
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-step photomechanical motion of a dibenzobarrelene crystal†
Takuya Taniguchi a,
Ayumi Kubotab,
Tatsuya Moritokic,
Toru Asahiad and
Hideko Koshima*d
a,
Ayumi Kubotab,
Tatsuya Moritokic,
Toru Asahiad and
Hideko Koshima*d
aDepartment of Advanced Science and Engineering, Graduate School of Advanced Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
bDepartment of Life Science and Medical Bioscience, School of Advanced Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
cDepartment of Materials Science and Biotechnology, Graduate School of Science and Engineering, Ehime University, 3 Bunkyo-cho, Matsuyama, Ehime 790-8577, Japan
dResearch Organization for Nano & Life Innovation, Waseda University, 513 Wasedatsurumaki-cho, Shinjuku-ku, Tokyo 162-0041, Japan. E-mail: hkoshima@aoni.waseda.jp
First published on 5th October 2018
Abstract
Photomechanical crystals are interesting from both basic and applied perspectives, and thus it is important to develop new examples. We investigated the photomechanical bending behaviour of a photochromic crystal of a dibenzobarrelene derivative. When a plate-like crystal was irradiated with ultraviolet (UV) light at 365 nm, two-step bending was observed. In the first step, the crystal quickly bent away from the light source, with an accompanying crystal colour change from colourless to purple. In the second step, under prolonged UV light, the bending returned slowly and then the crystal bent up towards the opposite direction, accompanied by an additional colour change to light yellow. Spectroscopic measurements and X-ray crystallographic analysis suggested that a long-lived biradical species is generated immediately upon UV light irradiation via a Norrish type II intramolecular hydrogen abstraction, and then the final photoproducts are formed under continuous UV exposure. X-ray crystallographic analysis before and after UV light irradiation for a few seconds revealed that the longitudinal axis (a axis) of the crystal elongated slightly after irradiation, which is consistent with the direction of the first-step bending. Based on these results, we propose that first-step bending could be induced by a biradical species, generated via a Norrish type II intramolecular hydrogen abstraction, and the second-step bending could originate from the formation of a mixture of final photoproducts under prolonged light irradiation.
Introduction
Materials that move by responding to external stimuli, such as light, heat, electricity, magnetic field, pressure, etc., have attracted substantial attention in recent years. Such mechanically responsive materials are expected to find applications as actuators and sensors for soft robots. Photomechanical polymers and gels have been well developed in previous research.1,2Contrary to their appearance as solid but fragile objects, in the past decade molecular crystals have been attractive because they exhibit mechanical motion induced by external stimuli. Typical photochemical reactions, such as pericyclic reactions,3,4 trans–cis isomerisation,5,6 enol–keto isomerisation,7 [2 + 2] cycloaddition,8,9 and [4 + 4] dimerisation,10–12 induce various kinds of photomechanical motion in molecular crystals. Recent advances in photomechanical crystals have been summarised in several reviews.13–18 Although many photomechanical crystals have been developed so far, the types of photochemical reactions that have been used to achieve photo-induced motion are limited. Thus, there is a need to explore other photochemical reaction systems to evaluate a wider variety of mechanical behaviour.
Dibenzobarrelene derivatives are well known to partake in various photochemical reactions in solution, such as tri-π-methane rearrangement, di-π-methane rearrangement, Norrish type II reactions, and [2 + 2] cycloaddition.19–21 The solid state photochemistry of dibenzobarrelenes has also been well-studied, by Scheffer and co-workers.22,23 Photochromism in the solid state of a dibenzobarrelene derivative was reported in detail by Ramaiah and co-workers, in which the photochromism is caused by a long-lived triplet biradical, formed from a Norrish type II intramolecular hydrogen abstraction initiated by ultraviolet (UV) light irradiation.24 However, photochromism in other dibenzobarrelene derivatives has not been well-studied.
In this study, we investigated the photochromism and bending behaviour of a similar dibenzobarrelene derivative, namely, 11,12-dibenzoyl-9,10-dihydro-9,10-dimethyl-9,10-ethenoanthracene (1), which was reported to exhibit a colour change from colourless to purple after UV light irradiation (Fig. 1).24 In terms of photomechanical motion, the plate-like crystal of compound 1 was observed to bend quickly with the colour change (colourless to purple) initiated by UV light irradiation. Following this, the bent crystal slowly bent up towards the opposite direction, with additional colour changes from purple to yellow under prolonged UV light exposure. The mechanism of the two-step photomechanical bending motion of the compound 1 crystal is discussed with respect to spectral and crystal structure changes.
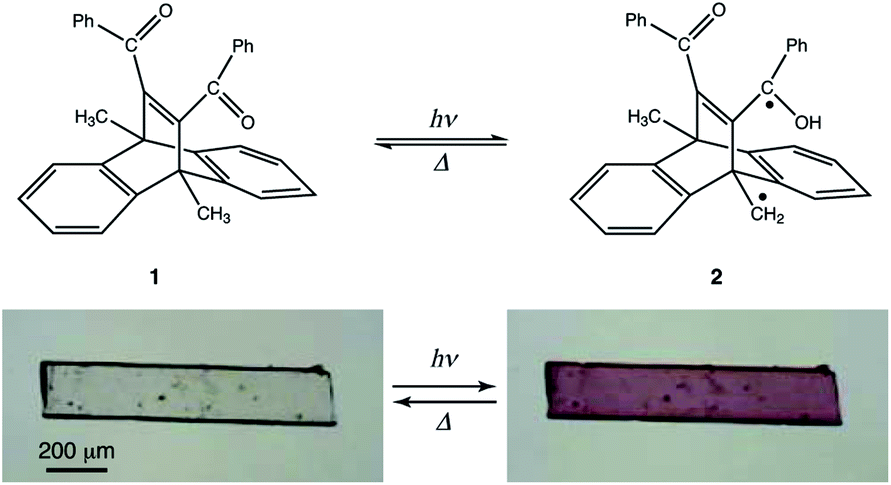 | ||
| Fig. 1 Photochromism of dibenzobarrelene 1 in the solid state. | ||
Experimental
Materials
Compound 1 was synthesised according to the same scheme used in previous reports.19,25 Compound 1 was purified by silica gel column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 10:1). After purification, plate-like crystals were obtained by evaporation of the solvent mixture. The compound was identified by nuclear magnetic resonance (NMR) and melting point measurements. The melting point was determined with the MP-S3 Micro Melting Point System (Yanako New Science, Inc.).
:ethyl acetate = 10:1). After purification, plate-like crystals were obtained by evaporation of the solvent mixture. The compound was identified by nuclear magnetic resonance (NMR) and melting point measurements. The melting point was determined with the MP-S3 Micro Melting Point System (Yanako New Science, Inc.).
Diffuse reflectance measurements
UV-Vis diffuse reflectance spectra of a powdered sample of compound 1 were measured with a Lambda 650 spectrometer (PerkinElmer). The powder was mounted on a glass plate and then covered with a quartz glass plate. In each experiment, the sample was picked up from the measurement holder, and then irradiated by UV light (365 nm, 40 mW cm−2) from the quartz glass side. Every measurement was performed in the range 200–800 nm at 1 nm resolution with a scan speed of 200 nm min−1.FT-IR measurements
Fourier-transform infrared (FT-IR) spectra of a powdered sample of compound 1 were measured with an attenuated total reflection (ATR) method using a Nicolet 6700 spectrometer (Thermo Fisher Scientific) in the range 500–4500 cm−1. The sample was irradiated by UV light (365 nm, 40 mW cm−2).X-ray crystallographic analysis
Single-crystal diffraction data for compound 1 were collected using an R-AXIS RAPID diffractometer (Rigaku) equipped with monochromatic Cu-Kα and Mo-Kα radiation sources. Measurements were performed at 223 and 293 K. The low temperature measurements were conducted to obtain the crystal structure after light irradiation. At first, measurements were taken before irradiation at 223 K. Following this, the crystal sample was irradiated with UV light for several seconds at 293 K while being rotated, as photochemical reactions are slow at low temperature. After irradiation, the structure was determined again at 223 K to preserve photoproducts in the crystal. The crystal structure at 293 K in the absence of UV light was also determined, to correlate with the bending behaviour observed at room temperature. Crystal structures were solved by using the direct method of SHELXD2013, and refined on F2 by the full-matrix least-squares method, SHELXL.26 Calculations were performed using the software package CrystalStructure (Rigaku).27Observation of bending behaviour
Following flash column chromatographic purification, thin plate-like crystals were obtained by solvent evaporation. A crystal was fixed at the tip of a glass needle with glue, and then irradiated by 365 nm UV light with a flux density of 40 mW cm−2 (UV-400; Keyence). Movies were recorded using a digital high-speed microscope (Keyence; VHX-5000).Results and discussion
Diffuse reflectance spectra
Diffuse reflectance spectra of a powdered sample of compound 1 were measured. Prior to UV light irradiation, the spectrum had a broad peak between 200–400 nm, and no peaks above 450 nm (Fig. 2a). Upon UV light irradiation for 0.1 s, a peak appeared at 574 nm, for which the peak intensity increased with irradiation time until 3 s, with a corresponding colour change from virtually colourless to purple (Fig. 2a). Difference spectra, obtained by subtracting the spectrum before UV light irradiation from each spectrum after light irradiation, highlight these spectral changes (Fig. 2b). The difference spectra exhibit three peaks at 340, 430, and 574 nm, all of which increased with irradiation time (Fig. 2b). The temporal behaviour of the peaks at 340 and 430 nm were similar, but in contrast to that of 574 nm (Fig. S1a†). The largest increase in intensity was for the 574 nm peak, which indicates the generation of a purple-coloured species, consistent with the colour of the powder after irradiation (Fig. S2a†). | ||
| Fig. 2 (a, c and e) Diffuse reflectance spectra and (b, d and f) difference spectra of a powdered sample of compound 1. (a–d) Changes induced by ultraviolet (UV) light irradiation; (a and b) in the initial 3 s, and (c and d) for 60 s of UV exposure. (e and f) Thermal relaxation after UV light exposure for 3 s. Black arrows indicate changes in peak intensities. Difference spectra were obtained by subtracting the spectrum before UV light irradiation from spectra obtained under or after irradiation. | ||
The absorption peak at 574 nm can be attributed to the extensive π-delocalisation of the biradical species between the triplet benzyl radical and the benzoyl groups, which are connected by the conjugated alkenyl double bond.24 The increase in peak intensity at 574 nm until 3 s after irradiation suggests that biradical 2 was produced immediately by a Norrish type II intramolecular hydrogen abstraction. The apparent half-life (τ1/2UV) of hydrogen abstraction from compound 1 to generate biradical 2 was estimated to be 0.12 s, based on the initial changes in peak intensity at 574 nm (Fig. S1b†). The peak at 430 nm can also be assigned to biradical 2, as reported previously.24,28,29
After stopping UV light irradiation for 3 s, the absorption peak at 574 nm decreased gradually with time, and its initial intensity returned completely within 3 days (Fig. 2e). However, the difference spectrum after 3 days indicates that spectral changes for the peaks at 340 and 400 nm clearly remained (Fig. 2f), especially at 340 nm, for which virtually no decrease was observed (Fig. S1g and h†); this suggests that final photoproducts were produced. This spectral behaviour indicates that biradical 2 largely reverted to initial compound 1, and that the remaining absorption peaks after 3 days should be assigned to π-conjugated final photoproducts produced by irreversible tri-π-methane and di-π-methane rearrangements, which are reported to occur in benzene solution (Fig. S3†).20 The photoproducts via the rearrangements in the crystals should be produced immediately upon UV irradiation based on the absorption increase at 340 nm by light irradiation (Fig. 2b). The half-life of thermal relaxation (τ1/2) from biradical 2 into compound 1 was estimated to be 30.6 min, based on the initial changes in peak intensity at 574 nm (Fig. S1c and d†); this is almost coincident with that (τ1/2 = 39.2 min) estimated at 430 nm (Fig. S1e and f†). The slow thermal relaxation of biradical 2 originates from the long lifetime of the triplet state biradical.
When the sample was irradiated for longer than 3 s, the peak intensity at 574 nm decreased gradually, but the intensities at 340 and 430 nm continued to increase with irradiation time (Fig. 2c). Difference spectra show these changes in the three peaks clearly (Fig. 2d). After irradiation for 60 s, the intensity at 430 nm increased the most, which is consistent with the observations of yellow-coloured material in the powder (Fig. S2b†).
The gradual decrease in the peak at 574 nm under prolonged UV light exposure might be due to the coupling of biradical 2 to produce cyclobutanol, which prohibits radical delocalisation (Fig. S3†). In terms of the photochemistry of benzophenone derivatives in the solid state, such cyclobutanol formation from biradical species has been well-documented.30–32 The continuous increase in the peak at 430 nm indicates that a biradical species is produced under light, but that the biradical cannot delocalise; this may be due to cyclisation. The continuous increase in the peak at 340 nm suggests that photoproducts of di-π-methane and tri-π-methane rearrangements are also continuously produced. Therefore, after prolonged light irradiation, the powdered sample could be a mixture of the initial compound, biradical 2, cyclobutanol, and the photoproducts from rearrangements (Fig. S3†).
FT-IR spectra
FT-IR spectra of a powdered sample of compound 1 were measured after UV light irradiation (Fig. 3). Before light irradiation, the spectrum had two peaks in the range 1600–1700 cm−1, at 1649 and 1664 cm−1, which were attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of compound 1. These two peaks decreased with increasing irradiation time, and a new peak appeared at 1658 cm−1, which increased upon prolonged UV light exposure. The temporal behaviour of these three peaks is summarised in Fig. S4.† The two peaks at 1649 and 1664 cm−1 continued to decrease, while the new peak at 1658 cm−1 increased for 60 s and then reached saturation (Fig. S4a and b†).
O stretching vibrations of compound 1. These two peaks decreased with increasing irradiation time, and a new peak appeared at 1658 cm−1, which increased upon prolonged UV light exposure. The temporal behaviour of these three peaks is summarised in Fig. S4.† The two peaks at 1649 and 1664 cm−1 continued to decrease, while the new peak at 1658 cm−1 increased for 60 s and then reached saturation (Fig. S4a and b†).
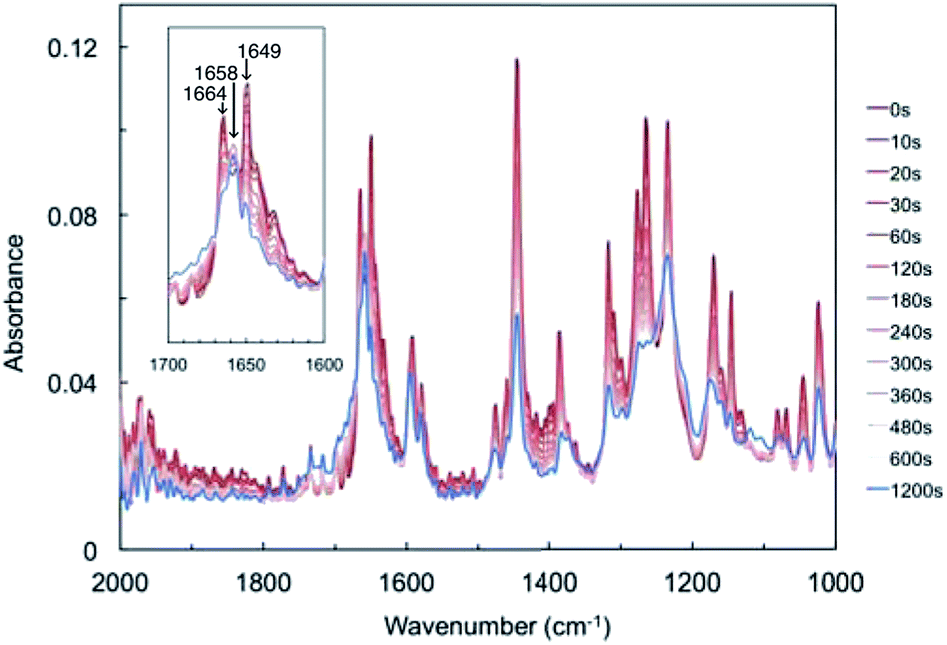 | ||
| Fig. 3 Dependence of Fourier-transform infrared (FT-IR) spectral changes on UV irradiation time. The inset shows enlarged spectra in the range of 1600–1700 cm−1. The peaks at 1649 and 1664 cm−1 can be attributed to the CO stretching vibration of compound 1, and the peak at 1658 cm−1 might be attributed to biradical 2, generated from hydrogen abstraction. | ||
The new peak at 1658 cm−1 might be attributed to biradical species 2. Here, the CO stretching vibrations of cyclobutanol and rearrangement photoproducts were not detected, probably due to low conversion rates or spectral overlap with the peaks of compound 1. These changes suggest that a Norrish type II intramolecular γ-hydrogen abstraction proceeded to decrease the CO bond strength of compound 1 to form biradical 2.
Crystal structure
X-ray crystallographic analysis was performed at 293 K (Fig. 4 and Table S1†). The ORTEP drawing of compound 1 is shown by thermal ellipsoids with 25% probability (Fig. 4a). The molecule has a disordered phenyl ring with an occupancy of 56% as its main conformation (Fig. 4a).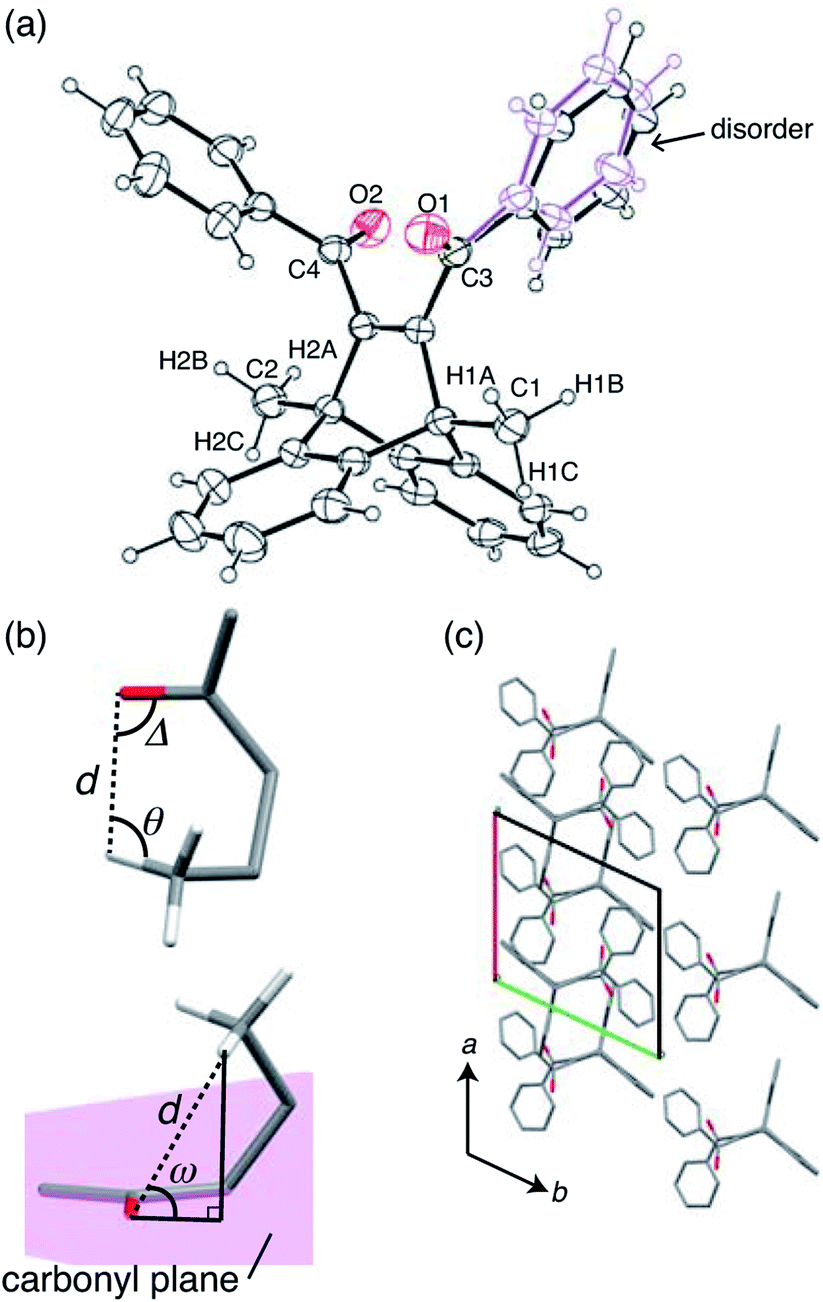 | ||
| Fig. 4 Crystal structure of compound 1 at 293 K. (a) Molecular conformation drawn by thermal ellipsoids with 25% probability. The main conformation of the disordered phenyl ring is drawn in black (the same colour as other carbons), and the minor conformation is drawn in purple. (b) Definition of geometric parameters for γ-hydrogen atom abstraction: d is the distance between the carbonyl oxygen and the γ-hydrogen, Δ is the CO⋯H angle, θ is the C–H⋯O angle, and ω is the angle by which the γ-hydrogen atom lies outside the mean plane of the carbonyl group. (c) Molecular arrangement of the (001) face, which is the largest face of the plate-like crystal of compound 1. Hydrogen atoms and the disordered phenyl ring are omitted for clarity. | ||
To analyse the intramolecular hydrogen abstraction reaction, we used four geometric parameters, d, Δ, θ, and ω (Fig. 4b), which were first introduced by Scheffer for the analysis of γ-hydrogen abstractions in the solid state.33 These parameters are defined as follows: d is the distance between the carbonyl oxygen and the γ-hydrogen atom, Δ is the CO⋯H angle, θ is the C–H⋯O angle, and ω is the angle by which the γ-hydrogen atom lies outside the mean plane of the carbonyl group (Fig. 4b). The previous paper reported that the favourable d is the sum of the van der Waals radii of hydrogen and oxygen atoms (2.72 Å), and that favourable angle geometries of ω, Δ, and θ are around 50°, 80°, and 115°, respectively.33
In Table 1, these parameters are summarised for compound 1. Here, only H1A and H2A (shown in Fig. 4a) were considered in the CO⋯H geometry, because the other methyl group hydrogen atoms are too far away from the carbonyl O1 and O2 atoms. The distances d of C1O1⋯H1A and C2O2⋯H2A are 2.656 and 2.874 Å, respectively, both of which are close to the favourable distance (2.72 Å). In terms of angle requirements, the values of ω, Δ, θ were 59.4°, 83.8°, and 111.3° for C1O1⋯H1A, and 63.6°, 72.7°, and 110.7° for C2O2⋯H2A, respectively. These angles are also similar to the favourable geometries. Based on this analysis, it is estimated that both carbonyl groups can abstract hydrogen atoms from methyl groups to produce biradical 2. The biradical species may cause further radical coupling, because the distances of both C1⋯C3 (2.92 Å) and C2⋯C4 (2.88 Å) allow for the formation of cyclobutanol, as reported in benzophenone crystals.30–32
| d (Å) | ω (°) | Δ (°) | θ (°) | |
|---|---|---|---|---|
| C1O1⋯H1A |
2.656 | 59.4 | 83.8 | 111.3 |
| C2O2⋯H2A |
2.874 | 63.6 | 72.7 | 110.7 |
| Favorable geometrics33 | 2.72 | 50–60 | 80 | 115 |
Bending behaviour
First, bending of the plate-like crystal was observed after a short period of UV light irradiation (Fig. 5). From the X-ray diffraction analysis, the plate-like crystal was found to have a (001) face as its largest face, with a longitudinal direction along the a axis (Fig. 5a). When the (001) face of the crystal (length: 4335 μm, width: 274 μm, thickness: 25 μm) was irradiated from the left by UV light (365 nm, 40 mW cm−2) for 1 s, the crystal bent away from the light source up to the tip, with a displacement angle (see the angle definition in Fig. S5a†) of 13.6°, and an accompanying crystal colour change from colourless to purple (Fig. 5b, c and Movie S1†). Gradually, the crystal bent up to 17.4° even after the UV irradiation was stopped (Fig. 5d). Then the bending returned partially to 10.6° (Fig. 5e) 100 min after the UV irradiation was stopped. The reasons that the crystal could not straighten back 100%, can be due to the formation of the photoproducts via tri-π-methane and di-π-methane rearrangements, and the partial reverse of biradical 2 back to compound 1.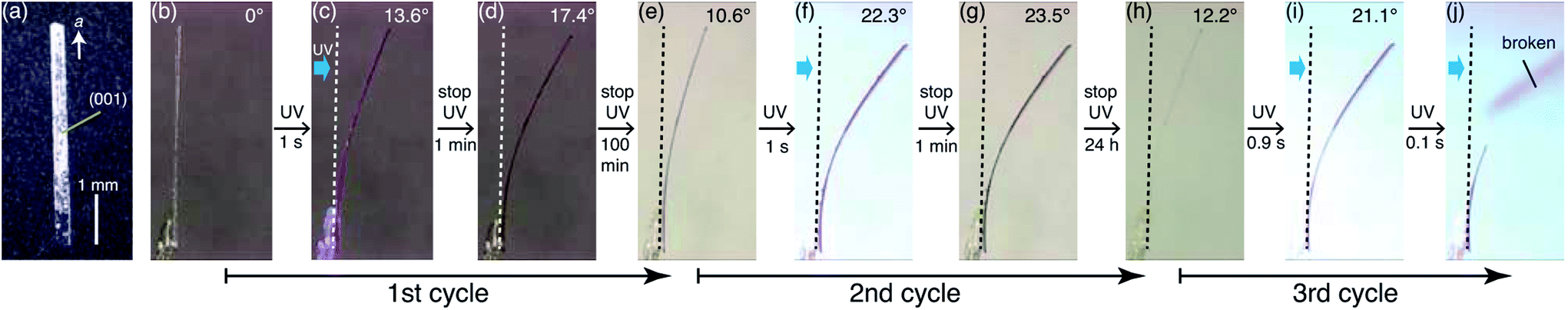 | ||
| Fig. 5 First step photomechanical bending of a plate-like crystal of compound 1 upon UV light irradiation for 1 s. (a) Top view of the plate-like crystal before light irradiation. (b–j) Snapshots of side views of the plate-like crystal in (b–d) the first stage of bending and relaxation, (e–f) the second stage of bending and relaxation, and (h–j) the third stage of bending. The angle at the upper right corner in each panel shows the tip displacement angle. Dotted lines indicate the initial position at the tip displacement angle (0°). UV radiation was always applied to the left side of the crystal. | ||
After a second application of UV radiation for 1 s, the crystal bent up to 22.3° (Fig. 5f). The crystal bent slightly to 23.5° after the UV light was stopped (Fig. 5g). The bending returned gradually, reaching 12.2° in 24 h, with an accompanying colour change to colourless (Fig. 5h). Upon a third dose of UV exposure for 1 s, the crystal bent to 21.1°, and broke into two pieces in the next moment (Fig. 5i and j). The temporal profile of the tip displacement angle is summarised in Fig. S5b.† The tip displacement angle at maximum bending decreased with the increase of crystal thickness (Fig. S6†).
A two-step bending motion was observed when another thin plate-like crystal (length: 525 μm, width: 134 μm, thickness: 8.6 μm) was submitted to prolonged (1 min) UV light irradiation (Fig. 6 and Movie S2†). In the first step, the crystal underwent bending upon initial UV light irradiation for 1 s (Fig. 6a and b). Under prolonged UV irradiation, the crystal returned to the straight form with decreasing purple colour after 9 s (Fig. 6c), and then bent up in the opposite direction with an additional colour change to light yellow after 50 s (Fig. 6d). The yellow-coloured bent crystal did not change its colour or shape after the UV irradiation was stopped, indicating that the second-step bending is irreversible.
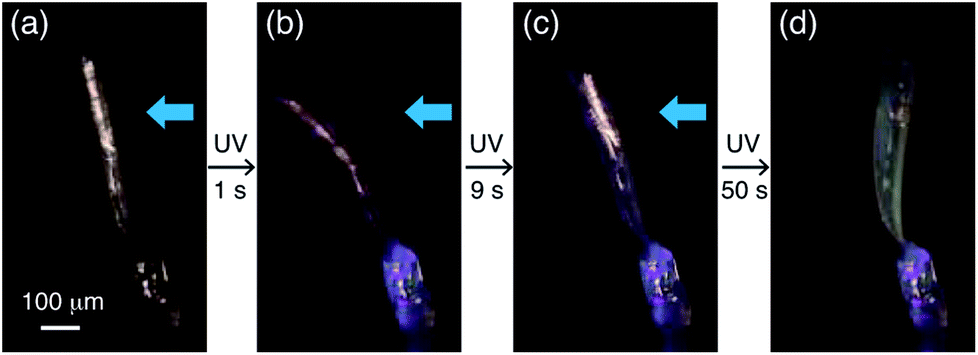 | ||
| Fig. 6 Two-step bending of a plate-like crystal of compound 1 under continuous UV light exposure for 1 min. | ||
Bending mechanism
The mechanism of the first-step bending is discussed based on the crystal structure change after short exposure to UV radiation (Table S1†). The initial X-ray diffraction measurement was performed on the crystal at 223 K before UV irradiation. Then, to clarify crystal structure changes due to biradical 2, the crystal was irradiated for several seconds at 293 K while being rotated, because photochemical reactions are slow at low temperature. After a short period of UV irradiation, the crystallographic analysis was performed again at 223 K, stabilising biradical 2 in the crystal.The molecular arrangement of the (001) plane before light irradiation is shown in Fig. 4c. The carbonyl CO group is aligned almost parallel to the a axis, which is the longitudinal direction of the crystal. After UV irradiation, biradical species 2 was not found, probably due to it not being present in sufficient quantities for structure determination, but the unit cell parameters changed slightly. The lengths of the a and b axes were elongated by 0.15% and 0.08%, respectively, and the c axis shortened by 0.19%. This lattice change could have been induced by biradical species 2, formed via an intramolecular γ-hydrogen abstraction, as reported by Hosoya and his co-worker.34 They determined the crystal structure of compound 1 after UV light irradiation at low temperature (90 K), and detected an electron density peak of the methylene radical (·CH2–) as the disorder of the methyl group (CH3) and the methylene radical. The abstracted hydrogen atom was also detected as a small electron density peak near the benzoyl oxygen atom with a distance of 0.967(2) Å, forming the benzyl-type radical. This result suggested that the γ-hydrogen atom transferred from the methyl group to the benzoyl oxygen atom, and that biradical 2 was produced during the hydrogen abstraction. Based on these results, biradical 2 might move away surrounding molecules along the a axis due to hydrogen transfer to the carbonyl CO, which is aligned almost parallel to the a axis. This molecular change should induce the first-step bending due to elongation of the a axis at the irradiated surface.
The second-step bending could have been induced by the formation of the photoproduct mixture via radical coupling of the biradical 2, di-π-methane rearrangement, and tri-π-methane rearrangement (Fig. S3†). The bending speed and colour change observed during the second-step bending are in reasonably good agreement with the measured changes in UV-vis and IR spectra.
Conclusions
A two-step bending motion of a plate-like crystal of compound 1 was observed under prolonged UV light irradiation. In the first step, the crystal bent away from the light source quickly upon initial light irradiation, with an accompanying colour change from colourless to purple. In the second step, under prolonged light irradiation, the crystal returned slowly to its straight form with decreasing purple colour, and then bent up in the opposite direction, accompanied by an additional colour change to light yellow. Based on the results of the spectroscopic measurements and X-ray crystallographic analysis, the first-step bending could have originated from the triplet biradical 2, generated by a Norrish type II intramolecular hydrogen abstraction. The subsequent second-step bending might have been induced by the photoproduct mixture via irreversible reactions under continuous UV exposure. Although the bending behaviour of crystals of compound 1 may not have practical applications, this work indicates that a typical photochemical reaction, namely, a Norrish type II intramolecular hydrogen abstraction, also induces photomechanical bending.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the JSPS Grant-in-Aid for Scientific Research B (17H03107), Challenging Exploratory Research (16K12918), Research Fellowship for Young Scientists, and WISE project research (Research for Energy Next) at Waseda University. T. T. also would like to thank the Leading Graduate Program in Science and Engineering at Waseda University.References
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- D. Kitagawa, H. Nishi and S. Kobatake, Angew. Chem., Int. Ed., 2013, 52, 9320–9322 CrossRef CAS PubMed.
- H. Koshima, N. Ojima and H. Uchimoto, J. Am. Chem. Soc., 2009, 131, 6890–6891 CrossRef CAS PubMed.
- T. Taniguchi, J. Fujisawa, M. Shiro, H. Koshima and T. Asahi, Chem.–Eur. J., 2016, 22, 7950–7958 CrossRef CAS PubMed.
- H. Koshima, K. Takechi, H. Uchimoto, M. Shiro and D. Hashizume, Chem. Commun., 2011, 47, 11423–11425 RSC.
- R. Medishetty, A. Husain, Z. Bai, T. Runčevski, R. E. Dinnebier, P. Naumov and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 5907–5911 CrossRef CAS PubMed.
- R. Medishetty, S. C. Sahoo, C. E. Mulijanto, P. Naumov and J. J. Vittal, Chem. Mater., 2015, 27, 1821–1829 CrossRef CAS.
- R. O. Al-Kaysi and C. J. Bardeen, Adv. Mater., 2007, 19, 1276–1280 CrossRef CAS.
- L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, J. Am. Chem. Soc., 2011, 133, 12569–12575 CrossRef CAS PubMed.
- H. Koshima, H. Uchimoto, T. Taniguchi, J. Nakamura, T. Asahi and T. Asahi, CrystEngComm, 2016, 18, 7305–7310 RSC.
- New Frontiers in Photochromism, ed. M. Irie, Y. Yokoyama and T. Seki, Springer, Heidelberg, 2013, ch. 1 and 2 Search PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- T. Kim, L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, ChemPhysChem, 2014, 15, 400–414 CrossRef CAS PubMed.
- P. Commins, I. T. Desta, D. P. Karothu, M. K. Panda and P. Naumov, Chem. Commun., 2016, 52, 13941–13954 RSC.
- C. V. Kumar, B. A. R. C. Murty, S. Lahiri, E. Chackachery, J. C. Scaiano and M. V. George, J. Org. Chem., 1984, 49, 4923–4929 CrossRef CAS.
- C. V. Asokan, S. A. Kumar, S. Das, N. P. Rath and M. V. George, J. Org. Chem., 1991, 56, 5890–5893 CrossRef CAS.
- D. Ramaiah, M. C. Sajimon, J. Joseph and M. V. George, Chem. Soc. Rev., 2005, 34, 48–57 RSC.
- P. R. Pokkuluri, J. R. Scheffer and J. Trotter, J. Am. Chem. Soc., 1990, 112, 3676–3677 CrossRef CAS.
- J. Chen, J. R. Scheffer and J. Trotter, Tetrahedron, 1992, 48, 3251–3274 CrossRef CAS.
- M. C. Sajimon, D. Ramaiah, C. H. Suresh, W. Adam, F. D. Lewis and M. V. George, J. Am. Chem. Soc., 2007, 129, 9439–9445 CrossRef CAS PubMed.
- J. J. Zhang and G. B. Schuster, J. Am. Chem. Soc., 1989, 111, 7149–7155 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- CrystalStructure, version 4.2.2., Rigaku Corporation, Tokyo, Japan, 2016 Search PubMed.
- R. D. Small Jr and J. C. Scaiano, Chem. Phys. Lett., 1977, 50, 431–434 CrossRef.
- L. J. Johnston and J. C. Scaiano, Chem. Rev., 1989, 89, 521–547 CrossRef CAS.
- H. Koshima, A. Maeda, N. Masuda, T. Matsuura, K. Hirotsu, K. Okada, H. Mizutani, Y. Ito, T. Y. Fu, J. R. Scheffer and J. Trotter, Tetrahedron: Asymmetry, 1994, 5, 1415–1418 CrossRef CAS.
- H. Koshima, D. Matsushige and M. Miyauchi, CrystEngComm, 2001, 33, 1–3 Search PubMed.
- H. Koshima, M. Fukano, N. Ojima, K. Johmoto and H. Uekusa, J. Org. Chem., 2014, 79, 3088–3093 CrossRef CAS PubMed.
- H. Ihmels and J. R. Scheffer, Tetrahedron, 1999, 55, 885–907 CrossRef CAS.
- S. Okabe and T. Hosoya, JPS Conf. Proc., 2015, 8, 031014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1860420–1860422. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06639f |
| This journal is © The Royal Society of Chemistry 2018 |