mode. By the same token, the mAb-coated microplate before and after the reaction with target AFP and (UA)SiNP–Arg–pAb (i.e., after each step, as control tests) was studied in Tris-HCl buffer (100 μL, pH 8.0, 5.0 mM) containing 0.5 mM arginine on the conductometric transducer (Fig. 2B). Column ‘a’ gave conductometric response of the mAb-coated microplate in the detection solution (≈7.82 μS). With the reaction of the mAb antibody with 10 ng mL−1 AFP, the conductivity of the resulting microplate was almost the same as that before the reaction (column ‘b’ vs. column ‘a’). After incubation with (UA)SiNP–Arg–pAb again, significantly, the conductivity greatly increased to ∼24.26 μS (column ‘c’), indicating that the use of (UA)SiNP–Arg–pAb could cause the change in the conductivity of the detection system. Moreover, the conductivity was determined in the (UA)SiNP–Arg–pAb-based system in the absence of the arginine substrate, and was found to be 8.12 μS (column ‘d’, as the control test). Therefore, the change of the detectable signal derived from the reaction of arginine with (UA)SiNP–Arg–pAb. In addition, we investigated whether the strong signal was achieved by the mAb-coated microplate toward non-specific adsorption of (UA)SiNP–Arg–pAb. In this regard, (UA)SiNP–Arg–pAb was used for direct incubation with the mAb-coated microplate. As seen from column ‘e’, the conductivity was close to that of the mAb-coated microplate alone (column ‘a’). On the basis of these results, we might come to a conclusion that our designed immunosensing system could be applied for the detection of target AFP.
Optimization of assay conditions
As mentioned above, the assay involves immunoreaction and conductometric measurements. So, the analytical properties of the conductometric immunoassay depend on the antigen–antibody reaction. A short incubation time is unfavorable for the conjugation of (UA)SiNP–Arg–pAb on the microplate, thus resulting in a weak change in the conductivity. Fig. 3A shows the effect of the immunoreaction time on the performance of the immunoassay from 10 min to 60 min at room temperature by using 10 ng mL−1 as an example (note: the immunoreactions of mAb with target AFP were paralleled with those of mAb–AFP with (UA)SiNP–Arg–pAb to avoid confusion). The conductivities increased with the increment of the immunoreaction time, and tended to level off after 45 min. Hence, an immunoreaction time of 45 min was selected for the detection of AFP.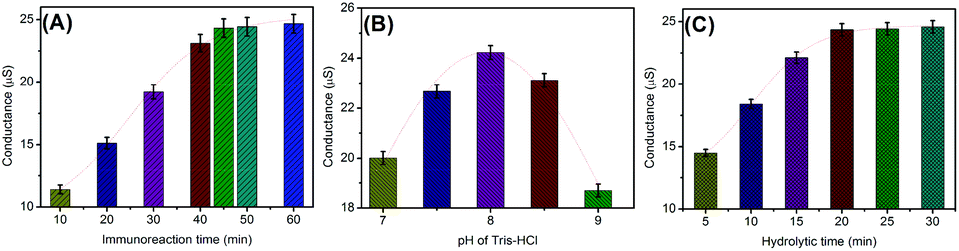 | ||
| Fig. 3 Effects of (A) immunoreaction time, (B) pH of Tris-HCl buffer and (C) hydrolytic reaction time on the conductometric responses of the bienzyme-based immunoassay (10 ng mL−1 AFP used in these cases). | ||
In this work, the change in the conductivity derived from the bienzymes toward the hydrolysis of arginine. To maintain the enzymatic bioactivity and adequately fulfil the catalytic potential of the bienzyme, a moderate pH detection solution would be preferable. Fig. 3B displays the dependence of the conductivities on pH of Tris-HCl buffer, and an optimum conductivity was obtained at pH 8.0. Higher or lower pH resulted in the decrease of the conductivity. Another factor that could contribute to the conductivity change was the conversion of bicarbonate to non-ionic carbonic acid at low pH. Thus, a pH 8.0 Tris-HCl buffer was chosen as the detection solution.
The produced amount of ammonia (NH4+) and bicarbonate (HCO3−) ions during this hydrolytic process will affect the readout on the conductometric transducer. Generally speaking, it takes some time to hydrolyze the substrate using natural enzymes. As shown in Fig. 3C, the maximum conductivity was acquired after 20 min. To ensure adequate hydrolysis of the added arginine molecules and save the assay time, 20 min was allowed for the hydrolysis of arginine by sandwiched (UA)SiNP–Arg–pAb.
Dose responses of conductometric immunoassay toward target AFP
Under aforementioned conditions, the developed conductometric immunosensing system was employed for the quantitative monitoring of target AFP standards with different concentrations on the mAb-coated microplate using (UA)SiNP–Arg–pAb as the secondary antibody. The conductometric measurements were carried out in Tris-HCl buffer (100 μL, pH 8.0, 5.0 mM) containing L-arginine (0.5 mM). Fig. 4A represents the conductometric responses of the immunosensing platform relative to AFP concentrations, giving a sigmoidal ‘S’ response relationship between the conductivity and the logarithm of the AFP level. A good linear relationship could be found within the range of 0.01–100 ng mL−1. The regression equation could be fit as y (μS) = 5.9125 × logC[AFP] + 18.1346 (ng mL−1, R2 = 0.9946, n = 9). The limit of detection (LOD) could be evaluated to be 4.8 pg mL−1 AFP based on the 3sB criterion (where sB is the standard deviation of the blank, n = 11), which was comparable with commercial human AFP ELISA kits from different companies [e.g., 0.5 ng mL−1 (cat#: ab108838 for Abcam), 0.087 ng mL−1 (cat#: ab193765 for Abcam), 0.35 ng mL−1 (cat#: ab108680 for Abcam), 2.0 ng mL−1 (cat#: ab108631 for Abcam), 0.046 ng mL−1 (DAFP001 for R&D Systems) and 0.25 ng mL−1 (cat#: RK00841 for Abclonal)], and other detection strategies (Table 1). Significantly, our strategy was capable of continuously carrying out all the steps within 2.0 h for one sample, including incubation, washing, enzymatic reaction and measurement, which is less than that of the commercialized ELISA kits (∼3.0 h).
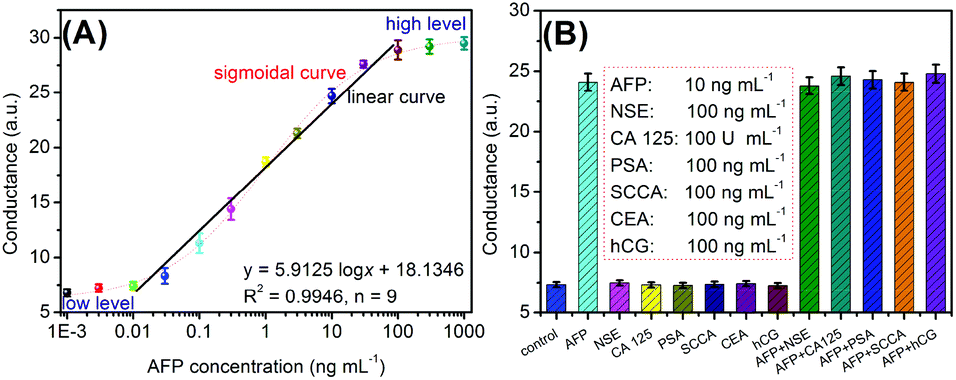 | ||
| Fig. 4 (A) Calibration plots of the bienzyme-based conductometric immunoassay toward target AFP with different concentrations; and (B) the specificity of the bienzyme-based conductometric immunoassay against neuro-specific enolase (NSE; 10 ng mL−1), cancer antigen 125 (CA 125; 100 U mL−1), prostate-specific antigen (PSA; 100 ng mL−1), squamous cell carcinoma antigen (SCCA; 100 ng mL−1), carcinoembryonic antigen (CEA; 100 ng mL−1), and human chorionic gonadotropin (hCG; 100 ng mL−1) (note: each data point represents the average value obtained from three measurements; the errors represent the 95% confidence interval of the mean for y-axis conductance). | ||
| Method | Materialsa | Linear range | LOD | Ref. |
|---|---|---|---|---|
| a HRP: horseradish peroxidase; Au: gold nanoparticles; MOF: metal organic framework. | ||||
| Impedimetric immunoassay | Peptide nanotubes | — | 1.0 ng mL−1 | 5 |
| Colorimetric immunoassay | Gold-hemin@MOF | 0.08–43 ng mL−1 | 20 pg mL−1 | 31 |
| Magnetic resonance detection | Manganese ferrite | 1.0–500 ng mL−1 | 0.3 ng mL−1 | 32 |
| Electrochemical aptasensor | Graphene oxide | 0.01–100 ng mL−1 | 3.0 pg mL−1 | 7 |
| Voltammetric immunoassay | Nanogold/dendrimer | 0.05–30 ng mL−1 | 20 pg mL−1 | 33 |
| Voltammetric immunoassay | MnO2/nanogold clusters | 0.01–10 ng mL−1 | 5.0 pg mL−1 | 34 |
| Fluorescent immunosensor | g-C3N4 nanosheets | 5.0–600 ng mL−1 | 430 pg mL−1 | 10 |
| Lateral flow device | Wax-printed paper | 0.1–100 ng mL−1 | 100 pg mL−1 | 35 |
| Electrochemical aptasensor | Graphene oxide/nanogold | 0.1–100 μg mL−1 | 50 ng mL−1 | 36 |
| Electrochemical immunoassay | HRP/nanogold | 0.02–1.0 × 105 pg mL−1 | 0.01 pg mL−1 | 37 |
| Electrochemical immunosensor | HRP/nanogold | 20–100 ng mL−1 | 0.64 ng mL−1 | 38 |
| Electrochemical immunosensor | HRP/Fe3O4/Au | 0.01–8.0 μg mL−1 | 1.78 ng mL−1 | 39 |
| Conductometric immunoassay | Bienzymes, silica beads | 0.01–100 ng mL−1 | 4.8 pg mL−1 | This work |
Reproducibility, specificity and stability
The reproducibility of the (UA)SiNP–Arg–pAb-based conductometric immunoassay was investigated using AFP standards at different concentrations within replicates of the assay by using sensing systems from a single batch and from different synthetic batches, respectively. As shown in Table 2, the relative standard deviations (RSDs) for the synthesis-to-synthesis reproducibility were slightly higher than those of the intra-assays. Significantly, all the RSDs were below 15%, indicating good reproducibility and precision for the batch preparation of conductometric immunosensing systems.| Conc. (ng mL−1) | Intra-assay (times; conc.: ng mL−1) | RSD (%) | Inter-assay (times; conc.: ng mL−1) | RSD (%) | ||||
|---|---|---|---|---|---|---|---|---|
| 1st | 2nd | 3rd | 1st | 2nd | 3rd | |||
| 0.1 | 0.11 | 0.096 | 0.098 | 7.47 | 0.098 | 0.13 | 0.12 | 14.11 |
| 10 | 9.83 | 9.12 | 10.17 | 5.52 | 11.31 | 8.98 | 10.02 | 11.55 |
| 50 | 52.35 | 56.12 | 48.43 | 7.35 | 48.61 | 53.78 | 57.12 | 8.06 |
Next, the specificity of conductometric immunosensing systems was evaluated against target AFP and other disease-related biomarkers, such as neuro-specific enolase (NSE), cancer antigen 125 (CA 125), prostate-specific antigen (PSA), squamous cell carcinoma antigen (SCCA), carcinoembryonic antigen (CEA), and human chorionic gonadotropin (hCG). Fig. 4B displays the conductometric responses of this system in the presence and absence of target AFP. The conductometric responses of this system were relatively low toward non-targets including NSE, CA 125, PSA, SCCA, CEA and hCG, which were almost the same as the background signal. Strong conductometric signals were achieved only if target AFP was present in the samples. Therefore, the selectivity of the conductometric immunoassay is satisfactory.
Furthermore, the storage stability of as-prepared (UA)SiNP–Arg–pAb was monitored for a six-month period by storing them at 4 °C when not in use. Following that, the bioconjugations were applied for the determination of 10 ng mL−1 AFP intermittently (every 15 days). The conductivities could preserve 98.6%, 95.4%, 94.1%, 92.4%, 90.7% and 88.7% of the initial signal at 1st, 2nd, 3rd, 4th, 5th and 6th month, respectively, suggesting good stability.
Analysis of human serum specimens
The accuracy of the conductometric immunoassay was investigated for the analysis of human serum specimens by using the commercial human AFP ELISA kit as the reference. In this regard, 10 serum specimens including AFP with different concentrations were initially collected from our hospital according to the rules of the ethical committee of Shan'xi Province (note: informed consent was obtained from all individual participants). Thereafter, these samples before and after dilution with the dilution solution in the kits were detected by using the conductometric immunoassay and the human AFP ELISA kit, respectively. The results are listed in Table 3. The RSD values in all cases between the two methods were less than 10.88%, suggesting a good method accuracy for the detection of target AFP in comparison with the commercial human AFP ELISA kit at the 0.05 significance level.| Sample typea | Sample no. | Method; concentration (ng mL−1, n = 3) | RSD (%) | |
|---|---|---|---|---|
| This method | ELISA kit | |||
| a Samples 7–10 were obtained by dilution of samples by 6 to 10, 20, 50 and 100-fold with the dilution solution, respectively. | ||||
| Undiluted | 1 | 76.8 | 79.2 | 2.18 |
| Undiluted | 2 | 34.6 | 32.1 | 5.31 |
| Undiluted | 3 | 56.5 | 58.4 | 2.34 |
| Undiluted | 4 | 38.9 | 39.8 | 1.62 |
| Undiluted | 5 | 23.6 | 21.5 | 6.59 |
| Undiluted | 6 | 15.4 | 14.7 | 3.29 |
| Diluted | 7 | 1.43 | 1.51 | 3.85 |
| Diluted | 8 | 0.61 | 0.54 | 8.61 |
| Diluted | 9 | 0.28 | 0.24 | 10.88 |
| Diluted | 10 | 0.14 | 0.16 | 9.43 |
Conclusions
In summary, this work reports on the proof-of-concept of advanced conductometric immunoassay for the detection of a liver cancer biomarker, AFP, in human serum samples. Compared with traditional conductometric enzyme immunoassays, the highlight of this work can be summarized as follows: (i) the immunoreaction was implemented in a low-cost microplate in sandwich-type assay mode, whereas the signal was achieved by using enzyme-coated nanometer-sized enzyme-doped silica beads for the hydrolysis of arginine; (ii) the conductometric responses were amplified by coupling with nano labels and enzyme labels; and (iii) introduction of a bienzyme in this system caused a change in the conductivity of the detection solution. So, a combination of bienzyme-based biochemical reactions with the high-loading nanobeads can provide a new idea for the development of high-efficiency conductometric immunoassays in the electrochemical sensing system. By controlling the coated and labeled antibodies, this system can be easily applied for the determination of other disease-related biomarkers. Compared to the commercially available AFP ELISA kits, however, our strategy has several disadvantages as follows: (i) the fabrication of bienzyme-based signal tags is tedious and more expensive than the signal tags consisting of HRP-labeled antibody for ELISA since the ELISA can also be feasibly performed using a portable detector; (ii) the conductivity is not stable in solution with a low ionic strength and the electrode must be adjusted using a standard solution; and (iii) the whole duration of the assay was relatively longer than that of the commercialized ELISA kit (∼60–90 min) because this system was capable of continuously carrying out all steps in more than 110 min for one sample including incubation, washing, enzymatic reaction and measurement. Therefore, future work should be focused on the design of the sensor and the improvement of the assay time.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No: 61335012, 61705177, 61727823, 61575156, 61775178, and 61505159), and the China Postdoctoral Science Foundation (Grant No: 2017M613107 and 2015M572570).References
- Y. Komaki, F. Komaki, D. Micic, A. Ido and A. Sakuraba, Gastrointest. Endoscopy, 2017, 86, 93–104 CrossRef PubMed.
- K. Nguyen, A. Nguyen and D. Dabir, Anticancer Res., 2017, 37, 3379–3383 CrossRef PubMed.
- R. Wong, A. Ahmed and R. Gish, Clin. Liver Dis., 2015, 19, 309–323 CrossRef PubMed.
- G. Mizejewski, Life Sci., 1994, 56, 1–9 CrossRef.
- M. Moazeni, F. Karimzadeh and A. Kermanpur, Biosens. Bioelectron., 2018, 117, 748–757 CrossRef CAS PubMed.
- M. Attia, A. Youssef, Z. Khan and M. Abou-Omar, Talanta, 2018, 186, 36–43 CrossRef CAS PubMed.
- S. Yang, F. Zhang, Z. Wang and Q. Liang, Biosens. Bioelectron., 2018, 112, 186–192 CrossRef CAS PubMed.
- Z. Yu, G. Cai, R. Ren and D. Tang, Analyst, 2018, 143, 2992–2996 RSC.
- Q. Li, J. Jin, F. Lou and D. Tang, Sci. China: Chem., 2018, 61, 750–756 CrossRef CAS.
- Y. Li, L. Dong, X. Wang, Y. Liu, H. Liu and M. Xie, Spectrochim. Acta, Part A, 2018, 196, 103–109 CrossRef CAS PubMed.
- G. Korotcenkov, V. Brinzari and B. Cho, Microchim. Acta, 2016, 183, 1033–1054 CrossRef CAS.
- F. Christy and P. Shrivatav, Crit. Rev. Anal. Chem., 2011, 41, 236–269 CrossRef CAS.
- J. Tang, J. Huang, B. Su, H. Chen and D. Tang, Biochem. Eng. J., 2011, 53, 223–228 CrossRef CAS.
- G. Korotcenkov and B. Cho, Sens. Actuators, B, 2017, 244, 182–210 CrossRef CAS.
- N. Jaffrezic-Renault and S. Dzyadevych, Sensors, 2008, 8, 2569–2588 CrossRef PubMed.
- R. Dewan and M. Roy, Phys. Chem. Liq., 2011, 49, 145–154 CrossRef CAS.
- T. Matsue, M. Nishizawa and I. Uchida, Nippon Kagaku Kaishi, 1995, 1995, 493–501 CrossRef.
- I. Kucherenko, D. Kucherenko, O. Soldatkin, F. Lagarde, S. Dzyadevych and A. Soldatkin, Talanta, 2016, 150, 469–475 CrossRef CAS PubMed.
- K. Liang, W. Mu, M. Huang, Z. Yu and Q. Lai, Electroanalysis, 2006, 18, 1505–1510 CrossRef CAS.
- O. Soldatkina, O. Soldatkin, T. Velychko, V. Prilipko, M. Kuibida and S. Dzyadevych, Bioelectrochemistry, 2018, 124, 40–46 CrossRef CAS PubMed.
- Q. Zhou, Y. Lin, K. Zhang, M. Li and D. Tang, Biosens. Bioelectron., 2018, 101, 146–152 CrossRef CAS PubMed.
- J. Shu and D. Tang, Chem. – Asian J., 2017, 12, 2780–2789 CrossRef CAS PubMed.
- Z. Qiu, J. Shu and D. Tang, Anal. Chem., 2018, 90, 1021–1028 CrossRef CAS PubMed.
- Z. Luo, L. Zhang, R. Zeng, L. Su and D. Tang, Anal. Chem., 2018, 90, 9568–9575 CrossRef CAS PubMed.
- Z. Qiu, J. Shu and D. Tang, Anal. Chem., 2017, 89, 5152–5160 CrossRef CAS PubMed.
- D. Knopp, D. Tang and R. Niessner, Anal. Chim. Acta, 2009, 647, 14–30 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, in Pierce Biotechnology, Thermo Fisher Scientific, Rockford, Illinois, USA, 2nd edn, 2008, pp. 798–800 Search PubMed.
- Y. Lin, Q. Zhou, Y. Lin, D. Tang, R. Niessner and D. Knopp, Anal. Chem., 2015, 87, 8531–8540 CrossRef CAS PubMed.
- Z. Zhang, G. Li, Z. Cheng and X. Zuo, J. Hazard. Mater., 2012, 229–230, 404–410 Search PubMed.
- D. Thangaraju, A. Durairajan, D. Balaji, S. Babu and Y. Hayakawa, Mater. Chem. Phys., 2012, 135, 1115–1121 CrossRef CAS.
- L. Zhang, C. Fan, M. Liu, F. Liu, S. Bian, S. Du, S. Zhu and H. Wang, Sens. Actuators, B, 2018, 266, 543–552 CrossRef CAS.
- T. Hu, Z. Wang, P. Lv, K. Chen and Z. Ni, Sens. Actuators, B, 2018, 266, 270–275 CrossRef CAS.
- Y. Shen, G. Shen and Y. Zhang, Microchim. Acta, 2018, 185, 346 CrossRef PubMed.
- W. Xiang, G. Wang, S. Cao, Q. Wang, X. Xiao, T. Li and M. Yang, Microchim. Acta, 2018, 185, 335 CrossRef PubMed.
- P. Preechakasedkit, W. Siangproh, N. Khongchareonporn, N. Ngamrojanavanich and O. Chailapakul, Biosens. Bioelectron., 2018, 102, 27–32 CrossRef CAS PubMed.
- G. Li, S. Li, Z. Wang, Y. Xue, C. Dong, J. Zeng, Y. Huang, J. Liang and Z. Zhou, Anal. Biochem., 2018, 547, 37–44 CrossRef CAS PubMed.
- X. Fang, J. Liu, J. Wang, H. Zhao, H. Ren and Z. Li, Biosens. Bioelectron., 2017, 97, 218–225 CrossRef CAS PubMed.
- Y. Yuan, S. Li, Y. Xue, J. Liang, L. Cui, Q. Li, S. Zhou, Y. Huang, G. Li and Y. Zhao, Anal. Biochem., 2017, 534, 56–63 CrossRef CAS PubMed.
- S. Li, J. Liang, Z. Zhou and G. Li, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 382, 022017 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |