DOI:
10.1039/C7AN01491K
(Paper)
Analyst, 2018,
143, 280-288
Separation of proteins from complex bio-matrix samples using a double-functionalized polymer monolithic column†
Received
8th September 2017
, Accepted 11th November 2017
First published on 14th November 2017
Abstract
A double-functionalized polymer monolithic column was fabricated within the confines of a stainless-steel column (50 mm × 4.6 mm i.d.) via a facile method using iron porphyrin, ionic liquid (1-allyl-3-methylimidazolium chloride) and 1,10-decanediol dimethacrylate as tri-monomers; ethylene dimethacrylate as a crosslinker; polyethylene glycol 400 and N,N-dimethylformamide as co-porogens; benzoyl peroxide and N,N-dimethyl aniline as the redox initiation system. Results obtained from scanning electron microscopy, nitrogen adsorption–desorption, and mercury intrusion porosimetry confirmed the uniform pore structure and the pore size distribution of macro-pores. The home-made monolith was further characterized by elemental analysis to investigate the elemental composition of Fe supplied by iron porphyrin, confirming the synthetic process. The resulting optimized monolithic column was used as the stationary phase in high performance liquid chromatography for separating proteins, such as mixture of standard proteins, egg white, and human plasma, exhibiting good selectivity and high performance. It is worth noting that the home-made double-functionalized polymer monolithic column shows excellent selectivity for fractionation separation of human plasma proteins, and it is a promising separation tool for complex bio-samples in proteomic research.
1. Introduction
Porous polymer-based monolithic materials have attracted substantial interest and have been applied in the field of bio-separation.1–5 It is should be emphasized that the best results have always been achieved with capillary columns, in which the contribution of radial diffusion to peak broadening is much smaller compared to the 4.6 mm and 8 mm i.d. analytical size columns used traditionally.6 However, the monolithic columns suffered from poor selectivity for protein separation, which was caused by the non-uniform pore structure and inappropriate chemical groups. Thus far, proteomics has become a fundamental component for biological study and is recognized as a key method for novel biomarker discovery and personalized therapy.7 As the most adopted material for clinical analysis, human plasma is a complex mixture composed of tens of thousands of different proteins, which along with their cleaved or modified forms, is a reflection of ongoing physiological and pathological events.8 Hence, in the postgenomic era of human biology, studies focusing on human plasma proteome appear to be a tantalizing and direct approach to building knowledge that would enable us to address many fundamental biomedical questions.9 In addition, separation of proteins from complex human plasma is a challenge in separation science because of the presence of multiple substances and the wide dynamic range of proteins. It is evident that these challenging tasks would not be accomplished with a single analytical tool but through the combination of separation and detection techniques,10 in which the separation method is the first and key process. Hundreds of human plasma proteins are divided into different fractions via different interactions, such as hydrogen bonding, electrostatic force, dipole–dipole interaction, and π–π interaction; hence, the so-called “fractionation separation” method can simplify the analysis of the abovementioned proteins identification.11 Thus far, the main methods for protein separation are two-dimensional electrophoresis (2DE) and capillary electrochromatography (CEC).12–17 On the one hand, the 2DE method, which provides high resolving capacity for proteins, is the preferred separation technology for first-generation proteomic platforms. However, it also has shortcomings, such as complex processes, poor reproducibility, insolubility with hydrophobic proteins, and difficulty in achieving automated analysis. The CEC method can offer a unique advantage, combining selectivity based on chromatographic interactions (hydrophobicity, ion-exchange, etc.) and electrophoretic mechanisms (charge and size). However, due to the existence of electrostatic and hydrogen bonds between proteins and capillary walls, the analyte would be adsorbed on the inner wall of the capillary, thus affecting the performance of separation. Ever since polymer-based monolithic columns were prepared in the 1990s18–20 it has proven to be an outstanding stationary phase for the rapid separation of proteins.6 In order to improve its selectivity for proteins in a complex bio-matrix, ionic liquids (ILs) and porphyrins were combined as co-monomers in the present study.
ILs possess some fascinating properties, including low volatility, good miscibility with most solvents, and good thermal stability, which make ILs more unique and useful for chromatographic analysis.21–24 Thus far, our group has employed ILs as functional monomers for preparing polymer monolithic columns, resulting in good protein separation performance.25–28 Porphyrin is a heterocyclic compound with a huge conjugated structure, which is easily combined with metal to form metal-porphyrin in nature because of the unshared pair of electrons in the N atoms. Metal-porphyrins, such as iron porphyrin (IP, the chemical structure is shown in Fig. S1 of the ESI†) play their biological function by assembling into supramolecular compounds with proteins, indicating a specific absorption between the metal-porphyrin and the protein.29–31
In this study, we combined the unique advantages of IP and ILs, aiming to develop a double-functionalized monolithic column via redox initiation. The home-made monolithic column exhibits a uniform skeleton porous structure. Furthermore, the resulting monolith was evaluated chromatographically and applied in the separation of proteins from complex bio-samples to study the effect of IL and IP on chromatographic separation with high performance liquid chromatography (HPLC).
2. Experimental
2.1. Chemicals and materials
IP, 1-methylimidazole, and allyl chloride were obtained from Aladdin-Reagent (Shanghai, China). 1,10-Decanediol dimethacrylate (DEDMA) was provided by DECO Composite Technology Co., Ltd (Guangzhou, China). Polyethylene glycol 400 (PEG400), benzoyl peroxide (BPO), N,N-dimethyl aniline (DMA), and N,N-dimethylformamide (DMF) were purchased from Tianjin Guangfu Fine Institute of Chemistry (Tianjin, China). Ethylene dimethacrylate (EDMA) was supplied by MAYA Reagent (Zhenjiang, China). Trifluoroacetic acid (TFA), HPLC-grade methanol, acetonitrile (ACN), and KBr were acquired from Kermel Chemical Reagent Factory (Tianjin, China). Standard proteins, including insulin, cytochrome C, lysozyme, myoglobin, and bovine serum albumin (BSA) were purchased from MAYA Reagent (Zhenjiang, China). Ultrapure water was used in all experiments and all mobile phases were filtered through a 0.45 μm membrane before use.
2.2. Instruments
HPLC experiments were performed on a 1100 system from Agilent Technologies (USA) at 25 °C. Elementary analyses were performed on a EURO EA elementary analyzer (Eurovector, Italy). Scanning electron microscopy (SEM) images were recorded on a Hitachi S-3400 SEM instrument (Hitachi High Technologies, Japan). The micro-, meso- and macro-pore porosities of monoliths were determined by nitrogen adsorption/desorption measurements on a TriStar II 3020 instrument (Micromeritics, USA) and an AutoPore IV 9500 instrument (Micromeritics, USA), respectively. The qualitative identification of protein was carried out on a LC/MS system composed of a NCS500 LC system (DIONEX, USA) and a Q Exactive mass spectrometer (Thermo Scientific, USA).
2.3. Preparation of poly(IP-co-IL-co-DEDMA) monoliths
The synthetic procedure for IL (1-allyl-3-methylimidazolium chloride, AMIM+Cl−) was described in detail in our previous study.28
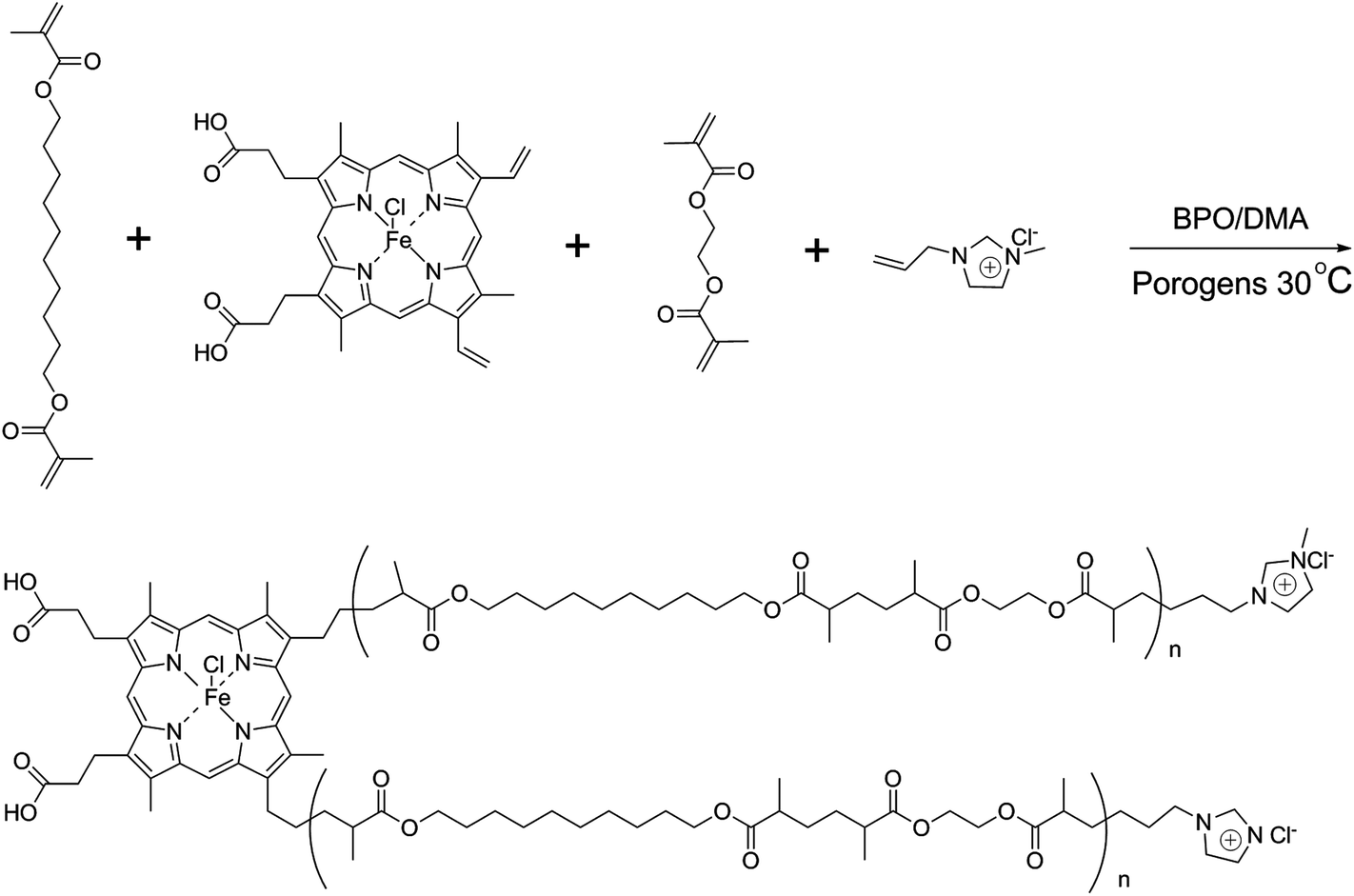
The process for preparing poly(IP-co-IL-co-DEDMA) monoliths was as follows: IP, IL and DEDMA were used as tri-monomers; EDMA was used as a crosslinker; PEG400 and DMF were used as co-porogens; BPO and DMA were used as initiators. All reagents, except for DMA, were added in a tube, followed by mixing and degassing by sonication for 15 min. Then, DMA was added to the mixture, followed by degassing with an ultrasonicator; then, the mixture was poured into a stainless-steel column (50 mm × 4.6 mm i.d.) with one end sealed. The pre-polymerization solution was allowed to react at room temperature for 2.5 h after sealing the other end of the column. The resulting monolithic column was washed online with methanol by an HPLC pump to remove the porogens, solvent, and other dissoluble components. The synthetic polymerization scheme is illustrated in Fig. 1.
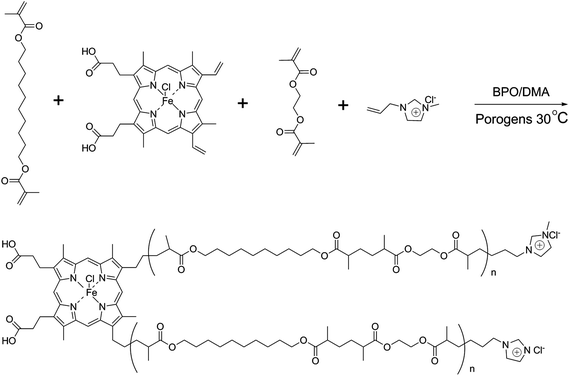 |
| | Fig. 1 Synthetic scheme of the poly(IP-co-IL-co-DEDMA) monolith. | |
2.4. Sample preparation
Fresh human blood was centrifuged at 4 °C and 4500 rpm for 15 min. The supernatant fluid was collected and stored at 4 °C before use.
All standard proteins were prepared with ultrapure water (2.0 mg mL−1) and stored at 4 °C before use.
Chicken egg was purchased locally and egg white was 2-fold diluted with 50 mmol L−1 phosphate buffer (pH 7.0). The mixture was oscillated for 1 min and then centrifuged at 4 °C and 4500 rpm for 15 min. The supernatant fluid was collected and stored at 4 °C.
All samples were filtered by a 0.22 μm syringe filter before use.
All experiments were in accordance with the ethical guidelines of biomedical research involving human subjects (for Trial Implementation), Ministry of Health, China, and approved by the institutional ethical committee of Hebei University. Informed consent was obtained from all subjects.
2.5. Calculation of permeability
Permeability of the resulting monolithic column was calculated by Darcy's Law,32
wherein F, η, L, r and ΔP are the flow rate of the mobile phase (m3 s−1), the dynamic viscosity of the mobile phase (Pa s), the effective length of the column (m), the inner radius of the column (m) and the back pressure of the column measured at room temperature (Pa), respectively. In this study, ACN was used as the mobile phase with η = 0.37 × 10−3 Pa s at 25 °C.
2.6. HPLC procedure
An Agilent 1100 series system was used for chromatographic experiments, and the home-made monoliths were used as stationary phases. In addition, 0.1% TFA in water (A) and 0.1% TFA in ACN (B) were used as mobile phases with gradient elution. The analytical flow rate was 1.0 mL min−1 and the wavelength was set at 280 nm. The chromatographic separations carried out at ambient temperature.
3. Results and discussion
3.1. Optimization of the preparative conditions
Table 1 shows part of the representative conditions of the pre-polymerization solution and the corresponding characters. It is well-known that porogens are important factors affecting the mechanical strength and permeability of the resulting monolithic columns. Considering the solubility of IP, DMF was adopted as a co-porogen together with PEG400, which were used as good and bad solvents for preparing the poly(IP-co-IL-co-DEDMA) monolith, respectively, and the feasible content of DMF was found to be 0.4 mL in the pre-polymerization solution. In addition, the results obtained from Monoliths A–C showed that the permeability increased from 1.24 × 10−14 to 2.65 × 10−14 m2; simultaneously, the back pressure decreased from 15 × 105 Pa to 7 × 105 Pa as the content of PEG400 increased from 0.6 mL to 0.8 mL. However, these results were obtained with the decrease in mechanical strength. This is because PEG400 was a bad solvent in the pre-polymerization solution, and increasing its content resulted in formation of numerous large pores, which resulted in high permeability and low back pressure. Considering both the permeability and mechanical strength of the resulting monoliths A, B and C, the proportion of porogens used in Monolith B was adopted as the optimized condition. Table 1 also illustrates the effect of co-monomer IP on the monoliths (Monoliths B, D and E). The results indicated that following the increase in content of IP, the column back pressure decreased and the permeability increased, accompanied with the decrease in mechanical strength. This was because the increase in amount of monomer decreased the ratio of crosslinker and monomers, which resulted in a low degree of crosslinking and low back pressure. Considering the permeability and mechanical strength of the resulting monoliths B, D and E, Monolith B was considered the most appropriate. In order to confirm the effect of the co-monomers, IP and IL, on the chromatographic separation, Monoliths F and G were fabricated via the same method as that for Monolith B, but without IP (Monolith F) or IL (Monolith G).
Table 1 Effect of the pre-polymerization solution component on the monoliths
| Monoliths labela |
Monomers |
Crosslinker |
Porogens |
Back pressureb (×105 Pa) |
Permeabilityc (×10−14 m2) |
| IP (g) |
IL (μL) |
DEDMA (mL) |
EDMA (mL) |
PEG400 (mL) |
DMF (mL) |
|
All monoliths were prepared via redox polymerization by using 0.01 g of BPO and 30 μL of DMA.
Back pressure was investigated by using ACN as the mobile phase with a flow rate of 1.0 mL min−1.
Permeability was calculated following Darcy's Law with ACN as the mobile phase (viscosity as 0.37 × 10−3 Pa s at 25 °C).
|
| A |
0.002 |
30 |
0.25 |
0.55 |
0.6 |
0.4 |
15 |
1.24 |
| B |
0.002 |
30 |
0.25 |
0.55 |
0.7 |
0.4 |
8 |
2.32 |
| C |
0.002 |
30 |
0.25 |
0.55 |
0.8 |
0.4 |
7 |
2.65 |
| D |
0.001 |
30 |
0.25 |
0.55 |
0.7 |
0.4 |
13 |
1.43 |
| E |
0.003 |
30 |
0.25 |
0.55 |
0.7 |
0.4 |
6 |
3.01 |
| F |
— |
30 |
0.25 |
0.55 |
0.7 |
0.4 |
14 |
1.33 |
| G |
0.002 |
— |
0.25 |
0.55 |
0.7 |
0.4 |
15 |
1.24 |
3.2. Characterizations of the prepared monoliths
The prepared monoliths were investigated by elemental analysis, SEM method, nitrogen adsorption–desorption, and mercury intrusion porosimetry and the obtained results are discussed as follows.
3.2.1. Element analysis of Monolith B.
Monolith B was assayed by elemental analysis, which determined 0.033% (wt%) of Fe, confirming the polymerization schedule.
3.2.2. SEM images of the prepared monoliths.
The morphologies of the prepared monoliths listed in Table 1 were investigated by SEM; Fig. 2 shows the morphology of Monolith B (SEM images of Monolith A and C–G are shown in Fig. S2 of ESI†). The pore structures of all these monoliths were created by particle accumulation. Based on the images of Monoliths A–C, as the porogen content in the pre-polymerization solution increased from 53.9% (Monolith A, wt%) to 58.6% (Monolith C, wt%), the pores clearly became larger due to the increase in content of bad solvent, PEG400. Monolith B exhibits a relatively uniform pore structure and proper pore size; thus, the porogen content of 56.4% was adopted as optimal. The SEM images obtained for Monoliths B, D and E showed the effect of co-monomer IP on the morphologies of resulting monoliths. Compared to Monolith B, Monolith D showed a compact pore structure due to the increased ratio of crosslinker and monomers. A high ratio of crosslinker and monomers would lead to a high cross-linking degree, resulting in compact pore structure, high back pressure, and low permeability. In contrast, compared to Monolith B, Monolith E, prepared by decreasing the ratio of crosslinker and monomers, exhibits a loose structure, low back pressure, and high permeability, but poor mechanical strength. The SEM image of Monolith F, which was IP-free, showed a similar morphology to that of Monolith D. The image of Monolith G displayed a relatively non-uniform structure without IL addition. These results indicated that Monolith B possessed the appropriate pore structure for the HPLC stationary phase.
 |
| | Fig. 2 SEM image of monolith B. | |
3.2.3. Pore size distribution of Monolith B.
As shown in Fig. 3, Monolith B was further investigated using a nitrogen adsorption–desorption instrument (a) and a mercury intrusion porosimeter (b), in order to obtain its pore type and pore size distribution. The results, shown in Fig. 3(a), indicated that the nitrogen adsorption–desorption isotherm was adjusted to type III. Poor absorption at low pressures (P/P0: 0–0.1) and the absence of a hysteresis loop at intermediate pressures (P/P0: 0.3–0.8) indicated that there were few micro- (pore size < 2 nm) or meso-pores (2 nm < pore size < 50 nm) in the monolithic material. According to the Brunauer–Emmett–Teller mechanism, the specific surface area obtained from the micro- and meso-pores was 11.02 m2 g−1, which confirmed the absence of micro- and meso-pores. Moreover, the micro- and meso-pore size distributions based on pore area and pore volume are shown in Fig. S3(a and b) of ESI.† In addition, as shown in the isotherm, the increase in adsorption at high pressure (P/P0: 0.9–1.0) confirmed that there were numerous macro-pores (pore size > 50 nm) in the monolithic material, which was feasible for the separation of bio-macromolecules. The macro-pores distribution was further assayed by mercury intrusion porosimetry and the results, shown in Fig. 3(b), indicated that the mode pore size, pore volume (the intrusion pore volume curve is shown in Fig. S4 of ESI†), and porosity was 1.02 μm, 1.66 mL g−1, and 65.81%, respectively. The specific surface area obtained from the macro-pores was 17.15 m2 g−1.
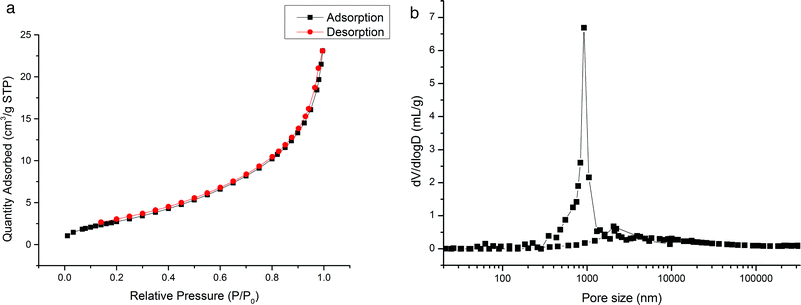 |
| | Fig. 3 Pore size distribution of the poly(IP-co-IL-co-DEDMA) monolith. (a) Nitrogen adsorption–desorption isotherm; (b) macro-pore size distribution by mercury intrusion porosimetry. | |
3.3. Chromatographic behavior of the monoliths
In order to study the effect of IP and IL on the chromatographic behavior of the resulting monoliths, Monoliths B, F and G were used as the stationary phases of HPLC to separate a mixture of standard proteins, egg white and human plasma, respectively.
3.3.1. Chromatographic separation of standard proteins.
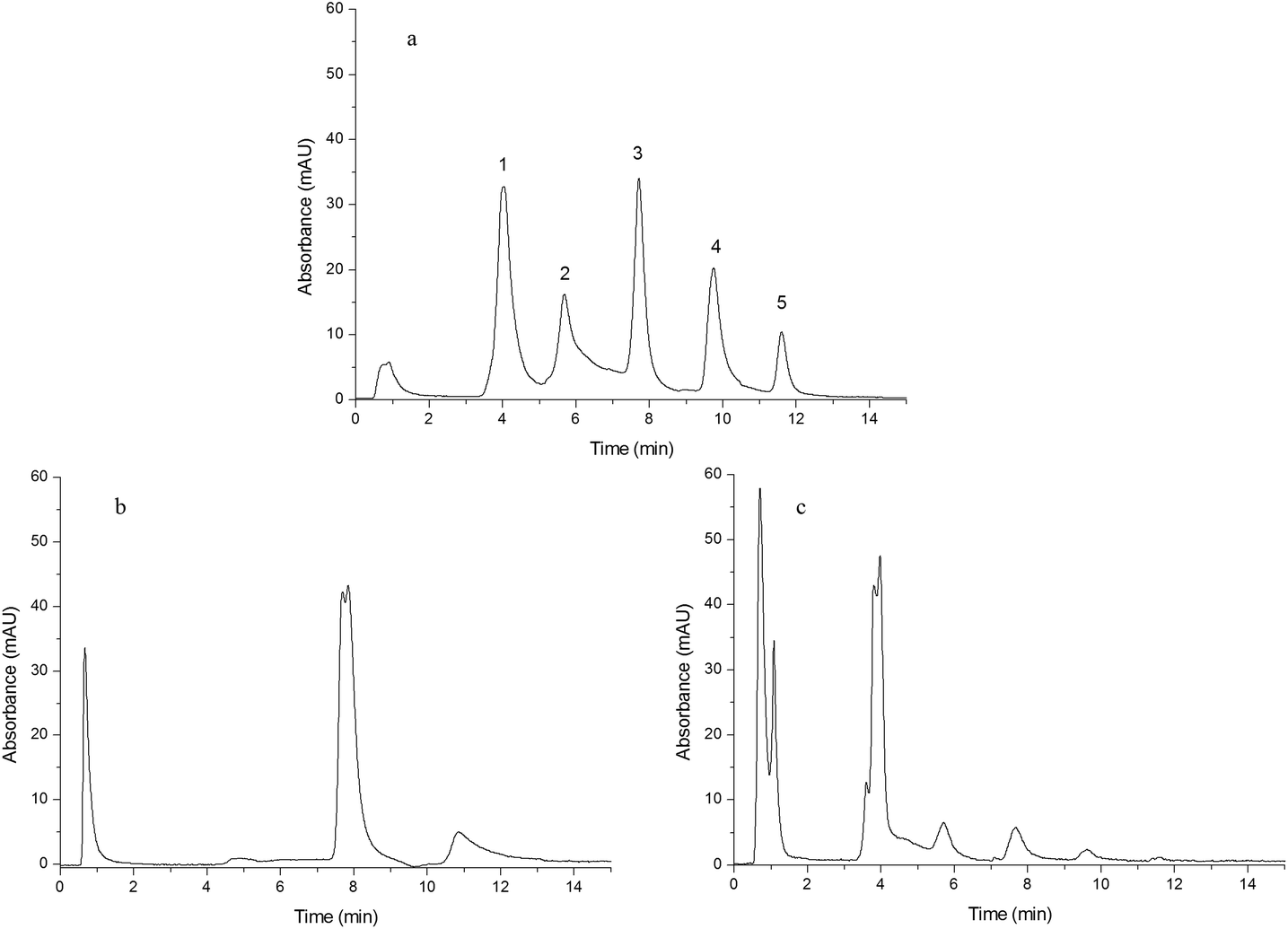
A mixture of five standard proteins was separated by Monoliths B (a), F (b) and G (c) and the resulting chromatograms are shown in Fig. 4(a–c). The five peaks, with the baselines separated in Fig. 4(a), were insulin (1), cytochrome C (2), lysozyme (3), myoglobin (4) and BSA (5). The chromatographic gradient was as follows: the mobile phase component linearly changed from 0% B to 33% B in 15 min. Compared to Monoliths F and G (Fig. 4(b) and (c)), Monolith B (Fig. 4(a)) exhibited high performance and good selectivity due to the combination of the double functional groups obtained from IP and IL. Thus, IL could improve the uniformity of the pre-polymerization solution and enhance the uniformity of the monolith, and IP could promote the selectivity for proteins.
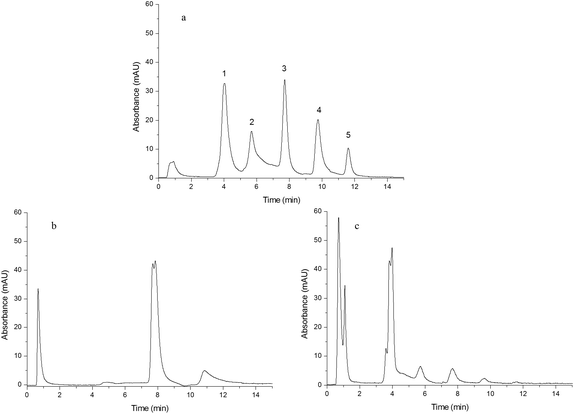 |
| | Fig. 4 Chromatograms obtained from separating a mixture of five standard proteins. Chromatograms were obtained from Monoliths B (a), F (b) and G (c). Experimental conditions: mobile phase A was 0.1% TFA aqueous solution and B was 0.1% TFA in ACN; the gradient was as follows: mobile phase linearly changed from 100% A to 67% A + 33% B in 15 min; UV detection was 280 nm; flow rate was 1.0 mL min−1; sample concentration was 2.0 mg mL−1; injection volume was 20 μL; the peaks in (a) were insulin (1), cytochrome C (2), lysozyme (3), myoglobin (4) and BSA (5). | |
3.3.2. Chromatographic separation of egg white.
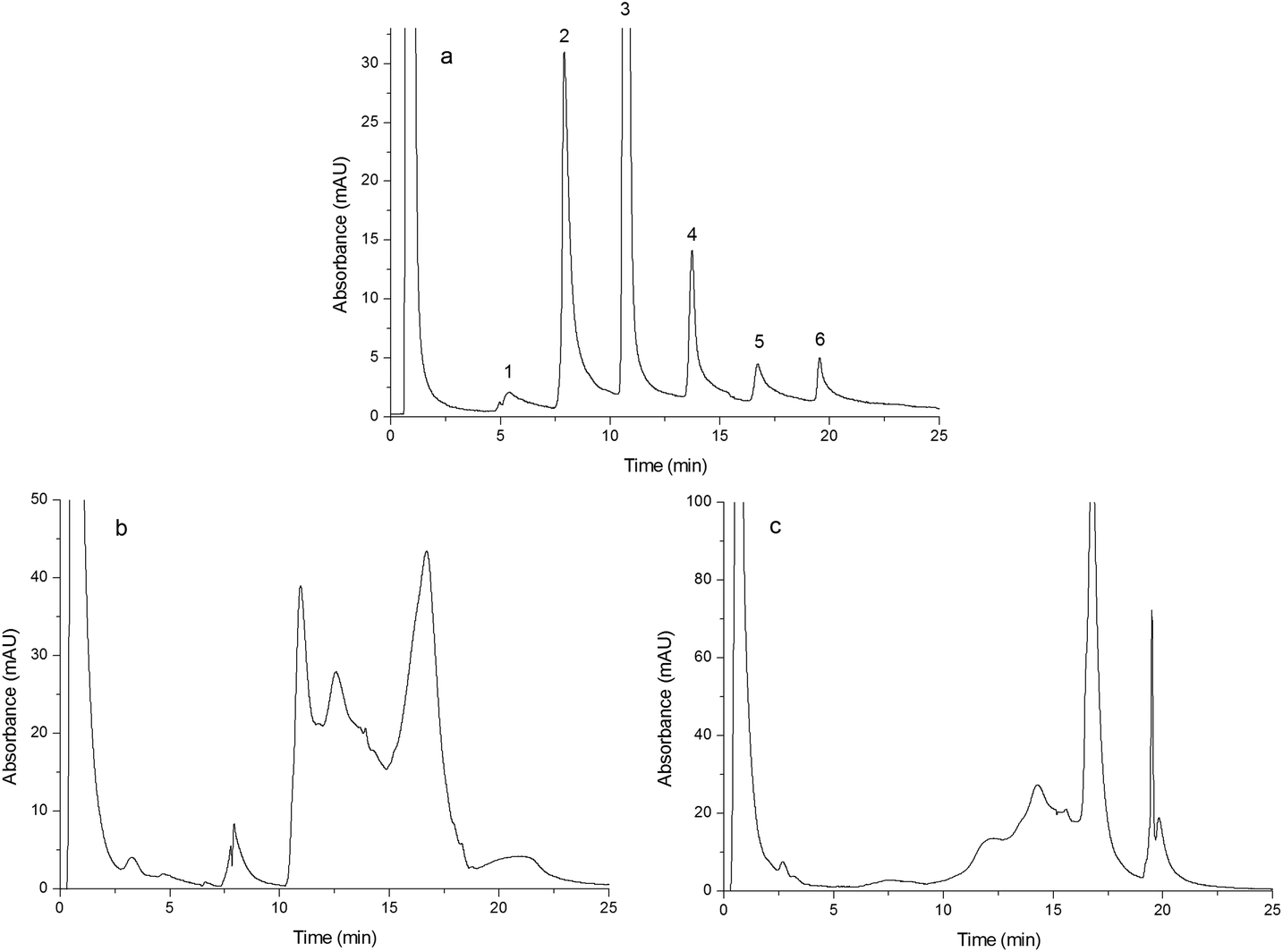
As shown in Fig. 5(a–c), egg white was separated by Monoliths B (a), F (b) and G (c), respectively. The chromatographic conditions were the same as those of Fig. 4, except for the gradient as follows: the mobile phase component linearly changed from 0% B to 40% B in 25 min. Compared to Fig. 5(b) and (c), Fig. 5(a) showed six peaks that were baseline separated and the third peak was identified as lysozyme, which further confirmed the merit of the double-functional monolith.
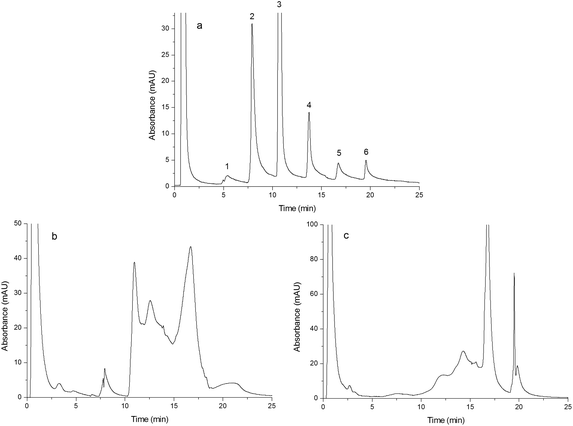 |
| | Fig. 5 Chromatograms obtained from separating egg white. Chromatograms were obtained from Monoliths B (a), F (b) and G (c). Experimental conditions were the same as those of Fig. 4 except that the gradient of the mobile phase linearly changed from 100% A to 60% A + 40% B in 25 min and the injection volume was 15 μL. | |
3.3.3. Chromatographic separation of human plasma.
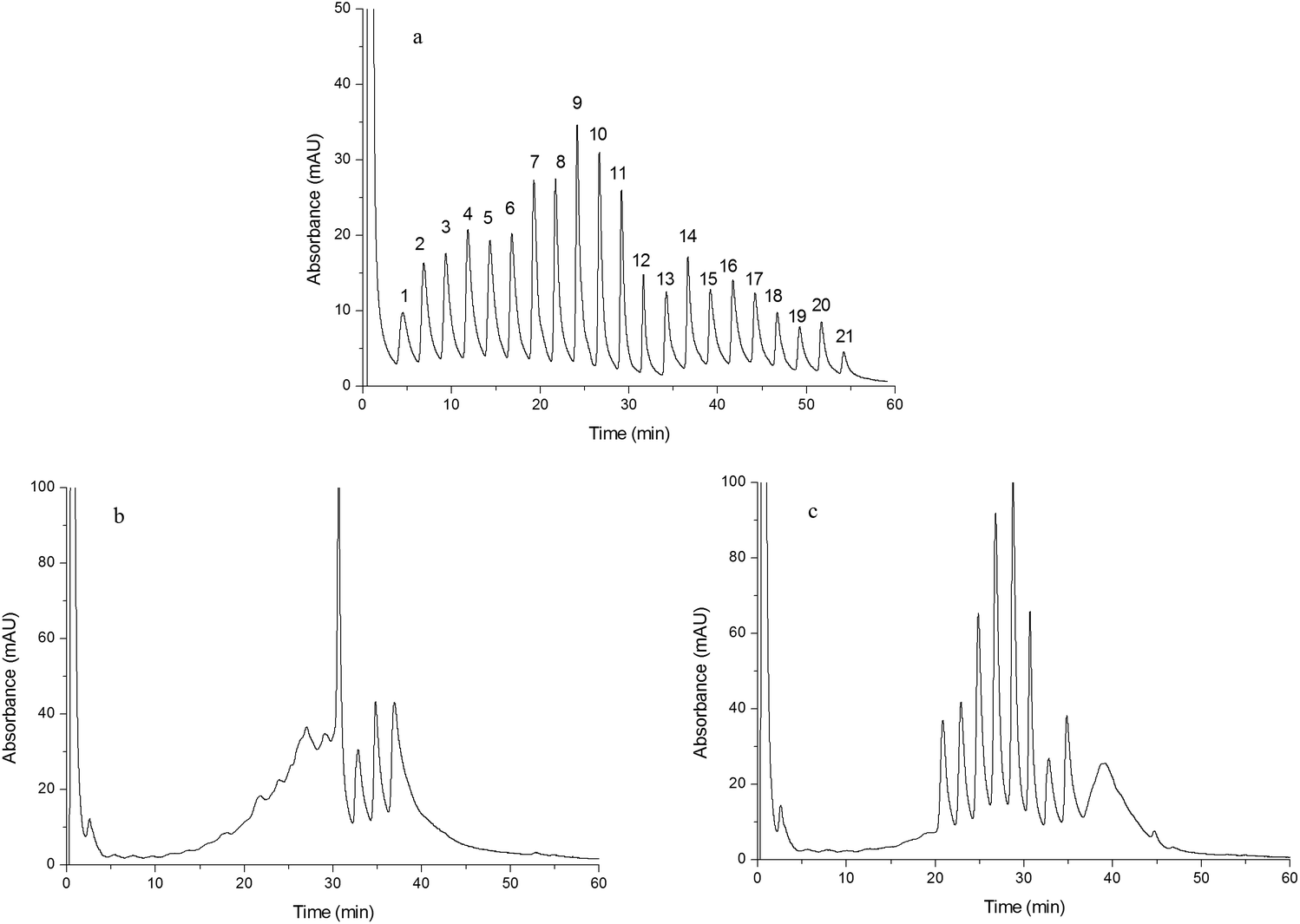
The chromatograms shown in Fig. 6(a–c) were obtained by separating human plasma using Monoliths B (a), F (b) and G (c) as the stationary phases, respectively. Compared to Monoliths F and G, Monolith B exhibited superior chromatographic separation of human plasma proteins. Twenty one baseline separated peaks obtained from Monolith B indicated that human plasma proteins could be divided into twenty one fractions by the double-functional monolith, which were caused by different interactions between proteins and the monolith, such as hydrogen bonding, electrostatic force, dipole–dipole interaction, π–π interaction, and ionic exchange. The uniformity and the chemical groups of the monolithic column both affected the chromatographic separation. On the one hand, the addition of IL improves the uniformity of the monolith and enhances its chromatographic performance. In addition, the cations obtained from IL displayed ionic exchange interactions with the proteins, resulting in more fractions in the fractionation separation. On the other hand, IP could supply chemical groups and interaction sites for the proteins. Thus, improved performance and enhanced selectivity could be obtained by the double-functionalized home-made monolith. The meaning of fractionation separation is as follows: each fraction contains relatively fewer types of proteins and those proteins in one fraction have similar structure or property because of their similar interactions with the monolith, which would simplify the following identification and analysis. Furthermore, in order to confirm the difference in the separated peaks, two peaks (peak 9 and peak 10 in Fig. 6a) were randomly selected and collected from the HPLC system and further identified using LC/MS. The obtained data were matched with “human_txid9606_110574.fasta” via “Proteome Discoverer”. The results shown in Fig. S5 of ESI† indicated that the unique peptide compositions of the two peaks differ from those of human plasma, confirming that the chromatographic separation was meaningful for “fractionation separation” of human plasma. However, the following study was expected to further probe the interaction mechanism and quantified identification.
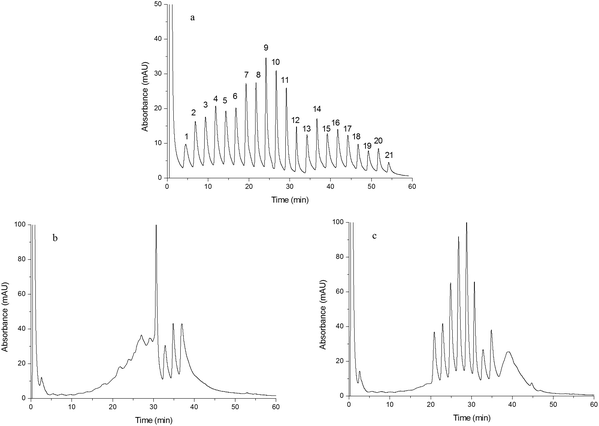 |
| | Fig. 6 Chromatograms obtained from separating human plasma. Chromatograms were obtained from Monoliths B (a), F (b) and G (c). Experimental conditions were the same as those of Fig. 4 except that the gradient of the mobile phase linearly changed from 100% A to 55% A + 45% B in 60 min and the injection volume was 15 μL. | |
3.3.4 Stability and life-span of monolith B.
The stability and life-span of the proposed monolith B were studied by using the monoliths prepared in different periods, including now, 1 month ago, 2 months ago, 5 months ago and 10 months ago to separate human plasma following the processes and conditions as those of Fig. 6, and the resulting chromatograms are shown in Fig. S6 of ESI.† The relative standard deviation (RSD, n = 5) values based on retention time and peak area for each peak were in the ranges of 0.85%–1.32% and 1.65%–1.99%, which illustrated good stability and at least a 10-month life-span for the proposed monolithic column. The monolith prepared 10 months ago was used to separate human plasma more than 100 times, and the values of RSD for the latest run-to-run (n = 5) were in the ranges of 0.96%–1.29% and 1.89%–2.12% based on the retention time and peak area of each peak, respectively, further indicating that the monolith exhibits good stability.
3.3.5 Advantages of the proposed column.
In the present study, the monolithic column prepared using a facile and fast method exhibited good selectivity and high performance for separating proteins from human plasma, which is attributed to the combination of three co-monomers: iron porphyrin, ionic liquid and 1,10-decanediol dimethacrylate. The monolith contained double functional groups: iron porphyrin groups and ionic liquid groups. Iron porphyrin, used as one of the monomers, facilitated the interactions between proteins and stationary phase, such as hydrogen bonding, dipole–dipole interaction, π–π interaction, and coordination interaction. Ionic liquid, used as a monomer, facilitated ionic exchange interactions and electrostatic interactions between the proteins and the stationary phase, which enriched the interaction sites and enhanced the selectivity of the monolith. In addition, 1,10-decanediol dimethacrylate, used as one of the co-monomers, supplied hydrophobic interactions between the proteins and the stationary phase. Furthermore, 1,10-decanediol dimethacrylate also played the role of the connecting arm, which could avoid the steric effect of iron porphyrin when separating proteins from bio-samples. Compared to our previous studies,33,34 more baseline separated peaks were obtained using the present monolith, indicating improved selectivity of the double-functionalized monolith. Compared to the post-modified monolithic column used in separating proteins,4,35,36 the present monolithic column not only had the advantage of a simple preparation process but also exhibited high selectivity in separating complex bio-samples due to the double-functionalized monolith supplying abundant types of interaction sites. Compared to silica-based capillary monolithic columns used in separating proteins,6,37,38 the present polymer-based monolithic column with 4.6 mm inner diameter had good chemical stability and it was easy to collect fractions for analysis and identification.
4. Conclusion
In this study, a double-functionalized poly(IP-co-IL-co-DEDMA) monolithic column based on ionic liquid and iron porphyrin was prepared via redox initiation within the confines of a 4.6 mm i.d. stainless steel column. The resulting monolith possessed a relatively uniform pore structure and exhibited enhanced selectivity for separating proteins in the “fractionation separation” of human plasma. All results indicated that the combination of ionic liquids and iron porphyrin could simultaneously improve the uniformity and selectivity of the monolithic column, suggesting the potential application of such double-functionalized monoliths in the efficient separation of complex bio-samples.
Abbreviations
| 2DE | Two dimensional electrophoresis |
| ACN | Acetonitrile |
| AMIM+Cl− | 1-Allyl-3-methylimidazolium chloride |
| BPO | Benzoyl peroxide |
| CEC | Capillary electrochromatography |
| DEDMA | 1,10-Decanediol dimethacrylate |
| DMA |
N,N-Dimethyl aniline |
| DMF |
N,N-Dimethylformamide |
| EDMA | Ethylene dimethacrylate |
| HPLC | High performance liquid chromatography |
| ILs | Ionic liquids |
| IP | Iron porphyrin |
| PEG400 | Polyethylene glycol 400 |
| SEM | Scanning electron microscope |
| TFA | Trifluoroacetic acid |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Numbers 21505030, 21575033); the Natural Science Foundation of Hebei Province (Grant Number B2015201024); and the Natural Science Foundation of Hebei University (Grant Number 2014-08).
References
- Y. Liang, C. Wu, Q. Zhao, Q. Wu, B. Jiang, Y. Weng, Z. Liang, L. Zhang and Y. Zhang, Anal. Chim. Acta, 2015, 900, 83–89 CrossRef CAS PubMed.
- J. C. Masini, Anal. Bioanal. Chem., 2016, 408, 1445–1452 CrossRef CAS PubMed.
- C. Liu, Q. Deng, G. Fang, X. Huang, S. Wang and J. He, ACS Appl. Mater. Interfaces, 2015, 7, 20430–20437 CAS.
- Y. Chen, N. Deng, C. Wu, Y. Liang, B. Jiang, K. Yang, Z. Liang, L. Zhang and Y. Zhang, Talanta, 2016, 154, 555–559 CrossRef CAS PubMed.
- J. C. Masini, J. Sep. Sci., 2016, 39, 1648–1655 CrossRef CAS PubMed.
- F. Svec and Y. Lv, Anal. Chem., 2015, 87, 250–273 CrossRef CAS PubMed.
- Z. Huang, L. Ma, C. Huang, Q. Li and E. C. Nice, Proteomics, 2017, 17, 1–3 Search PubMed.
- L. A. Liotta, M. Ferrari and E. Petricoin, Nature, 2003, 425, 905 CrossRef CAS PubMed.
- M. Molloy and F. Witzmann, Briefings Funct. Genomics Proteomics, 2002, 1, 23–39 CrossRef CAS.
- E. Nägele, M. Vollmer, P. Hörth and C. Vad, Expert Rev. Proteomics, 2004, 1, 37–46 CrossRef PubMed.
- D. K. Swertfeger, H. Li, S. Rebholz, X. Zhu, A. S. Shah, W. S. Davidson and L. J. Lu, Mol. Cell. Proteomics, 2017, 16, 680–693 CAS.
- F. Weiland, C. M. Zammit, F. Reith and P. Hoffmann, Electrophoresis, 2014, 35, 1893–1902 CrossRef CAS PubMed.
- D. Sharma and D. Bisht, Electrophoresis, 2016, 37, 1187–1190 CrossRef CAS PubMed.
- F. Lin, S. Yu, L. Gu, X. Zhu, J. Wang, H. Zhu, Y. Lu, Y. Wang, Y. Deng and L. Geng, Microchim. Acta, 2015, 182, 2321–2328 CrossRef CAS.
- A. M. Al-Hossaini, L. Suntornsuk and S. M. Lunte, Electrophoresis, 2016, 37, 2297–2304 CrossRef CAS PubMed.
- C. Liu, Q. Deng, G. Fang, H. Liu, J. Wu, M. Pan and S. Wang, Anal. Chim. Acta, 2013, 804, 313–320 CrossRef CAS PubMed.
- C. Liu, Q. Deng, G. Fang, X. Feng, H. Qian and S. Wang, Anal. Bioanal. Chem., 2014, 406, 7175–7183 CrossRef CAS PubMed.
- S. Hjerten, J. L. Liao and R. Zhang, J. Chromatogr. A, 1989, 473, 273–275 CrossRef CAS.
- T. B. Tennikova, M. Bleha and F. Svec, J. Chromatogr. A, 1991, 555, 97–107 CrossRef CAS.
- F. Svec and J. Fréchet, Anal. Chem., 1992, 64, 820–822 CrossRef CAS.
- P. Sun and D. W. Armstrong, Anal. Chim. Acta, 2010, 661, 1–16 CrossRef CAS PubMed.
- Z. Mao and Z. Chen, J. Chromatogr. A, 2017, 1480, 99–105 CrossRef CAS PubMed.
- T. Wang, Y. Chen, J. Ma, X. Zhang, L. Zhang and Y. Zhang, Analyst, 2015, 140, 5585–5592 RSC.
- B. Singco, C. Lin, Y. Cheng, Y. Shih and H. Huang, Anal. Chim. Acta, 2012, 746, 123–133 CrossRef CAS PubMed.
- J. Qin, L. Bai, J. Wang, Y. Ma, H. Liu, S. He, T. Li and Y. An, Anal. Methods, 2014, 7, 218–225 RSC.
- R. Guo, D. Zhang, X. Zhu, L. Tang, X. Zhang, L. Bai and H. Liu, Chromatographia, 2017, 80, 23–30 CAS.
- L. Bai, J. Wang, H. Zhang, S. Liu, J. Qin and H. Liu, Anal. Methods, 2015, 7, 607–613 RSC.
- H. Zhang, L. Bai, Z. Wei, S. Liu, H. Liu and H. Yan, Talanta, 2016, 149, 62–68 CrossRef CAS PubMed.
- K. Oohora, Y. Kihira, E. Mizohata, T. Inoue and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 17282–17285 CrossRef CAS PubMed.
- M. Biesaga, K. Pyrzyńska and M. Trojanowicz, Talanta, 2000, 51, 209–224 CrossRef CAS PubMed.
- J. Kopečná, I. Cabeza de Vaca, N. B. P. Adams, P. A. Davison, A. A. Brindley, C. N. Hunter, V. Guallar and R. Sobotka, J. Biol. Chem., 2015, 290, 28477–28488 CrossRef PubMed.
- C. Martin, J. Coyne and G. Carta, J. Chromatogr. A, 2005, 1069, 43–52 CrossRef CAS PubMed.
- X. Jiang, D. Zhang, X. Li, X. Wang, L. Bai, H. Liu and H. Yan, J. Pharm. Biomed. Anal., 2017, 138, 14–21 CrossRef CAS PubMed.
- Z. Wei, D. Zhang, C. Li, L. Bai, H. Liu and H. Yan, Anal. Methods, 2017, 9, 2596–2602 RSC.
- M. Catalá-Icardo, S. Torres-Cartas, S. Meseguer-Lloret, C. Gómez-Benito, E. Carrasco-Correa, E. F. Simó-Alfonso, G. Ramis-Ramos and J. M. Herrero-Martínez, Anal. Chim. Acta, 2017, 960, 160–167 CrossRef PubMed.
- K. Du, Q. Zhang, S. Dan, M. Yang, Y. Zhang and D. Chai, J. Chromatogr. A, 2016, 1443, 111–117 CrossRef CAS PubMed.
- Z. Liu, J. Ou, Z. Liu, J. Liu, H. Lin, F. Wang and H. Zou, J. Chromatogr. A, 2013, 1317, 138–147 CrossRef CAS PubMed.
- L. Terborg, J. C. Masini, M. Lin, K. Lipponen, M. Riekolla and F. Svec, J. Adv. Res., 2015, 6, 441–448 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01491k |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Haiyan
Liu
ab and
Hongyuan
Yan
*b
*ab,
Haiyan
Liu
ab and
Hongyuan
Yan
*b