
Functionalized gold nanoparticle-enhanced competitive assay for sensitive small-molecule metabolite detection using surface plasmon resonance†
Yong
Cao
 ab,
Bethany
Griffith
b,
Prasanna
Bhomkar
b,
David S.
Wishart
bcd and
Mark T.
McDermott
*ab
ab,
Bethany
Griffith
b,
Prasanna
Bhomkar
b,
David S.
Wishart
bcd and
Mark T.
McDermott
*ab
aDepartment of Chemistry, 11227 Saskatchewan Drive. and University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: mark.mcdermott@ualberta.ca
bNational Institute for Nanotechnology, 11421 Saskatchewan Drive, Edmonton, Alberta T6G 2M9, Canada
cDepartment of Computing Science, 2-21 Athabasca Hall, University of Alberta, Edmonton, Alberta T6G 2E8, Canada
dDepartment of Biological Sciences, CW 405, University of Alberta, Edmonton, Alberta T6G 2E9, Canada
First published on 6th November 2017
Abstract
The development of sensing systems for the measurement of small molecules is an active area of research. A sensor based approach for the measurement of metabolites can potentially provide the simplicity and portability required for widespread use. Rapid detection and quantitation of small-molecule metabolites can potentially emerge as an effective way to link the metabolite profile to the disease state. Surface plasmon resonance (SPR) combined with molecular recognition elements to deliver high specificity is a sensing platform that has been widely applied for a large range of biomolecules. However, direct detection of small molecules with SPR challenges the refractive index based detection mechanism. The work described here combines a periplasmic binding protein for recognition with target modified gold nanoparticles (AuNPs) in a competitive assay format for folic acid (FA) detection. Specifically, a SPR imaging substrate containing immobilized folate binding protein (FBP) is used to measure the adsorption of FA conjugated AuNPs. The immobilization of the FBP and the binding of the FA conjugated AuNPs are characterized and optimized. It is shown that free FA in solution can be quantitatively measured by competition for the surface binding sites with the functionalized AuNPs. We demonstrate that the dynamic range can be lowered from micromolar to nanomolar by simply decreasing the concentration of FA conjugated AuNPs, thus lowering the limit of detection to 2.9 nM. This type of competitive assay can be applied to a range of small molecules, which paves the way for future multiplexed analysis of metabolites using SPR.
Introduction
The development of sensor based approaches for the detection and quantitative measurement of small molecules (MW < 1000 Da) is impacting areas such as environmental monitoring1,2 and human health.3,4 For example, the measurement of small-molecule metabolites is a research area with increasing interest due to the promise of using the information for diagnostic purposes.4–8 Currently, the major advances in the identification and quantification of metabolites are being driven by work that uses nuclear magnetic resonance9–12 (NMR) and mass spectrometry10,11,13,14 (MS). These powerful techniques are producing and will continue to produce metabolite profiles8 that can be linked to the disease state. It is expected that the diagnosis and treatment of an expanded number of diseases will be greatly aided by the determination of the quantities of metabolites in a panel. In this regard, facile, rapid and quantitative sensing platforms for small-molecule metabolites will be important. The work presented here describes the development of a small molecule assay on a detection platform that satisfies the requirements noted above.Surface plasmon resonance (SPR) is an optical sensing technique first developed in 1990s.1,15 It can provide label-free, real-time, sensitive, and quantitative information of bio-molecular interactions.1 The technique is based upon the detection of a change in dielectric constant within a few hundreds of nanometers1 of a metal–solution interface and has been broadly applied to measure larger biomolecules such as proteins16–21 and nucleic acids.22–25 It is widely known that the direct detection of low molecular weight species with SPR is very challenging due to the small refractive index change induced by the adsorption of a monolayer of small molecules. There have been reports of the direct detection of small molecules but such demonstrations are sporadic and these measurements generally suffer from low signal intensity and poor sensitivity.26,27 The challenges of measuring small molecules with SPR could be addressed by the application of nanomaterials as labels.28,29 For example, metal nanoparticles,16,25,30 magnetic nanoparticles,31,32 quantum dots,33 and carbon-based nanomaterials22,34 have been used to amplify the signal in different assays, but most of the time such demonstrations are limited to the detection of proteins and nucleic acids, not metabolites. In addition, studies have shown that an indirect detection format,35,36 either inhibition or competition, can be utilized to measure targets like steroid hormones,37,38 toxins,39 drugs,40 and explosive residues.41 Based on the previous work, a pathway for the inclusion of SPR as a sensing platform for metabolites may be realized by employing metal nanoparticle reagents in a competitive format.
In this work, we have developed a gold nanoparticle (AuNP) enhanced competitive assay to detect and quantitate folic acid (FA) using SPR imaging (SPRi). Folic acid, which is used in the prevention and treatment of folate deficiency and megaloblastic anemia42 has been used as the target for a wide range of sensing strategies. Electrochemical techniques,43 surface enhanced Raman scattering (SERS)44 as well as SPR45 have been used to measure FA. The physiological levels of FA in human blood range from a few nanomolar (nM) to less than 50 nM.42,46 Our assay incorporates an immobilized layer of the folate binding protein (FBP) as the sensing surface and 10 nm FA-functionalized AuNPs (FA-AuNP) as a competitive reagent. The FBP was tagged with polyhistidine (HIS6) for facile immobilization onto the SPRi chip surface. The relatively large size of the AuNP as well as the previously reported plasmonic coupling between the particle and the SPR substrate47,48 enable the measurement of very low surface coverage of the adsorbed AuNPs. We demonstrate the applicability of this competitive assay to detect and quantitate FA in both the micromolar and nanomolar concentration ranges.
Experimental
Materials
BSA (bovine serum albumin), folic acid (meeting USP testing specifications), DTSP (3,3′-dithiodipropionic acid di(N-hydroxysuccinimide ester)), and DMSO (dimethyl sulfoxide, ≥99.9%) were all purchased from Sigma-Aldrich and used as received. HBS-P buffer (0.01 M HEPES, pH 7.4, 0.15 M NaCl, 0.005% v/v surfactant P20) and nickel chloride (NiCl2) solution (0.5 mM) were purchased from GE Healthcare. 10 nm citrate-capped gold nanoparticle (AuNP) stock solution (5.7 × 1012 particles per mL, 9.47 nM) was purchased from Ted Pella, Inc. Anhydrous ethyl alcohol was obtained from Commercial Alcohols. Deionized (DI) (18 M)/filtered H2O was from a NANOpure water purification system (Barnstead International, Dubuque, IA). 2-{2-[2-(1-Mercaptoundec-11-yloxy)-ethoxy]-ethoxy}-ethoxy-nitrilotriacetic acid (HS-(CH2)11-EG3-NTA) was purchased from ProChimia Surfaces. (Tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-dimethylchlorosilane was purchased from United Chemical Technologies, Inc. Polyhistidine-tagged folic acid binding protein (FBP) and polyhistidine-tagged periplasmic binding protein for glutamine (GBP) were expressed in E. coli. The complete amino-acid sequences and detailed expression and purification process for the two binding proteins are provided in the ESI.† Purified proteins were received in stock solutions of 18.3 μM in phosphate buffered saline (PBS, pH 7.4) for FBP and 34.2 mM in PBS buffer for GBP.Fabrication of a SPRi chip with multiple gold (Au) spots
An SF-10 glass sheet (100 mm × 100 mm × 1 mm) was purchased from Schott Glass. The sheet was cut into small square substrates (18 mm × 18 mm). The substrates were cleaned using hot piranha solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 30% H2O2:H2SO4) followed by thorough rinsing with DI water. Substrates were blown dry using Ar gas. [Warning: Piranha solution should be handled with extreme care; it is a strong oxidant and reacts violently with many organic materials. It also presents an explosion danger. All work should be performed under a fume hood with appropriate personal safety equipment.] The cleaned and dried substrates were mounted to a mask that exposes nine 2 mm diameter round spots and placed into the chamber of a thermal evaporator (Torr International Inc., New Windsor, NY). Metal films of chromium (2 nm) and gold (42 nm) were sequentially coated on the glass substrate through the mask. After removal from the evaporator, the substrates were then exposed to a vapor of (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-dimethylchlorosilane under reduced pressure for 24 h to create a hydrophobic background on the glass surface. The SPRi chips were stored in a desiccator under vacuum at room temperature until use.
:4 30% H2O2:H2SO4) followed by thorough rinsing with DI water. Substrates were blown dry using Ar gas. [Warning: Piranha solution should be handled with extreme care; it is a strong oxidant and reacts violently with many organic materials. It also presents an explosion danger. All work should be performed under a fume hood with appropriate personal safety equipment.] The cleaned and dried substrates were mounted to a mask that exposes nine 2 mm diameter round spots and placed into the chamber of a thermal evaporator (Torr International Inc., New Windsor, NY). Metal films of chromium (2 nm) and gold (42 nm) were sequentially coated on the glass substrate through the mask. After removal from the evaporator, the substrates were then exposed to a vapor of (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-dimethylchlorosilane under reduced pressure for 24 h to create a hydrophobic background on the glass surface. The SPRi chips were stored in a desiccator under vacuum at room temperature until use.
Preparation of 10 nm folic acid conjugated gold nanoparticles (FA-AuNPs)
We first added 10 μL of 10 mM DTSP in DMSO to 1 mL of 10 nm citrate-capped AuNP solution, and incubated the mixed solution for 30 min at room temperature. After incubation, 10 μL of 10 mM FA in DMSO solution was added to the mixed solution. The final solution was left overnight (∼17 h) at room temperature. Then the solution was centrifuged for 45 min at 14000 rpm (Eppendorf a5417R microcentrifuge). The supernatant was carefully removed to avoid any loss of the particles. The AuNP pellet was re-dispersed in 1 mL DI water as a stock FA-AuNP solution (9.47 nM based on the initial AuNP concentration). The suspended FA-AuNP solution was stored at 4 °C until use.
UV-vis spectroscopy
Extinction spectra of 10 nm citrate-capped AuNP solution, DTSP-AuNP solution, and FA-AuNP solution were obtained by UV-Vis measurements. 500 μL of each AuNP solution was added to a quartz micro-cuvette for the measurement. All solutions were measured in transmission mode in a double-beam PerkinElmer Lambda 35 instrument. DI water was used as a reference for each measurement.Raman spectroscopy
Raman spectra were collected with a Renishaw inVia Raman microscope fitted with a liquid cell accessory for solution samples. Spectra were collected using 633 nm excitation from a HeNe laser with 10 mW of power through a 50× objective. The integration time was 50 s. 500 μL of 100 μM folic acid solution and 500 μL of 9.47 nM FA-NP solution were measured separately.SPRi measurement of FBP immobilization
Prior to the modification, the homemade SPRi chip was rinsed with pure ethanol and DI water and dried under an argon gas flow. For the chip modification, the four spots at the corner of the square substrate were used as the control group and the remaining five spots in the middle were the experimental group. A 2.5 μL droplet of 1 mg mL−1 BSA solution was added to the control spots and a 2.5 μL droplet of 2 mM HS-(CH2)11-EG3-NTA solution was added to the experimental spots. The chips with the solution droplets were stored in a humid Petri dish for 7 h at room temperature. Then, the chip was rinsed with DI water and dried under an argon gas flow. Each spot of the chip was next activated by adding 2.5 μL of 0.5 mM NiCl2 solution over 1 h in the Petri dish. After rinsing and drying, the chip was mounted to the SPRi instrument (Horizon SPRimager, GWC Technologies, Madison, MI) to measure the adsorption of FBP onto the SH-NTA self-assembled monolayer (SAM). The apparatus has been described in detail elsewhere.49,50 FBP solutions at five different concentrations (91.5 nM, 183 nM, 366 nM, 915 nM, and 1830 nM) were prepared from the stock FBP solution (18.3 μM). All FBP solutions were sequentially exposed to the chip surface from the most diluted solution to the least diluted solution, and a real-time SPRi sensorgram was recorded and analyzed.SPRi measurement of 10 nm FA-AuNP binding
FBP and GBP were immobilized on the SPRi chip as described above to study the binding of the 10 nm FA-AuNPs. 2.5 μL of 3.42 μM GBP solution and 1.83 μM FBP solution were added to the corresponding control and experimental spots after the formation of SH-NTA SAMs and activation with Ni2+. After overnight (∼17 h) incubation, the chip was rinsed with DI water and dried under an argon gas flow. Seven 10 nm FA-AuNP solutions with dilution factors 1/2, 1/3, 1/5, 1/10, 1/25, 1/50, and 1/100 from the 9.47 nM 10 nm FA-AuNP stock solution were sequentially flowed into the instrument and a real-time SPRi sensorgram was obtained.Competitive assays
All assays were carried out on chips containing FBP (target) and GBP (control) spots. As described below, two concentration ranges of FA were studied. For the higher (μM) range, a solution of 9.47 nM (before mixing) 10 nm FA-AuNPs was mixed with FA standard solutions of 2, 20, 50, and 100 μM in a 1:1 volume ratio. The solutions were flowed into the instrument for measurements separately and an SPRi sensorgram was obtained for each mixed solution. For the physiologically relevant concentration range (nM), we decreased the concentration of 10 nm FA-AuNP solution to 379 pM (before mixing). The NP solution was mixed with FA standard solutions of 5, 10, 20, 30, 40, and 50 nM concentrations. The solutions were flowed into the instrument continuously from the most concentrated to the least concentrated and an SPRi sensorgram was obtained.
All Langmuir isotherms in this work were obtained by fitting the experimental data using the one-site ligand-binding model included in SigmaPlot (Systat Software, Inc., San Jose, CA). All error bars shown in this work represent the standard deviation of triplicate measurements.
Results and discussion
A variety of assay formats have been developed for recognition based measurement of small molecules for SPR using antibodies and aptamers.27,35–39 The competitive format for folic acid (FA) demonstrated in this work is shown in Fig. 1. A layer of polyhistidine labeled folate binding protein (FBP) is immobilized onto the gold SPRi chip through complexation with Ni2+ and a nitrilotriacetic acid (NTA)-terminated monolayer. A sample solution containing FA will be spiked with AuNPs conjugated to FA. The free FA in solution then competes with the AuNPs for FBP binding sites. The binding of the relatively large AuNPs to the SPR chip can easily be measured. The relationship between the concentration of free FA and the SPRi signal is expected to result in a calibration curve with a negative slope as shown in Fig. 1. | ||
| Fig. 1 Schematic illustration of the folic acid (FA) functionalized gold nanoparticle (FA-AuNP) enhanced competitive assay for the detection of FA. Polyhistidine tagged folic acid binding protein (FBP) is immobilized onto the chip surface via Ni2+-NTA coordination chemistry. FA-AuNPs and FA compete for surface binding sites provided by FBP. | ||
The binding between FA and FBP has been previously characterized.51–53 Thus, the location of the binding site is known, enabling the expression of FBP in E. coli with the HIS6 tag in a region of the protein that ensures the optimum orientation upon immobilization. NTA functional groups combined with Ni2+ are commonly used to capture or immobilize histidine tagged proteins.54 Thus, the SPRi chip was modified with an HS-(CH2)11-EG3-NTA self-assembled monolayer (SAM). The immobilization of the FBP to the chip was optimized by measuring the amount of FBP adsorbed with SPRi for five different solution concentrations (91.5 nM, 183 nM, 366 nM, 915 nM, and 1.83 μM). Fig. 2A contains a continuous flow SPRi sensorgram for the adsorption of FBP onto the chip surface. FBP exhibits specific adsorption onto the NTA-terminated monolayer (NTA SAM), while insignificant adsorption is observed for BSA control spots. A plot of the SPR signal vs. FBP solution concentration is shown in Fig. 2B. These data are fit to a Langmuir adsorption isotherm with a reasonable correlation (R2 = 0.9803). The dissociation constant (Kd) for the FBP adsorption was determined from this fit to be 47 ± 5 nM, which has a similar order of magnitude to the value reported in the literature (14 ± 1 nM).54 The results of the experiment shown in Fig. 2 allow us to conclude that the FBP is being immobilized via the expected complexation interaction and to control the surface density of FBP through the solution concentration.
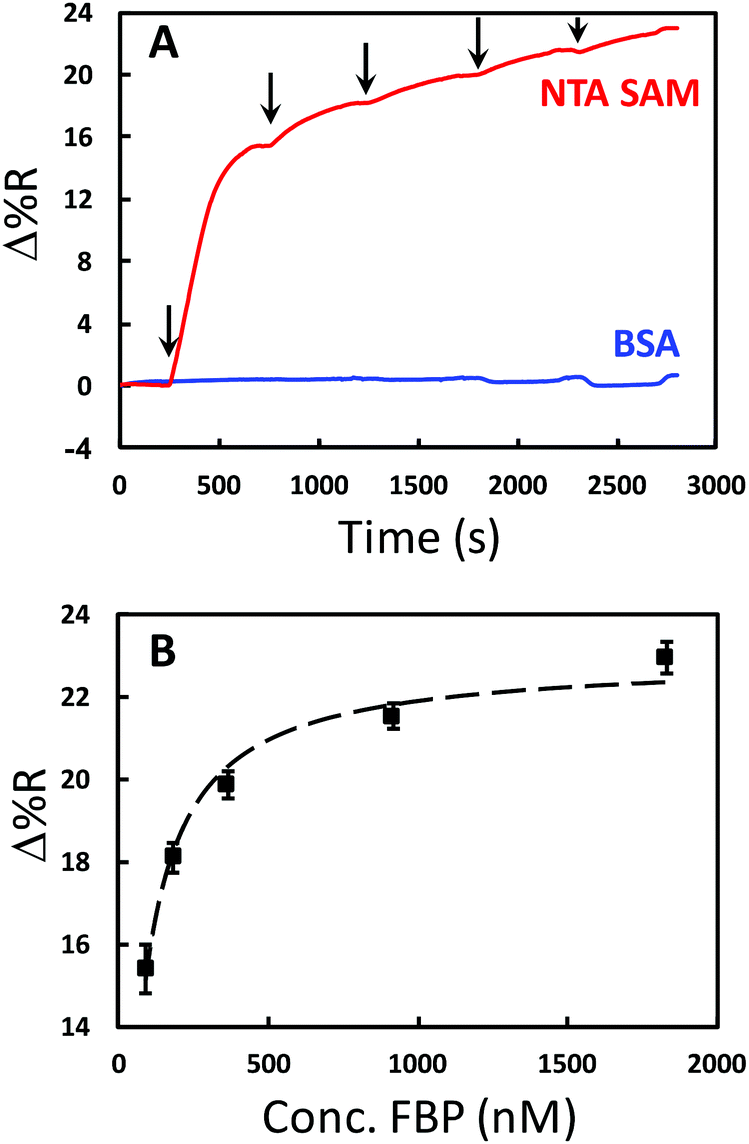 | ||
| Fig. 2 Results tracking the immobilization of polyhistidine tagged folic acid binding protein (FBP) onto the SPRi chip surface. (A) Typical continuous SPRi sensorgrams for FBP adsorption for different concentrations (91.5 nM, 183 nM, 366 nM, 915 nM, and 1830 nM) to NTA SAMs (red) and a BSA control surface (blue). The arrows indicate the injection of each concentration. (B) A plot of SPR signal vs. FBP solution concentration for adsorption onto NTA SAM. The symbols are the data points and the dashed line is the least-squares fit of the Langmuir adsorption isotherm equation (R2 = 0.9803). | ||
The FA conjugated AuNPs (FA-AuNPs) were prepared through a 2-step reaction. First, a monolayer of the bifunctional ligand DTSP was adsorbed onto 10 nm AuNPs. This ligand has been used previously to conjugate antibodies to AuNPs.55 FA was then linked to the AuNP via an amide bond formed from the reaction of the free amine on the FA molecule and the succinimide ester group on the DTSP molecule. The as-prepared FA-AuNPs were characterized by UV-Vis spectroscopy as well as Raman spectroscopy. Fig. 3A shows the extinction spectra of the initial 10 nm citrate-AuNPs, the DTSP modified AuNPs, and the FA-AuNPs. All spectra exhibit typical localized surface plasmon resonance (LSPR) peaks characteristic of 10 nm AuNPs. The extinction maxima of the LSPR band red shift from 518 nm (citrate-AuNP) to 520 nm (DTSP-AuNP) to 522 nm (FA-AuNP), indicative of a variation of the dielectric constant of the surface layer due to each modification step. The extinction spectra at higher wavelength (Fig. S1†) indicate negligible aggregation of the modified NPs. Fig. 3B shows the Raman spectra of FA in solution and a solution of the FA-AuNPs. Bands characteristic of FA are clearly observed in the spectra of the FA-AuNPs consistent with the association of FA with the AuNPs. The UV-Vis results combined with the Raman results confirm the successful conjugation of FA to the 10 nm AuNP surface. The FA-AuNPs showed colloidal stability when diluted in buffer solution, which is critical for a useful reagent in a competitive assay format.
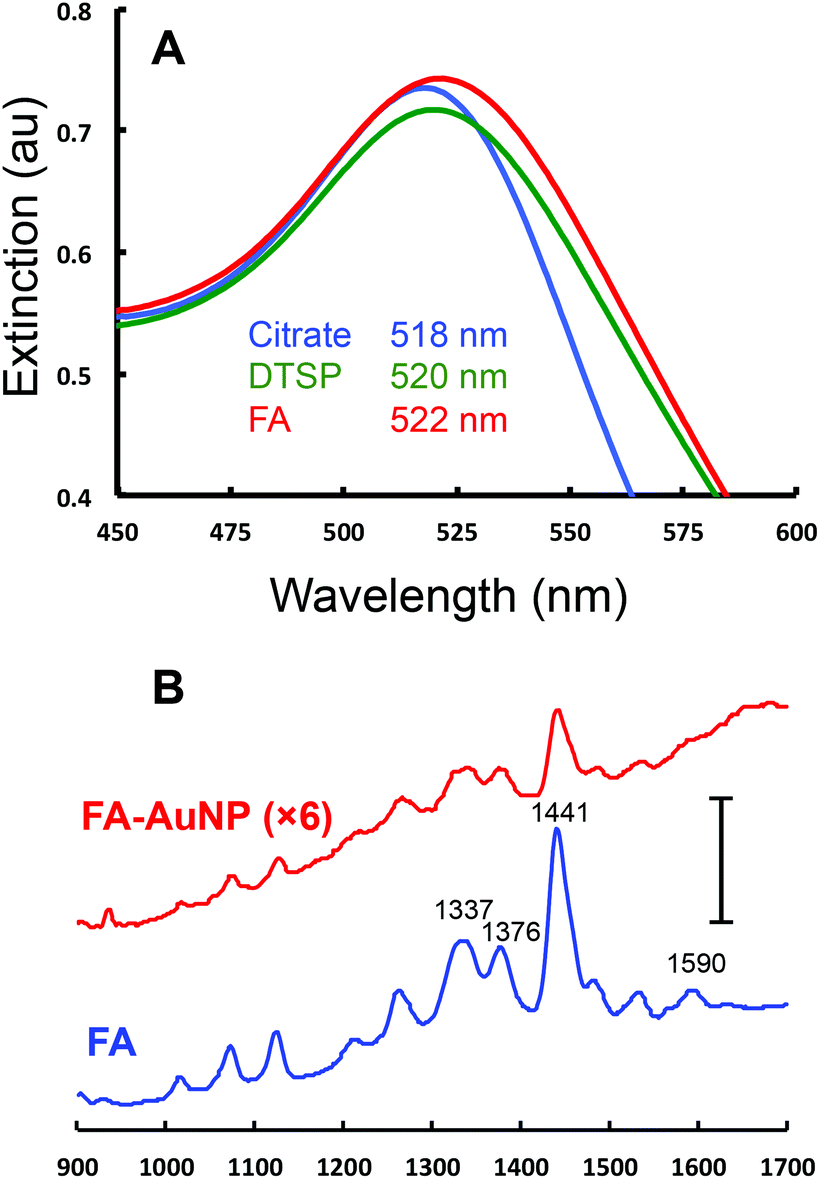 | ||
| Fig. 3 UV-Vis and Raman characterization of 10 nm AuNPs. (A) Extinction spectra for the as-purchased 10 nm citrate capped AuNP (blue), the AuNP after modification with DTSP-AuNP (green), and FA-AuNP (red). The axes are expanded around the maximum extinction for clarity and λmax values for the three spectra are listed. The full range extinction spectra are included in Fig. S1.† (B) Raman spectra of FA in solution (100 μM in water, blue) and a solution of 9.47 nM of FA-AuNPs (red, signal ×6). The scale bar is equivalent to 40000 counts. | ||
The binding of FA-AuNPs to FBP immobilized on the chip surface was studied to aid in the design of a competitive assay. The chip contained 9 spots modified with either FBP or the control protein (Fig. S2†). The control spots in this experiment were modified identically to the FBP spots with histidine tagged glutamine binding protein (GBP). Part A of Fig. 4 shows a continuous SPRi sensorgram for various concentrations of FA-AuNPs interacting with the chip. An increase in the SPRi signal is observed at the FBP spots with each successive higher concentration. As shown in Fig. 4A, insignificant adsorption of the FA-AuNPs was observed at the GBP spots. Thus, the FA-AuNPs bind specifically to the immobilized FBP. Fig. 4B shows a plot of the baseline corrected SPRi signal vs. the concentration of FA-AuNPs and the corresponding Langmuir fit. Despite the rather poor fit, the Langmuir model yields a Kd value of 0.8 ± 0.2 nM. Fitting the results in Fig. 4B with the 4-parameter logistical model yields an R2 value of 0.999 and a Kd of 0.3 nM. The binding of large polymeric nanoparticles conjugated with FA to immobilized FBP was previously studied.56 The density of the FA on the polymer particles was relatively low (<15% of the surface covered) and the Kd for this system was reported to be 800 nM. We estimate a coverage of FA of at least 80% and speculate that the interaction between our FA-AuNPs and the immobilized FBP may involve multivalent interactions resulting in a tighter binding and a lower Kd, but this is not examined further in this work. Scanning electron microscopy (SEM) analyses of the chips following FA-AuNP adsorption reveal that very low coverage of the AuNPs is being detected at a lower concentration (Fig. S3†). For example, we estimate a coverage of 1.3 × 109 particles per cm2 resulting from the adsorption from 94.7 pM solutions. A coverage as low as 2 × 107 particles per cm2 has been detected in the initial reports of AuNP enhanced SPR.48 This lays a solid foundation for the following analytical performance of the competitive assay.
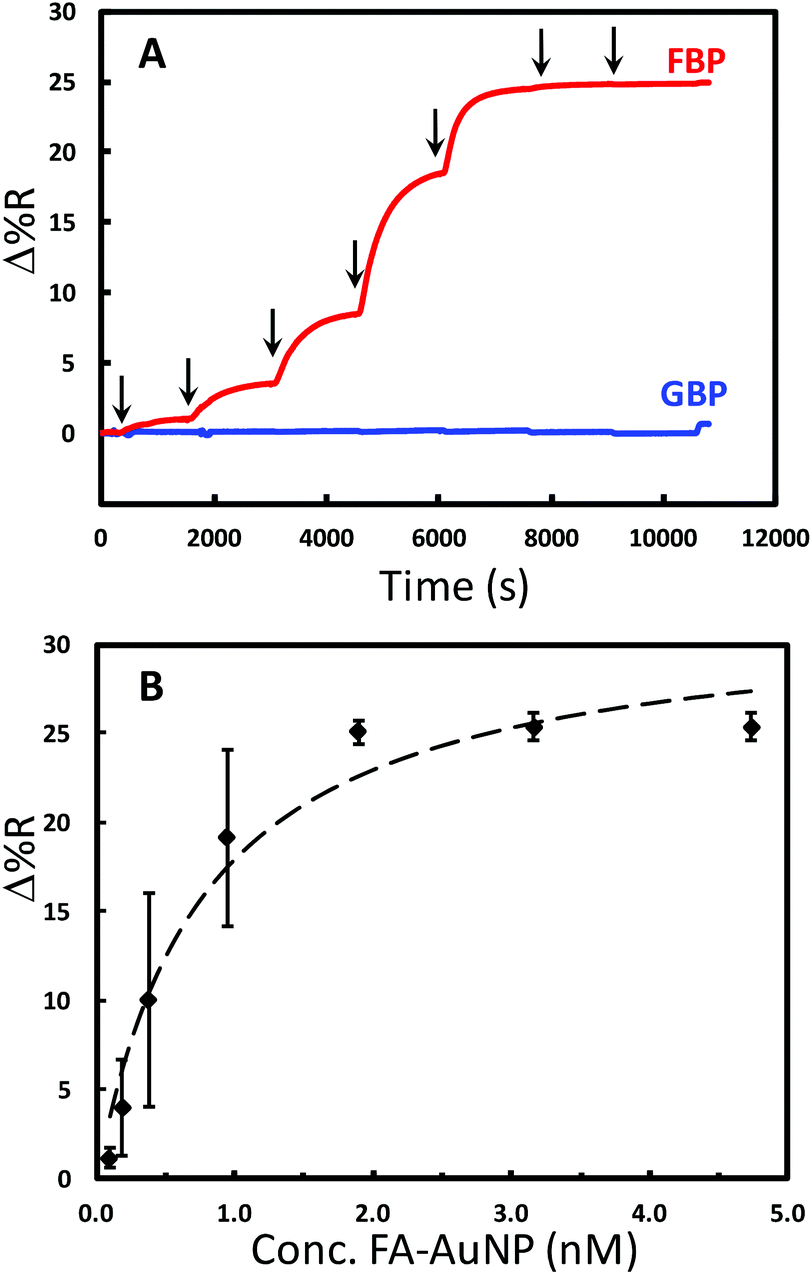 | ||
| Fig. 4 Binding of 10 nm FA-AuNPs to FBP. (A) Typical continuous SPRi sensorgrams for the adsorption of FA-AuNPs at seven concentrations (94.7 pM–4.74 nM) onto the FBP surface (red) and GBP control surface (blue). The arrows indicate the injection of each concentration. (B) A plot of SPR signal vs. FA-AuNP solution concentration for adsorption onto FBP. The symbols are the data points and the dashed line is the least-squares fit of the Langmuir adsorption isotherm equation (R2 = 0.9648). | ||
As noted above, FA has been the subject of many measurement studies. A variety of techniques have quantified FA in the concentration range of pM to hundreds of μM.43–45 The physiological concentration range of FA is 10–50 nM in blood. The proposed competitive assay was first tested to detect and quantitate FA in the μM concentration range as a proof-of-principle to ensure that it works for FA detection. Solutions containing a fixed concentration of FA-AuNPs (4.74 nM, after mixing) and various concentrations of free FA (2, 20, 50, and 100 μM) were allowed to compete with the FBP on the chip. The SPRi sensorgrams for these experiments exhibit a maximum signal for solutions with no free FA present and lower signals for solutions of higher free FA concentrations (Fig. S4A†). Fig. 5A shows a plot of the SPRi signal vs. free FA concentration in the concentration range that provided the highest sensitivity. This plot is analogous to a dose–response curve in drug screening or enzyme inhibition assays.57 The plot is linear with the expected negative slope and serves as a calibration curve for free FA in solution, which can be used to successfully quantitate FA in the range of 2 to 100 μM. The ability of the FA-AuNPs to preferentially compete for the FBP sites over free FA is illustrated in Fig. 5A. A reasonable signal for FA-AuNP adsorption is observed at the highest concentration of FA (100 μM) which represents a ∼20000× excess. This is consistent with stronger binding of the FA-AuNPs due to multivalent interactions noted above.
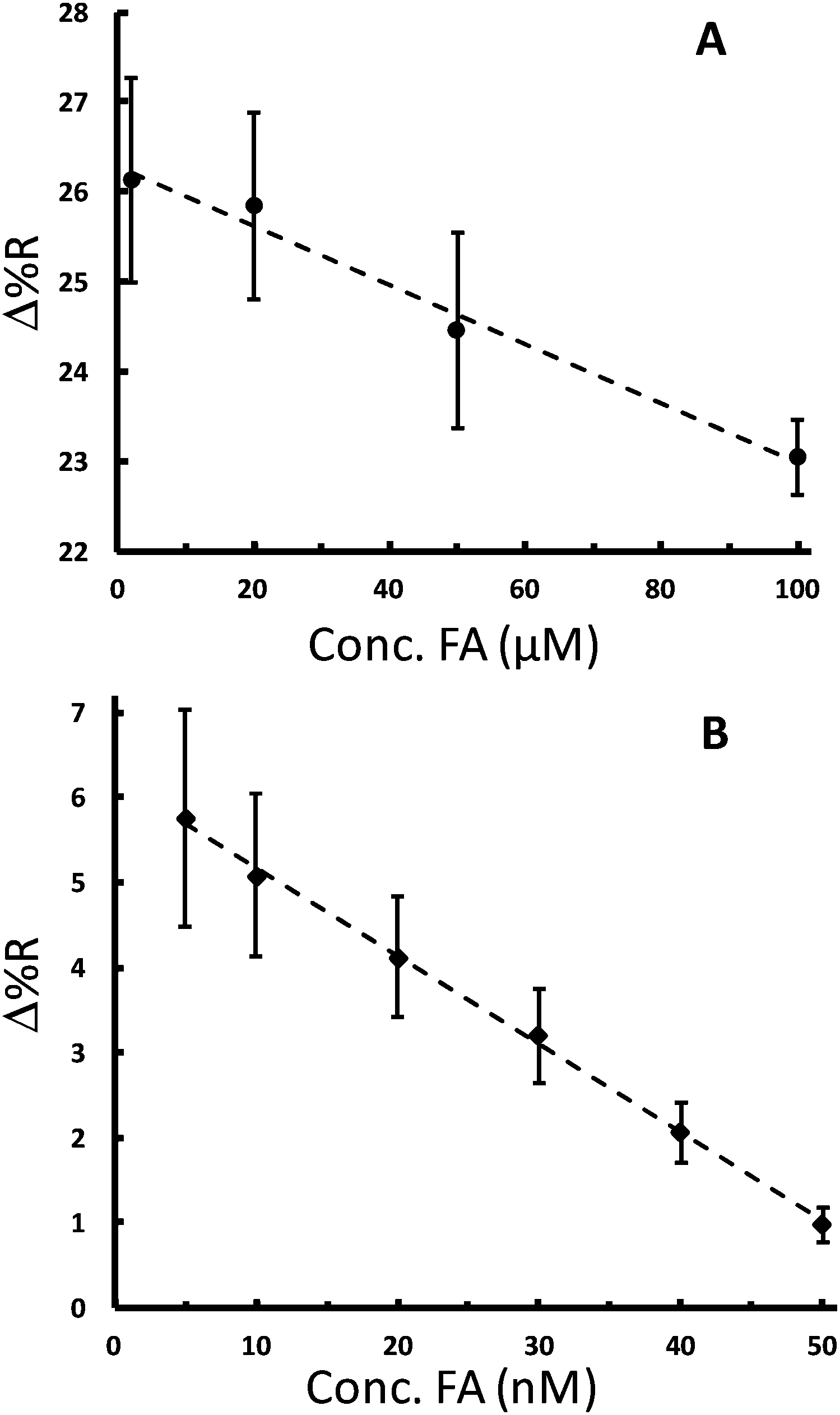 | ||
| Fig. 5 Calibration curves for the determination of FA using the AuNP based competitive assay. (A) Plot of SPRi signal vs. concentration of free FA for the assay where the concentration of FA-AuNPs was 4.74 nM (after 1:1 mixing). (B) Plot of SPRi signal vs. concentration of free FA for the assay where the concentration of FA-AuNPs was 189 pM (after 1:1 mixing). In both plots, the symbols represent the mean of at least 3 repetitive measurements and the error bars are the standard deviation. The dashed line through the data is the linear least-squares fit. | ||
It is known from the application of other competitive assays that the dose–response curve can be shifted to lower target concentrations by decreasing the concentration of the competitor.58,59 Thus, the quantitative concentration range can be varied in our competitive assay by changing the concentration of the FA-AuNP reagent. Qualitatively, considering Fig. 4B, a concentration of 4.74 nM is a concentration that results in the saturation coverage of the AuNPs. Thus, a relatively high concentration of free FA would be required to compete for the FBP sites. Lowering the concentration of the FA-AuNPs to the sub-nM range should provide higher sensitivity as suggested by the slope of the isotherm shown in Fig. 4B. Accordingly, we incorporated a fixed concentration of the FA-AuNPs 189 pM (after mixing) into solutions with free FA concentrations in the range of 5 to 50 nM. Analysis of the corresponding sensorgrams (Fig. S4B†) yielded a plot as shown in Fig. 5B. A linear curve is obtained in the range of 5 to 50 nM, which contains the physiological concentration range of interest. The limit of detection is determined to be 2.9 nM. A previous investigation that employed an antibody based competitive assay with SPR detection measured folic acid in fortified food from 16 nM to 1269 nM.45 Our study shows that small molecule functionalized AuNPs are flexible reagents for competitive assays with a tunable concentration range.
Conclusions
We have developed a SPRi-based competitive assay for detection and quantitation of FA. The assay is flexible and is able to quantitate FA at physiological concentrations. The introduction of FA-AuNPs overcomes the sensitivity challenge of detecting small molecules with SPR due to the mass effect as well as the coupling effect of the functionalized AuNPs. The FA-AuNPs are stable in buffer, and exhibit strong and specific binding to the FA binding protein, thanks to the chemistries used to immobilize the protein and conjugate FA to the AuNP surface. The competitive assay shows excellent analytical performance to detect and quantitate FA in the physiological concentration range, with a linear range from 5 nM to 50 nM and a limit of detection of 2.9 nM. In addition, the dynamic range can be adjusted by simply changing the concentration of the FA-AuNP solution used for the competition. Most importantly, this competitive assay can be applied to other small-molecule metabolites as well, which paves the way for future multiplexed detection of metabolites using the SPRi platform.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the NanoFab at the University of Alberta for dicing saw training and the National Institute for Nanotechnology for SEM characterization. This work was supported by Alberta Innovate Health Solutions (AIHS) and the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery Grant to MTM).Notes and references
- J. Homola, Chem. Rev., 2008, 108, 462–493 CrossRef CAS PubMed.
- F. Long, A. Zhu and H. Shi, Sensors, 2013, 13, 13928–13948 CrossRef CAS PubMed.
- J. R. Lee, J. Choi, T. O. Shultz and S. X. Wang, Anal. Chem., 2016, 88, 7457–7461 CrossRef CAS PubMed.
- M. Hilvo, I. de Santiago, P. Gopalacharyulu, W. D. Schmitt, J. Budczies, M. Kuhberg, M. Dietel, T. Aittokallio, F. Markowetz, C. Denkert, J. Sehouli, C. Frezza, S. Darb-Esfahani and E. I. Braicu, Cancer Res., 2016, 76, 796–804 CrossRef CAS PubMed.
- T. Seviour, L. E. Doyle, S. J. Lauw, J. Hinks, S. A. Rice, V. J. Nesatyy, R. D. Webster, S. Kjelleberg and E. Marsili, Chem. Commun., 2015, 51, 3789–3792 RSC.
- C. W. Lam, C. Y. Law, K. H. Sze and K. K. To, Clin. Chim. Acta, 2015, 438, 24–28 CrossRef CAS PubMed.
- M. H. Park and K. Igarashi, Biomol. Ther., 2013, 21, 1–9 CrossRef CAS PubMed.
- A. R. Fernie, R. N. Trethewey, A. J. Krotzky and L. Willmitzer, Nat. Rev. Mol. Cell Biol., 2004, 5, 763–769 CrossRef CAS PubMed.
- S. Ravanbakhsh, P. Liu, T. C. Bjorndahl, R. Mandal, J. R. Grant, M. Wilson, R. Eisner, I. Sinelnikov, X. Hu, C. Luchinat, R. Greiner and D. S. Wishart, PLoS One, 2015, 10, e0124219 Search PubMed.
- S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem, P. Liu, Z. T. Dame, J. Poelzer, J. Huynh, F. S. Yallou, N. Psychogios, E. Dong, R. Bogumil, C. Roehring and D. S. Wishart, PLoS One, 2013, 8, e73076 CAS.
- N. Psychogios, D. D. Hau, J. Peng, A. C. Guo, R. Mandal, S. Bouatra, I. Sinelnikov, R. Krishnamurthy, R. Eisner, B. Gautam, N. Young, J. Xia, C. Knox, E. Dong, P. Huang, Z. Hollander, T. L. Pedersen, S. R. Smith, F. Bamforth, R. Greiner, B. McManus, J. W. Newman, T. Goodfriend and D. S. Wishart, PLoS One, 2011, 6, e16957 CAS.
- C. K. Larive, G. A. Barding Jr. and M. M. Dinges, Anal. Chem., 2015, 87, 133–146 CrossRef CAS PubMed.
- D. Chen, X. Su, N. Wang, Y. Li, H. Yin, L. Li and L. Li, Sci. Rep., 2017, 7, 40543 CrossRef CAS PubMed.
- S. Zhao, X. Luo and L. Li, Anal. Chem., 2016, 88, 10617–10623 CrossRef CAS PubMed.
- M. Couture, S. S. Zhao and J. F. Masson, Phys. Chem. Chem. Phys., 2013, 15, 11190–11216 RSC.
- B. Wu, R. Jiang, Q. Wang, J. Huang, X. Yang, K. Wang, W. Li, N. Chen and Q. Li, Chem. Commun., 2016, 52, 3568–3571 RSC.
- A. Patra, T. Ding, G. Engudar, Y. Wang, M. M. Dykas, B. Liedberg, J. C. Kah, T. Venkatesan and C. L. Drum, Small, 2016, 12, 1174–1182 CrossRef CAS PubMed.
- M. Bockova, X. Chadtova Song, E. Gedeonova, K. Levova, M. Kalousova, T. Zima and J. Homola, Anal. Bioanal. Chem., 2016, 408, 7265–7269 CrossRef CAS PubMed.
- H. R. Jang, A. W. Wark, S. H. Baek, B. H. Chung and H. J. Lee, Anal. Chem., 2014, 86, 814–819 CrossRef CAS PubMed.
- M. Maisonneuve, C. Valsecchi, C. Wang, A. G. Brolo and M. Meunier, Biosens. Bioelectron., 2015, 63, 80–85 CrossRef CAS PubMed.
- J. Martinez-Perdiguero, A. Retolaza, L. Bujanda and S. Merino, Talanta, 2014, 119, 492–497 CrossRef CAS PubMed.
- O. Zagorodko, J. Spadavecchia, A. Y. Serrano, I. Larroulet, A. Pesquera, A. Zurutuza, R. Boukherroub and S. Szunerits, Anal. Chem., 2014, 86, 11211–11216 CrossRef CAS PubMed.
- W. J. Zhou, Y. Chen and R. M. Corn, Anal. Chem., 2011, 83, 3897–3902 CrossRef CAS PubMed.
- S. Scarano, M. L. Ermini, M. M. Spiriti, M. Mascini, P. Bogani and M. Minunni, Anal. Chem., 2011, 83, 6245–6253 CrossRef CAS PubMed.
- L. He, M. D. Musick, S. R. Nicewarner, F. G. Salinas, S. J. Benkovic, M. J. Natan and C. D. Keating, J. Am. Chem. Soc., 2000, 122, 9071–9077 CrossRef CAS.
- S. Li, M. Yang, W. Zhou, T. G. Johnston, R. Wang and J. Zhu, Appl. Surf. Sci., 2015, 355, 570–576 CrossRef CAS.
- W. Liang, S. Wang, F. Festa, P. Wiktor, W. Wang, M. Magee, J. LaBaer and N. Tao, Anal. Chem., 2014, 86, 9860–9865 CrossRef CAS PubMed.
- S. Zeng, D. Baillargeat, H. P. Ho and K. T. Yong, Chem. Soc. Rev., 2014, 43, 3426–3452 RSC.
- O. R. Bolduc and J. F. Masson, Anal. Chem., 2011, 83, 8057–8062 CrossRef CAS PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayad, Plasmonics, 2007, 2, 107–118 CrossRef CAS.
- R. P. Liang, G. H. Yao, L. X. Fan and J. D. Qiu, Anal. Chim. Acta, 2012, 737, 22–28 CrossRef CAS PubMed.
- J. Wang, A. Munir, Z. Zhu and H. S. Zhou, Anal. Chem., 2010, 82, 6782–6789 CrossRef CAS PubMed.
- L. Malic, M. G. Sandros and M. Tabrizian, Anal. Chem., 2011, 83, 5222–5229 CrossRef CAS PubMed.
- M. Singh, M. Holzinger, M. Tabrizian, S. Winters, N. C. Berner, S. Cosnier and G. S. Duesberg, J. Am. Chem. Soc., 2015, 137, 2800–2803 CrossRef CAS PubMed.
- J. Mitchell, Sensors, 2010, 10, 7323–7346 CrossRef CAS PubMed.
- J. Wang and H. S. Zhou, Anal. Chem., 2008, 80, 7174–7178 CrossRef CAS PubMed.
- H. Yockell-Lelievre, N. Bukar, K. S. McKeating, M. Arnaud, P. Cosin, Y. Guo, J. Dupret-Carruel, B. Mougin and J. F. Masson, Analyst, 2015, 140, 5105–5111 RSC.
- M. Miyashita, T. Shimada, H. Miyagawa and M. Akamatsu, Anal. Bioanal. Chem., 2005, 381, 667–673 CrossRef CAS PubMed.
- J. P. Meneely, M. Sulyok, S. Baumgartner, R. Krska and C. T. Elliott, Talanta, 2010, 81, 630–636 CrossRef CAS PubMed.
- S. S. Zhao, N. Bukar, J. L. Toulouse, D. Pelechacz, R. Robitaille, J. N. Pelletier and J. F. Masson, Biosens. Bioelectron., 2015, 64, 664–670 CrossRef CAS PubMed.
- D. R. Shankaran, T. Kawaguchi, S. J. Kim, K. Matsumoto, K. Toko and N. Miura, Anal. Bioanal. Chem., 2006, 386, 1313–1320 CrossRef CAS PubMed.
- J. A. Greenberg, S. J. Bell, Y. Guan and Y. H. Yu, Rev. Obstet. Gynecol., 2011, 4, 52–59 Search PubMed.
- S. Akbar, A. Anwar and Q. Kanwal, Anal. Biochem., 2016, 510, 98–105 CrossRef CAS PubMed.
- W. Ren, Y. X. Fang and E. K. Wang, ACS Nano, 2011, 5, 6425–6433 CrossRef CAS PubMed.
- M. B. Caselunghe and J. Lindeberg, Food Chem., 2000, 70, 523–532 CrossRef.
- A. Lermo, S. Fabiano, S. Hernandez, R. Galve, M. P. Marco, S. Alegret and M. I. Pividori, Biosens. Bioelectron., 2009, 24, 2057–2063 CrossRef CAS PubMed.
- J. J. Mock, R. T. Hill, A. Degiron, S. Zauscher, A. Chilkoti and D. R. Smith, Nano Lett., 2008, 8, 2245–2252 CrossRef CAS PubMed.
- L. A. Lyon, M. D. Musick and M. J. Natan, Anal. Chem., 1998, 70, 5177–5183 CrossRef CAS PubMed.
- B. P. Nelson, A. G. Frutos, J. M. Brockman and R. M. Corn, Anal. Chem., 1999, 71, 3928–3934 CrossRef CAS.
- V. Kanda, J. K. Kariuki, D. J. Harrison and M. T. McDermott, Anal. Chem., 2004, 76, 7257–7262 CrossRef CAS PubMed.
- I. G. Campbell, T. A. Jones, W. D. Foulkes and J. Trowsdale, Cancer Res., 1991, 51, 5329–5338 CAS.
- P. C. Elwood, J. Biol. Chem., 1989, 264, 14893–14901 CAS.
- E. Sadasivan and S. P. Rothenberg, J. Biol. Chem., 1989, 264, 5806–5811 CAS.
- S. Knecht, D. Ricklin, A. N. Eberle and B. Ernst, J. Mol. Recognit., 2009, 22, 270–279 CrossRef CAS PubMed.
- G. Wang, R. J. Lipert, M. Jain, S. Kaur, S. Chakraboty, M. P. Torres, S. K. Batra, R. E. Brand and M. D. Porter, Anal. Chem., 2011, 83, 2554–2561 CrossRef CAS PubMed.
- B. Stella, S. Arpicco, M. T. Peracchia, D. Desmaele, J. Hoebeke, M. Renoir, J. D'Angelo, L. Cattel and P. Couvreur, J. Pharm. Sci., 2000, 89, 1452–1464 CrossRef CAS PubMed.
- D. S. Auld, M. W. Farmen, S. D. Kahl, A. Kriauciunas, K. L. McKnight, C. Montrose and J. R. Weidner, in Assay Guidance Manual, ed. G. S. Sittampalam, N. P. Coussens, K. Brimacombe, A. Grossman, M. Arkin, D. Auld, C. Austin, J. Baell, B. Bejcek, T. D. Y. Chung, J. L. Dahlin, V. Devanaryan, T. L. Foley, M. Glicksman, M. D. Hall, J. V. Hass, J. Inglese, P. W. Iversen, M. Lal-Nag, Z. Li, J. McGee, O. McManus, T. Riss, O. J. Trask Jr., J. R. Weidner, M. Xia and X. Xu, Bethesda, MD, 2004, pp. 129–154 Search PubMed.
- O. B. McManus, M. L. Garcia, D. Weaver, M. Bryant, S. Titus and J. B. Herrington, in Assay Guidance Manual, ed. G. S. Sittampalam, N. P. Coussens, K. Brimacombe, A. Grossman, M. Arkin, D. Auld, C. Austin, J. Baell, B. Bejcek, T. D. Y. Chung, J. L. Dahlin, V. Devanaryan, T. L. Foley, M. Glicksman, M. D. Hall, J. V. Hass, J. Inglese, P. W. Iversen, M. Lal-Nag, Z. Li, J. McGee, O. McManus, T. Riss, O. J. Trask Jr., J. R. Weidner, M. Xia and X. Xu, Bethesda, MD, 2004, pp. 413–426 Search PubMed.
- C. Bee, Y. N. Abdiche, D. M. Stone, S. Collier, K. C. Lindquist, A. C. Pinkerton, J. Pons and A. Rajpal, PLoS One, 2012, 7, e36261 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01680h |
| This journal is © The Royal Society of Chemistry 2018 |