
Engineering the biosynthesis of fungal nonribosomal peptides
Liwen
Zhang†
 a,
Chen
Wang†
a,
Kang
Chen
a,
Weimao
Zhong
b,
Yuquan
Xu
*a and
István
Molnár
*bc
a,
Chen
Wang†
a,
Kang
Chen
a,
Weimao
Zhong
b,
Yuquan
Xu
*a and
István
Molnár
*bc
aBiotechnology Research Institute, The Chinese Academy of Agricultural Sciences, 12 Zhongguancun South Street, Beijing 100081, P. R. China. E-mail: xuyuquan@caas.cn
bSouthwest Center for Natural Products Research, University of Arizona, 250 E. Valencia Rd., Tucson, AZ 85706, USA
cVTT Technical Research Centre of Finland, P.O. Box 1000, FI-02044 VTT, Espoo, Finland. E-mail: istvan.molnar@vtt.fi
First published on 7th July 2022
Abstract
Covering: 2011 up to the end of 2021.
Fungal nonribosomal peptides (NRPs) and the related polyketide–nonribosomal peptide hybrid products (PK–NRPs) are a prolific source of bioactive compounds, some of which have been developed into essential drugs. The synthesis of these complex natural products (NPs) utilizes nonribosomal peptide synthetases (NRPSs), multidomain megaenzymes that assemble specific peptide products by sequential condensation of amino acids and amino acid-like substances, independent of the ribosome. NRPSs, collaborating polyketide synthase modules, and their associated tailoring enzymes involved in product maturation represent promising targets for NP structure diversification and the generation of small molecule unnatural products (uNPs) with improved or novel bioactivities. Indeed, reprogramming of NRPSs and recruiting of novel tailoring enzymes is the strategy by which nature evolves NRP products. The recent years have witnessed a rapid development in the discovery and identification of novel NRPs and PK–NRPs, and significant advances have also been made towards the engineering of fungal NRP assembly lines to generate uNP peptides. However, the intrinsic complexities of fungal NRP and PK–NRP biosynthesis, and the large size of the NRPSs still present formidable conceptual and technical challenges for the rational and efficient reprogramming of these pathways. This review examines key examples for the successful (and for some less-successful) re-engineering of fungal NRPS assembly lines to inform future efforts towards generating novel, biologically active peptides and PK–NRPs.
 Liwen Zhang | Liwen Zhang is an associate professor at the Biotechnology Research Institute, Chinese Academy of Agricultural Sciences, Beijing, China. She received her B.Sc. in Environmental Sciences from Peking University, China in 2008, and her Ph.D. in Agricultural and Environmental Science from University of Georgia, United States in 2013. She joined Prof. Yuquan Xu's group in 2014 to study the combinatorial biosynthesis of fungal secondary metabolites. By combining genomics, transcriptomics and metabolomics, she identifies bioactive secondary metabolites and their biosynthetic genes from Hypocrealean fungi, attempting to “upgrade” the biosynthetic pathways in order to produce agricultural chemicals with higher efficiency, lower toxicity and less environmental disturbance. |
 Chen Wang | Chen Wang received his B.Sc. (2010) in applied chemistry from Lanzhou University, and Ph.D. in biochemistry and molecular biology from the University of Chinese Academy of Sciences (2018). He is currently an associate professor at the Biotechnology Research Institute, Chinese Academy of Agricultural Sciences, Beijing, China. His research focuses on the discovery and biosynthesis of microbial bioactive natural products. |
 Kang Chen | Kang Chen received his B.Sc. (2017) in traditional Chinese medicine from China Pharmaceutical University, and M.Sc. in medicinal chemistry of natural products from China Pharmaceutical University (2020). He is currently a PhD student of Prof. Yuquan Xu at the Chinese Academy of Agricultural Sciences, Beijing, China. His research focuses on the discovery and biosynthesis of microbial bioactive natural products. |
 Weimao Zhong | Weimao Zhong received his B.Sc. (2012) in phytochemistry from Hunan Agriculture University, M.Sc. (2015) and Ph.D. (2019) in marine natural product chemistry from the University of Chinese Academy of Sciences. In 2020, he joined The University of Arizona to conduct postdoctoral research on combinatorial biosynthetic chemistry and microbial biotransformation with Prof. István Molnár and Prof. A. A. Leslie Gunatilaka. He is interested in the discovery and biosynthesis of natural products from microorganisms. |
 Yuquan Xu | Yuquan Xu is a professor at the Chinese Academy of Agricultural Sciences (CAAS), Beijing, China. He received his B.Sc. and Ph.D. in molecular biology from the China Agriculture University. He conducted postdoctoral research on fungal polyketide and nonribosomal peptide biosynthesis with Prof. István Molnár at the Southwest Center for Natural Products Research, University of Arizona (2005–2008 & 2011–2013) and the discovery of microbial natural products with MALDI-TOF mass spectrometry with Prof. Pieter Dorrestein at the University of California, San Diego (2009–2010). In 2014, he joined CAAS to study the biosynthesis of fungal natural products for agricultural applications. His research interests include the discovery of bioactive natural products and the engineering of biosynthetic pathways. |
 István Molnár | After his studies (M.Sc. at ELTE, Hungary, and Ph.D. from Hiroshima University, Japan, with Prof. Yoshikatsu Murooka), István Molnár conducted postdoctoral research with Prof. Peter Leadlay at Cambridge University (U.K.) on the biosynthesis of rapamycin. He investigated antibiotic biosynthesis at the Institute for Drug Research (Hungary) and studied epothilone, soraphen, and emamectin biosynthesis at Syngenta Biotechnology (NC). As a Professor at the Southwest Center for Natural Products Research at the University of Arizona (2004–2021), his group devised combinatorial biosynthesis with cyclooligomer depsipeptides and benzenediol lactones. As a Research Professor at VTT Research Centre, Finland (2022–), he concentrates on finding synthetic microbiology solutions for the sustainable production of chemicals of industrial interest. |
1. Introduction
Nonribosomal peptide (NRP) synthetases (NRPSs) assemble numerous structurally complex peptide natural products (NPs) with a wide range of bioactivities.1 NRPs are short linear, cyclic or branched cyclic peptides that also include the lipopeptides if they contain a fatty acid side chain,2 the glycopeptides if they are decorated by carbohydrate units,3 and the depsipeptides if they incorporate hydroxy acid residues and feature both amide and ester bonds.4,5 NRPs also form composite, hybrid metabolites with other NPs using a sequential or convergent biosynthetic logic.6 Fungi are a prolific source of NRPs that constitute, or inspired the design of, novel drugs with great pharmacological or agricultural importance, such as the antibacterial penicillins and cephalosporins, the antifungal echinocandins,2,7 the immunosuppressant cyclosporines,2,8 or the insecticidal PF1022A2 (Fig. 1). In contrast to peptides produced via the ribosomal route, NRPSs often incorporate non-canonical, non-proteinogenic amino acids such as α-, β-, or γ-amino acids, D-amino acids and other amino acid analogues, as well as various hydroxy- or ketocarboxylic acids. The structural diversity of NRPs is further expanded by the various ring topologies, and the modifications of the peptide backbone and the side chains, afforded by the NRPS or additional enzymes.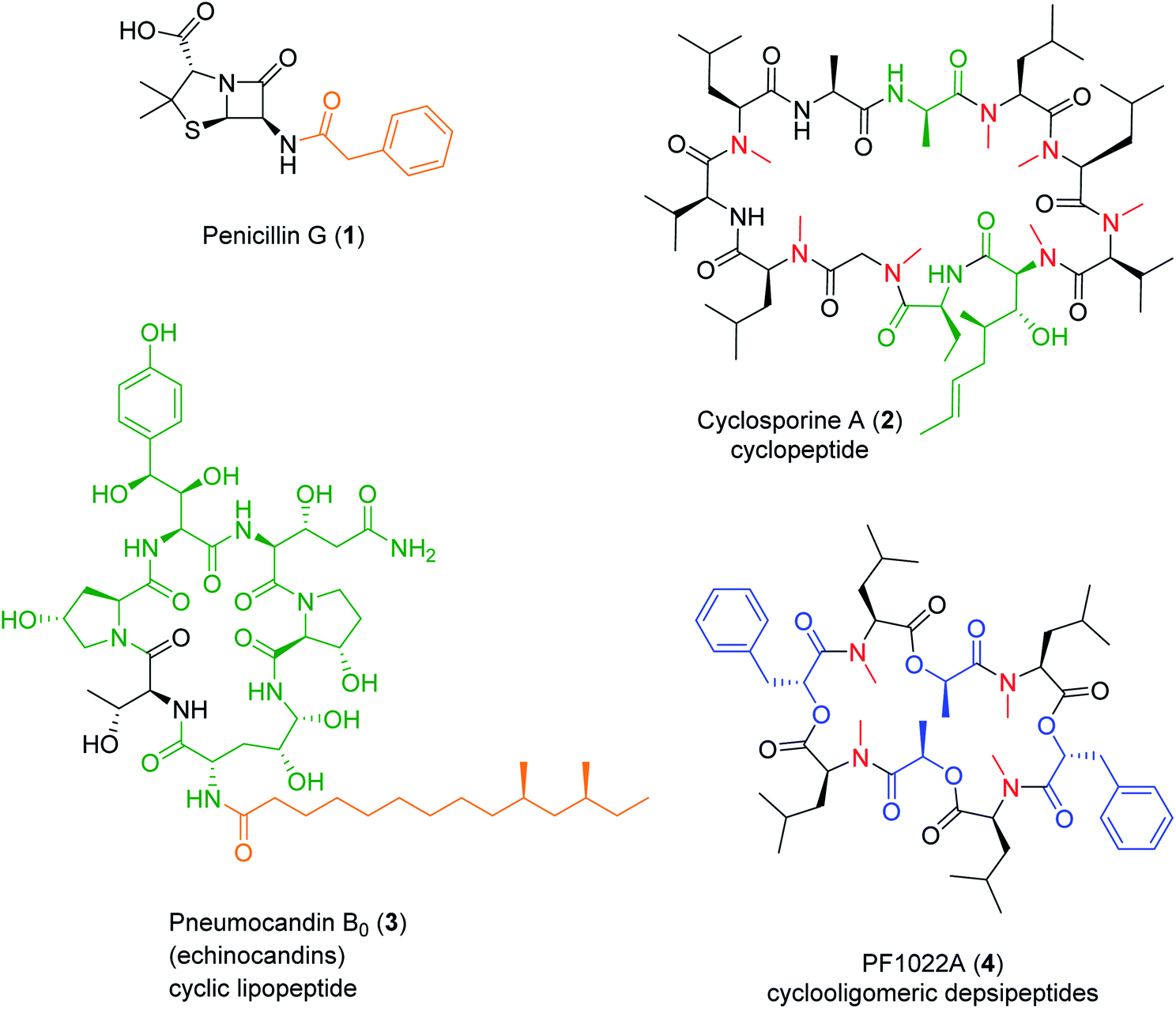 | ||
| Fig. 1 Structures of representative, clinically or agriculturally important fungal NRPs. Four determinants of structural diversity introduced by the NRPS are highlighted: carboxylic acid N-caps (orange), non-proteinogenic amino acids (green), hydroxy acids (blue), and N-methylation (red). | ||
Nature employs multiple ways to diversify NRPs, including duplication, recruitment, deletion, or mutation of the genes that encode the biosynthetic enzymes. Fittingly, NRP biosynthetic gene clusters (BGCs) are often located in dynamic regions of fungal chromosomes such as in loci near the telomere, or on accessory chromosomes.9,10 Accessory chromosomes are of particular interest due to their capacity for horizontal transfer between strains and their dynamic “crosstalk” with core chromosomes.11 The resulting structural complexity of NRPs makes them especially well-suited to interact with a variety of biological targets, including some that have been considered to be undruggable by other types of molecules.12 The structural modifications of the NRPs may also improve their properties as pharmaceutical drug candidates (e.g., N-alkylation for enhancing metabolic stability and intestinal permeability). Thus, NRPSs represent a promising target for synthetic biology to engineer the production of bioactive uNPs (unnatural products, i.e., NP analogues and de novo obtained small molecule products that are not present in nature, but produced in an amenable host organism through biosynthetic manipulation).
1.1 Fungal nonribosomal peptides and their biosynthesis
Fungal NRPSs are multifunctional, modular megaenzymes that synthesize NRPs through the sequential condensation of amino acid and hydroxycarboxylic acid building blocks.13 Elongation modules typically comprise an adenylation domain (A) that selects and activates an incoming building block; a condensation domain (C) that forms an amide or ester linkage between the activated amino acid or hydroxycarboxylic acid substrate and the peptidyl intermediate from the preceding module; and a thiolation domain (T, also known as the peptidyl carrier protein domain, PCP) whose thiol-containing phosphopantetheine arm tethers the activated amino acid and shuttles the growing NRP intermediate to the next NRPS module (Fig. 2). The final peptide chain is released from the last NRPS module as a linear or a cyclic NRP through hydrolysis or cyclization, typically catalyzed by a terminal condensation (CT) domain.14 Further product structure diversification is afforded by modification domains integrated into the NRPS, and tailoring enzymes acting in trans or after the release of the peptide.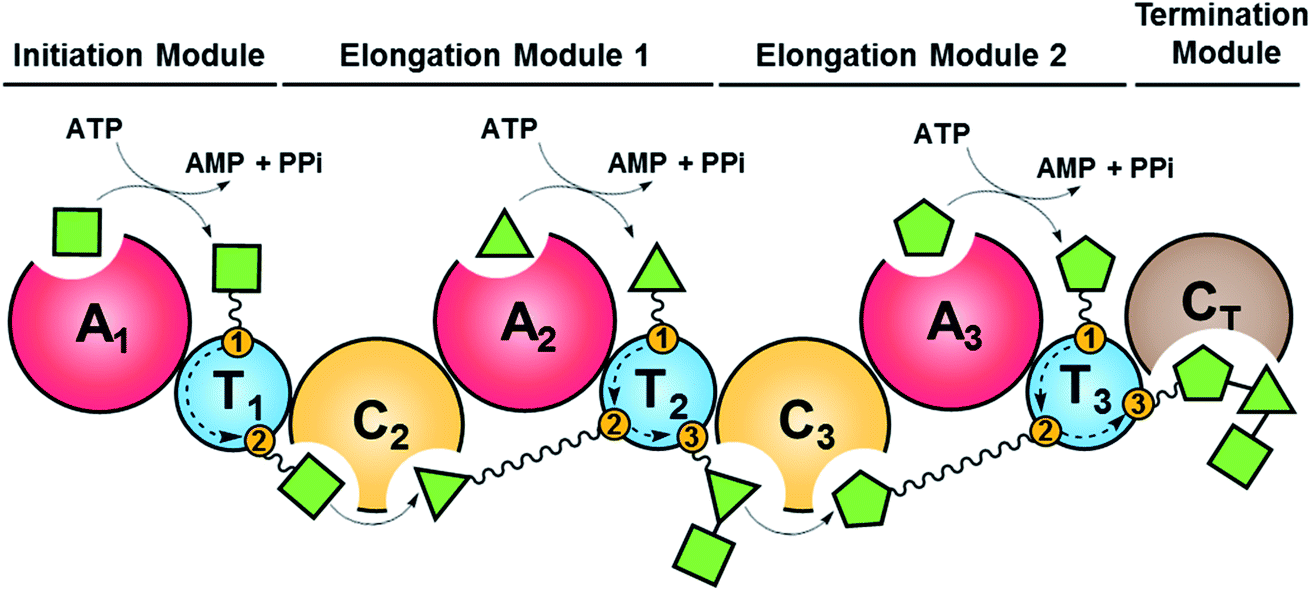 | ||
| Fig. 2 Schematic representation of fungal NRP biosynthesis catalyzed by modular NRPSs. The amino acid substrates are activated by the adenylation domain (A) through the formation of aminoacyl–AMP intermediates. These intermediates are captured by the thiol group of the flexible 4′-phosphopantetheine arm (wavy line) tethered to a thiolation domain (T, yellow circle 1). Condensation domains (C) catalyze successive peptide bond formation between the thioester intermediates loaded onto adjacent T domains (yellow circles 2 and 3 in T domains). Initiation modules often omit a C domain. Elongation modules are followed by a termination module featuring a terminal condensation domain (CT), or much less frequently a Dieckmann cyclase domain (D, also known as R, reductive release domain) or a thioesterase domain (TE) that catalyzes the release of the peptide from the NRPS by hydrolysis or regiospecific intramolecular cyclization. In contrast, the release of the mature peptide by hydrolysis or cyclization is typically catalyzed by a terminal TE domain in bacteria. NRPS modules may also contain additional domains that modify the loaded substrate or the bound intermediates through epimerization (E), N-methylation (N-MT), oxidation etc.15 The released peptide can subsequently be modified by tailoring enzymes, further increasing structural diversity. | ||
Fungal NRPSs assemble peptides in two modes. Linearly working NRPSs (type A) such as those that synthesize the marketed drugs cyclosporine and echinocandins use each of their constituent modules sequentially and only once during the biosynthetic cycle.16 In contrast, iterative NRPSs such as those that yield enniatin and beauvericin use some of their domains (type C) or complete modules (type B) in an iterative, recursive fashion following a biosynthetic metaprogram that is difficult to predict from sequence data alone.17
Chemical total synthesis of NRPs with their diverse structural modifications often faces significant challenges, including the poor availability of the constituent non-proteinogenic amino acids, inefficient regio- and stereospecific cyclization protocols, and low compatibility among different synthetic steps.18 Thus, the production of such compounds at a commercial scale typically necessitates in vivo biosynthesis (fermentation), while the generation of NRP-based uNPs with improved or novel bioactivities may require the reprogramming of the NRPS assembly line.
To date, a variety of reprogramming approaches have been demonstrated to produce NRP-based uNPs, mainly with bacterial NRPS assembly lines. These approaches include precursor-directed biosynthesis,19 mutasynthesis,20 site-directed mutagenesis of A domains,21 and combinatorial biosynthesis,22 the latter of which mainly includes module and domain exchanges. Similar approaches are gradually adapted to fungal NRPSs. However, two major obstacles remain to be addressed. First, our mechanistic understanding of NRPSs is still incomplete. This includes lingering questions about the intrinsic programming of substrate selectivity in the A and C domains; the extrinsic programming of domain–domain interactions through protein interfaces; and the determinants of the overall metaprogram of the whole NRPS assembly line. The second obstacle is technical, and stems from the lack of convenient and high-throughput techniques to genetically manipulate a large variety of filamentous fungi, or to transfer and manipulate the enormous fungal NRPS megaenzymes in heterologous hosts.
1.2 Scope of the review
In this highlight, we examine key examples for the re-engineering of fungal NRP assembly lines to generate uNPs. We list various attempts for the manipulation of the genes encoding the NRPSs and the tailoring enzymes, including mutasynthesis. We also emphasize the gaps in our knowledge and methodologies that must be bridged to achieve the desired engineering outcomes. We limit our discussions to works that generated unnatural NRPs including novel analogues. Approaches that lead to the increased production of natural NRP products, such as the heterologous expression or the activation of silent NRP gene clusters, have been reviewed elsewhere.23,24 This review covers the period of 2011 to the end of 2021.2. Methodologies that enable fungal NRP pathway engineering
2.1 Activation and heterologous expression
The rapid development of DNA sequencing technologies and bioinformatics tools has opened new possibilities to identify potentially novel chemical entities from genomic sequence information (reviewed elsewhere25–32). Sensitive chemical detection techniques33,34 have further sped up the dereplication process by decreasing the required sample amounts and increasing precision, and have been integrated into widely used systems such the NMR-based machine learning algorithm or the mass-spectrum-based Global Natural Products Social (GNPS) molecular networking tool.35–37Even with these advancements, far fewer compounds are routinely detected in fungal cultures than predicted from genome surveys, due to a lack of expression of the relevant BGCs under general laboratory conditions. Activation of these “cryptic” BGCs through the manipulation of global or local regulation cascades in the native hosts25,29,30 may remediate this situation, and lead to the production of novel natural products. Activation of multi-gene pathways in the native hosts has been greatly accelerated by the application of CRISPR/Cas9 gene editing techniques.31,38 Epigenetic remodeling of chromatin structure by the deletion/overexpression of global histone modifying proteins, both writers (which add modifications to histone tails) and erasers (which remove modifying groups), has been used extensively to activate cryptic BGCs in fungi. For example, deletion of the histone deacetylase (HDAC)-encoding gene hdaA increased expression in 42 of 68 BGCs in Calcarisporium arbuscular, revealing two novel cyclic peptides and two novel meroterpenoids;39 while deletion of the chromatin reader-encoding gene sntB in A. flavus allowed the production of depsipeptide aspergillicins.40 Similarly, deletion or overexpression of genes for other global regulators such as VeA and LaeA orthologues also affect the production of NPs.31 More examples of new NP discovery by manipulation of regulatory elements were summarized in previous reviews.10,23,25,29,41,42
Another frequently used strategy is the expression of the BGC in a heterologous chassis.43 Considering the lack of molecular tools for most native producers and the increasing availability of metagenomic sequences for pathway discovery, heterologous expression of cryptic BGCs is becoming an increasingly prominent strategy, as discussed in previous reviews.27,44–47 In addition to Saccharomyces cerevisiae,43,48–51 other chassis such as Aspergilli,45,52,53Fusarium54 and Penicillium55 spp. have also been developed. These filamentous fungi may correctly recognize and splice introns, bypassing the need for predicting and assembling large intron-free genes as is necessary when yeast is used as the host.44,47 Filamentous fungi may also offer a larger variety of precursors available for NP biosynthesis. However, host enzymes may modify or degrade heterologous products, and the detection and purification of the engineered compounds may be more complicated in the more complex metabolomic background of filamentous fungi. Both the increasingly sophisticated manipulation of native producers, and the more facile transfer of BGCs to heterologous chassis are critical for engineering the production of nonribosomal peptide uNPs by synthetic biology.
2.2 Precursor-directed biosynthesis and mutasynthesis
Much of the early attempts to biosynthetically generate novel NRP analogues focused on precursor-directed biosynthesis (PDB) or mutasynthesis. In both cases, different building blocks are fed to an NRP-producing organism, a wild-type strain as in PDB or an engineered strain as in mutasynthesis. If the substrate specificity of the NRPS is broad enough, the modified precursors can be incorporated into the final peptide product. With PDB, the competition of new precursors with the native building blocks leads to a mixture of wild-type and modified products. This approach requires only a limited understanding of the biosynthetic machinery, but may provide information of the biosynthetic mechanisms. In contrast, mutasynthesis supplies alternative precursors to a strain where the biosynthesis of the native precursor had been disrupted. As opposed to PDB, mutasynthesis produces only uNPs, exemplified by the biosynthesis of cyclooligomer depsipeptide analogues.56 However, mutasynthesis may substitute only those NRP building blocks that are not essential for the primary metabolism of the producer organism.2.3 Combinatorial biosynthesis and enzyme engineering
Combinatorial biosynthesis generates novel NP assembly lines by mixing genes, partially or as a whole, from different biosynthetic pathways.31 The increasing availability of information on the structure and catalytic mechanisms of bacterial NRPSs has also empowered the synthesis of uNPs through engineering the substrate specificity of these enzymes, primarily by altering the A domain “specificity code”57,58 or by exchanging whole A domains.59–63 However, A domain engineering of fungal NRPSs has been much less productive. While successful construction of bacterial NRP assembly lines by deleting or inserting entire NRPS modules have been described, analogous approaches are reported more rarely for fungal systems.64 Up till recently, the various algorithms for the prediction of the A domain “specificity code” have been trained on datasets derived mainly from bacterial NRPSs.27 Encouragingly, the latest versions of the gene cluster predictor, antiSMASH, incorporated improved algorithms and training sets for A domain substrate prediction from both bacteria and fungi. This may eventually promote the engineering of fungal NRPs.65Along with the substrate selectivity of the A domains, the specificity of the C domains was also found to play a critical role in the determination of the sequence of the NRPs. In addition to peptide bond formation, specific C domains perform β-lactam formation, dehydration, hydrolysis, cycloaddition, Pictet–Spengler cyclization, Dieckmann condensation and recruitment of auxiliary enzymes.66,67 Considering that C domains clade into various subtypes during phylogenetic analysis and that these groups may be recognized by specific conserved motifs, prediction of C domain catalytic functions from genome sequence information alone is increasingly feasible.68 Future structural, biochemical, and bioinformatics studies of fungal NRPSs will undoubtedly facilitate the use of combinatorial biosynthesis to generate novel NRPS-based uNPs in a more predictable and reliable fashion.13
2.4 In vitro biosynthesis and chemoenzymatic synthesis
In vitro reconstitution and engineering of NRPS biosynthetic pathways is a promising strategy to synthesize complex molecules, as it may combine the distinct advantages of enzymes (e.g., high selectivity and efficiency, mild reaction conditions) with the rich diversity of compounds from organic synthesis.69,70 Early examples of chemoenzymatic synthesis used solid-phase peptide synthesis (SPPS) to prepare linear peptide precursors activated as N-acetylcysteamine (SNAC) thioesters, and cyclized these peptides with high stereo- and regioselectivity using dissected TE domains from bacterial NRPS assembly lines. This strategy was applied to several synthetically challenging but clinically important bacterial NRPs.71,72 To synthesize a library of uNP cyclic peptides, the substrate flexibility of the TE domain of the tyrocidine NRPS was exploited, affording over 300 linear tyrocidine analogues.73 Another chemoenzymatic strategy used a ketoreductase (KR) domain-containing NRPS module to catalyze ester bond formation between chemically synthesized SNAC-activated precursors and 2-ketoisocaproic acid, generating cryptophycin analogs.74 The recent demonstration of NRP production in cell-free protein synthesis systems holds out the prospect of significantly accelerating uNP scaffold generation, although the relatively high cost of this method may limit its application for large scale production.75,76In fungi, the power of chemoenzymatic synthesis was mainly utilized to identify biosynthetic pathways or to characterize the biocatalytic functions and mechanisms of NRPS domains.14,77–79 In a remarkable example, the full-length polyketide synthase–nonribosomal peptide synthetase (PKS–NRPS) hybrid enzyme ApdA (aspyridone synthetase, 439 kDa) was purified from an S. cerevisiae chassis to examine the programming rules of both the PKS and NRPS modules.49 Often, such studies with reconstituted enzymes also generate shunt products, some of which are new to nature.14 In some studies, generation of uNPs was explicitly attempted, such as when a dissected NRPS module from a PKS–NRPS of Aspergillus terreus was reconstituted in vitro and was provided with different amino acids and free thiols to produce >60 different thiopyrazine compounds,80 as discussed in Section 3.3.
3. Engineering fungal NRP pathways
3.1 Cyclooligomer depsipeptide synthetases
Depsipeptides feature both amino acid and hydroxycarboxylic acid residues.5 In bacteria, the hydroxycarboxylic acid residues usually derive from α-ketocarboxylic acids that are stereoselectively reduced by dedicated ketoreductase (KR) domains integrated into the appropriate module of the NRPS. The KR domain and its N-terminal neighbor, the intact A domain that adenylates the α-keto acid substrate are bracketed by a split “pseudo Asub” domain that plays an important structural role.4 In contrast, fungal NRPSs directly select and incorporate preformed α-hydroxycarboxylic acids.81 Among depsipeptides, cyclooligomer NRPs are characterized by sequential repeats of dipeptidol motifs in an overall cyclic configuration. Members of this group display a wide variety of biological activities; some are marketed drugs. Their structures, biosynthesis and bioactivities were thoroughly reviewed.56 Representative cyclic hexadepsipeptides are the beauvericins (with antitumor, antibiotic, antifungal, insecticidal, and anthelmintic activities) and the enniatins (displaying insecticidal, phytotoxic, anthelmintic, antibiotic, antifungal, and cytostatic activities). Cyclooligomeric octadepsipeptides include the PF1022 compounds (with prominent anthelmintic activity) and bassianolide (with insecticidal and acetylcholine-induced smooth muscle contraction-inhibiting activities; Fig. 3). These compounds display an alternating sequence of various D-hydroxycarboxylic acids (inserted by module 1) and N-methyl-L-amino acids (incorporated and N-methylated by module 2; Fig. 3). Currently, these cyclooligomer depsipeptide synthetases are the most studied examples for the engineering of fungal NRP assembly lines to produce uNPs: precursor-directed biosynthesis, mutasynthesis and chimeric NRPSs have all been extensively tested with enzymes from this group. Manipulation of the cyclooligomer depsipeptide NRPSs also contributed to the exchange unit (XU) concept for the generation of hybrid assembly lines from noncognate modules, and this concept is now finding broader applications in the creation of chimeric synthetases from other NRP structural classes.82–84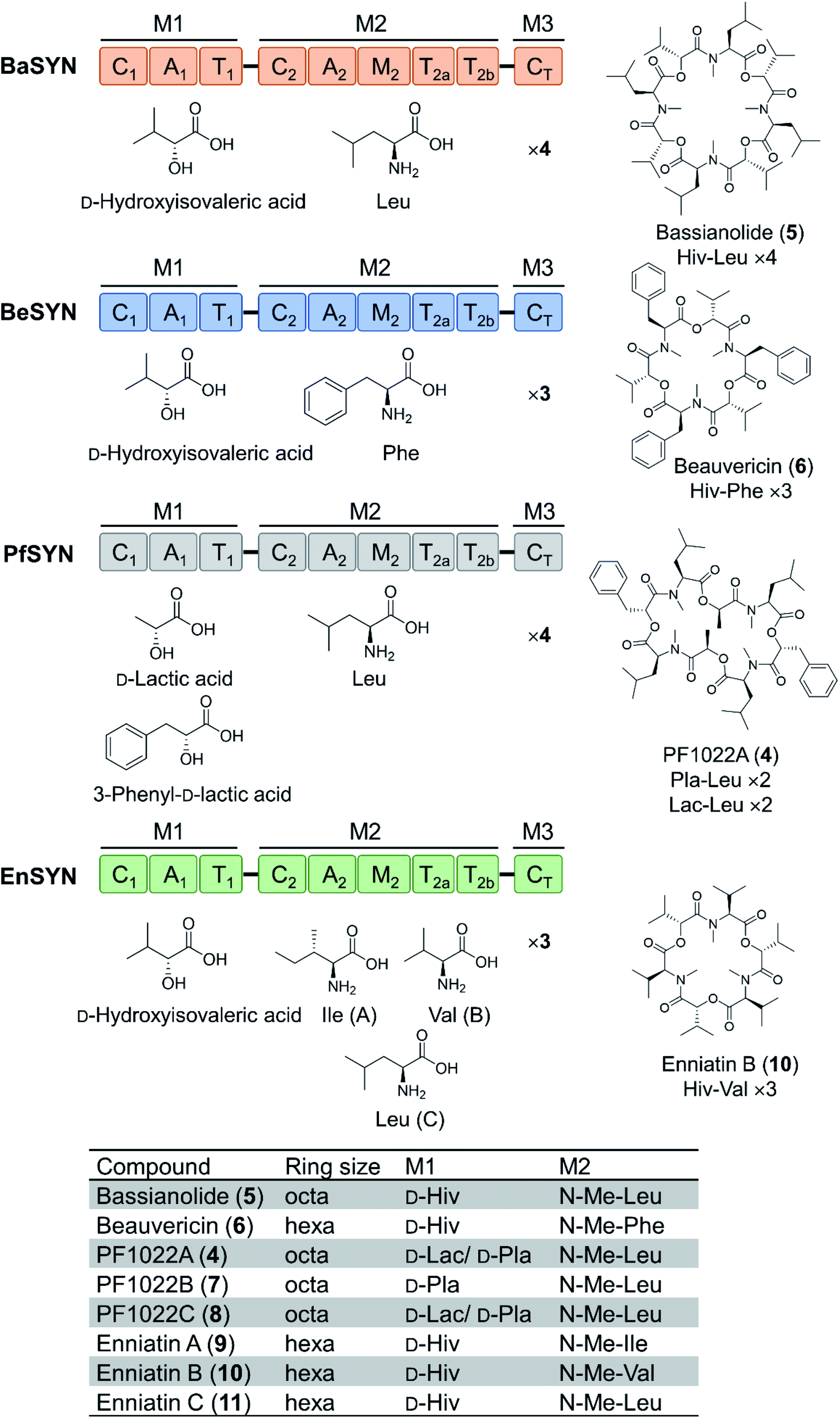 | ||
| Fig. 3 Domain and module architectures, substrate selectivities and product structures of representative cyclooligomer depsipeptide NRPSs.56 The table summarizes the structural features of naturally occurring fungal cyclooligomer depsipeptides. Hiv, D-hydroxyisovaleric acid; Pla, 3-phenyl-D-lactic acid; Lac, D-lactic acid. | ||
Early attempts to engineer the production of uNP cyclooligomer depsipeptides utilized precursor-directed biosynthesis85–89 and mutasynthesis20,90,91 as detailed in a previous review.56 Thus, supplying amino acid analogues to growing cultures or purified NRPS enzymes led to the gradual replacement of the aminoacyl constituents of the peptides by other (N-methyl)-L-amino acids.86 In a pioneering attempt at precursor supply engineering combined with mutasynthesis, three genes for p-aminophenylpyruvate biosynthesis from Streptomyces venezuelae were incorporated into a chorismate mutase-deficient strain of Rosellinia sp. Using its native phenyllactate dehydrogenase enzyme, the fungus produced p-aminophenyllactate, and its oxidation product p-nitrophenyllactate. These two in situ-produced alternative building blocks were then incorporated into PF1022 analogues to replace the native D-phenyllactate constituents (Fig. 4).90
 | ||
| Fig. 4 PF1022 analogues where D-phenyllactate was substituted with in situ produced hydroxycarboxylic acids.90 | ||
The naturally occurring PF1022 and enniatin NRPSs were challenged with a set of >30 aliphatic or aromatic α-D-hydroxycarboxylic acids to generate new analogues.87,88 These experiments revealed a surprising promiscuity for the PF1022 synthetase, with some noncognate precursor analogues turning out to be even better substrates than the native ones (Fig. 5). In contrast, the enniatin synthetase was found to be more selective, as it did not accept aromatic α-D-hydroxycarboxylic acids.
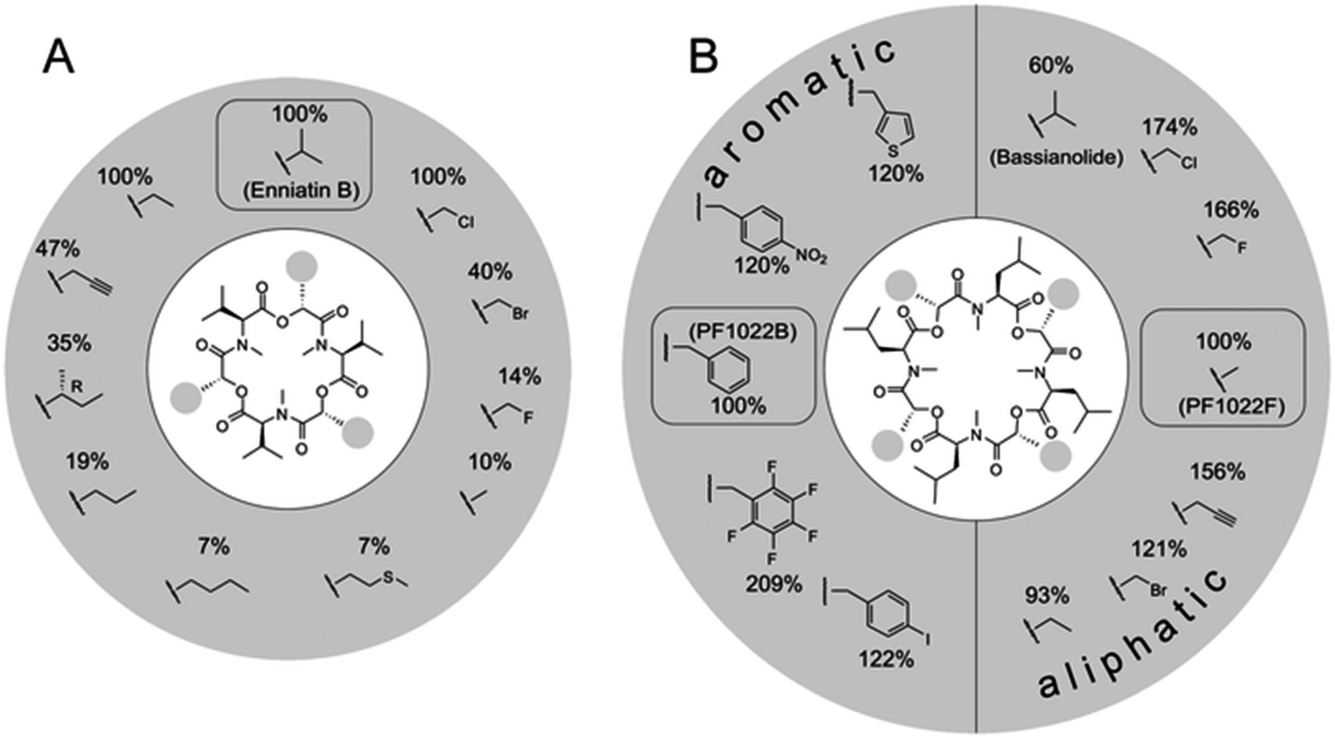 | ||
| Fig. 5 Substrate tolerance of the PF1022 and the enniatin synthetases towards α-D-hydroxycarboxylic acids. Incorporation of different synthetic 2-hydroxycarboxylic acids into: (A) enniatin; and (B) PF1022. The percentages describe the kcat,app in comparison to the natural substrate (enniatin: D-hydroxyisovaleric acid; PF1022: D-phenyllactic acid for the aromatic, and D-lactic acid for the aliphatic precursors, respectively). Figure reprinted with permission from Süssmuth et al.56 | ||
In vivo precursor-directed biosynthesis was also used to produce analogues of beauvericin that were evaluated for their cytotoxicity and directional cell migration (haptotaxis) inhibitory activity.89 Knockout of the novel gene encoding ketoisovalerate reductase (KIVR) in the producer fungus eliminated the production of the natural precursor D-hydroxyisovaleric acid, and allowed the mutasynthesis of 15 unnatural beauvericin congeners from noncognate hydroxycarboxylic acids fed to the culture. Some of these uNP beauvericin congeners displayed increased antiproliferative activity.91 Finally, 11 new beauvericin analogues were produced by in vitro chemoenzymatic and in vivo whole cell biocatalytic syntheses using either a B. bassiana ΔkivR mutant or an E. coli strain expressing the bbBeas beauvericin synthetase gene.20
Further expansion of the cyclooligomer depsipeptide structure space required advances in fungal NRPS engineering. First, whole modules were exchanged among the highly homologous cyclooligomer depsipeptide synthetases.92,93 Module swapping between the beauvericin and bassianolide synthetases showed that product formation requires the maintenance of the N-terminal linker region of the C2 domain of the second module (Fig. 6).92 Chimeric enzymes constructed from the hydroxycarboxylic acid-activating module of the PF1022 synthetase and the aminoacid-activating modules of the enniatin and beauvericin synthetases were also expressed in E. coli and Aspergillus niger, and afforded new cyclodepsipeptides (Fig. 6).93 These experiments also showed that the assembly lines could be recombined using different switchover positions, thus paving the way for the combinatorial biosynthesis of fungal NRPs.92–94
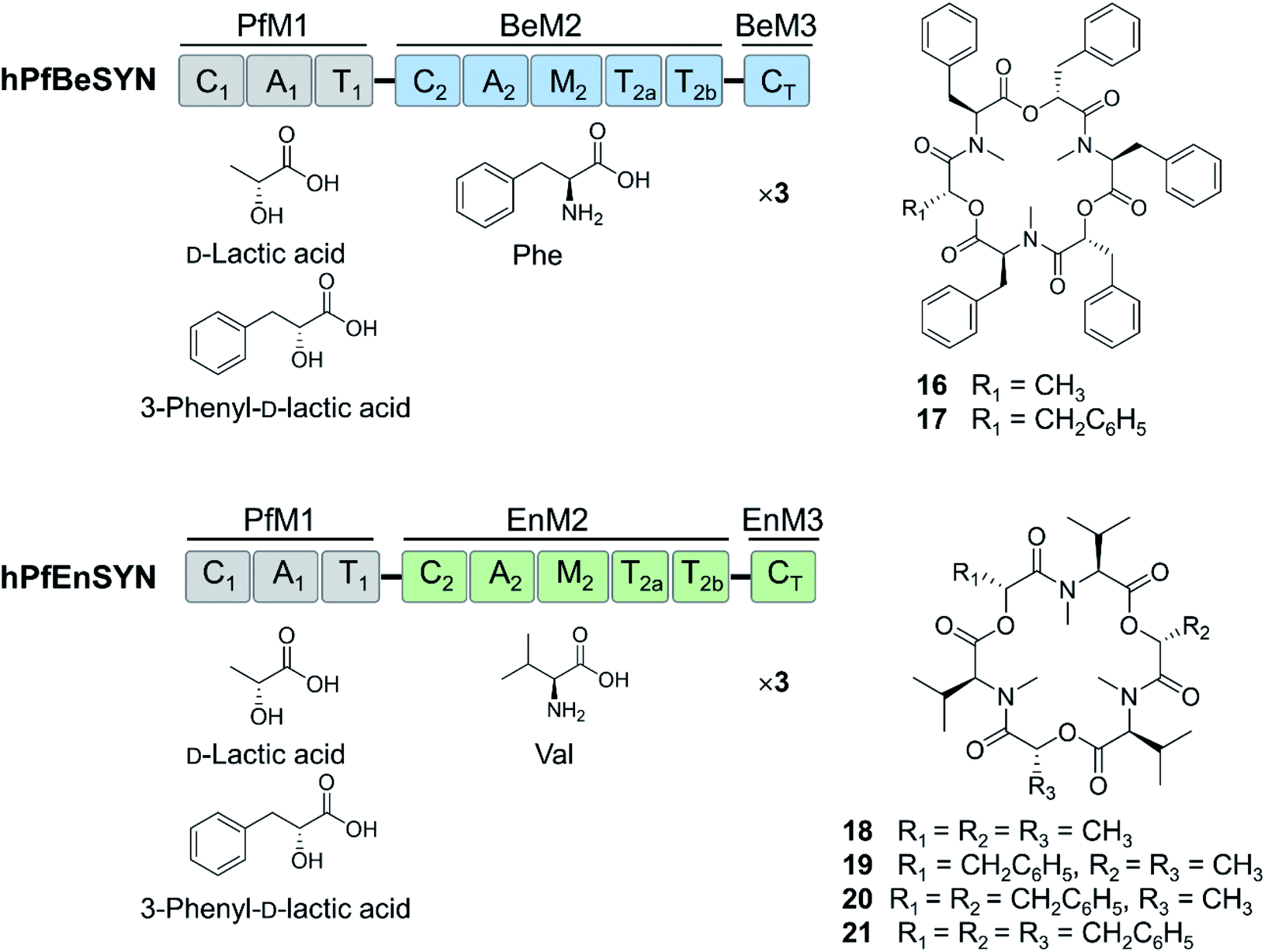 | ||
| Fig. 6 Hybrid cyclooligomer depsipeptide NRPSs generated by module swapping.93 Fusion of the first module of the octadepsipeptide-synthesizing PF1022 synthetase (PfM1) with the second and the third (termination) modules of the beauvericin (BeM2 and BeM3) or the enniatin (EnM2 and EnM3) synthetases generated chimeric enzymes that produced uNP hexadepsipeptides. | ||
Nevertheless, these combinatorial engineering attempts were limited to the four known, highly homologous cyclodepsipeptide synthetases, which consequently restricted the structural diversity of the uNPs. For further structural diversification, the programming of these iterative fungal NRPSs had to be deciphered. Two groundbreaking studies in 2017 revealed that fungal cyclooligomer depsipeptide NRPSs adopt a radically different strategy compared to their bacterial counterparts.78,94 Bacterial cyclooligomer depsipeptide synthetases had previously been described to follow a parallel (recursive) logic (Fig. 7A), whereby dipeptidol monomers are assembled first from the precursors, and these monomers are then oligomerized by the C-terminal thioesterase (TE) domain that controls the ultimate chain length (degree of oligomerization).72,95 Unexpectedly, assembly of cyclooligomer depsipeptides in fungi follows a linear (looping) model (Fig. 7B), involving the stepwise incorporation of the precursors. Surprisingly, the CT and C2 domains of the fungal synthetases take turns to incorporate the two biosynthetic precursors into the growing depsipeptide chain that shuttles between the T1 and T2a/T2b domains. When the peptide chain reaches its preordained length, the CT domain releases the product by cyclization. This proposed dual function sets the terminal CT domains of cyclooligomer depsipeptide synthetases apart from “canonical” CT domains in fungal linear assembly lines that perform only the termination step.14
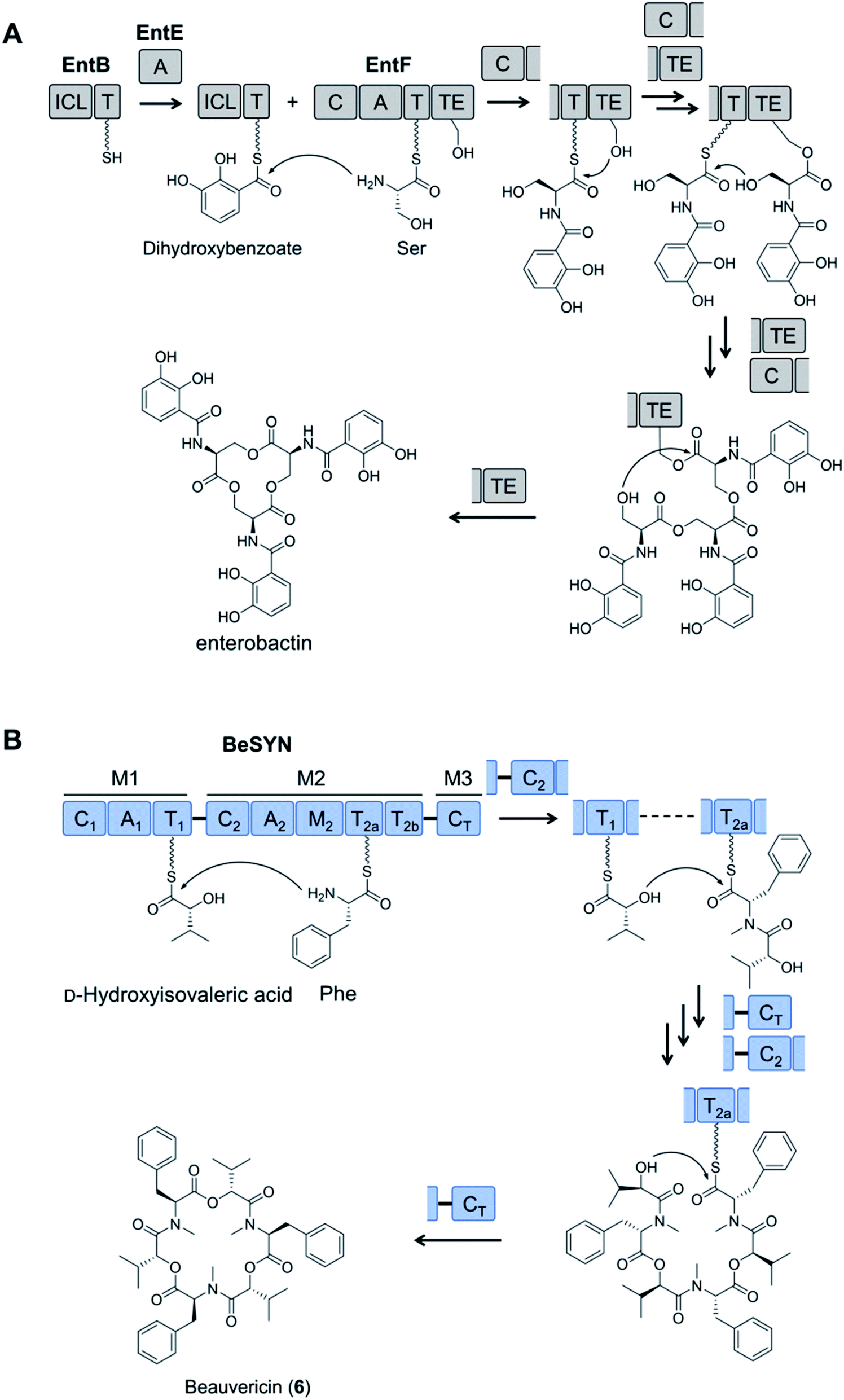 | ||
| Fig. 7 Comparison of the assembly mechanisms of cyclooligomer depsipeptide synthetases. (A) The parallel (recursive) logic seen in bacterial enzymes;78,94 and (B) the linear (looping) assembly mechanism of fungal cyclooligomer depsipeptide synthetases.72,95 | ||
Swapping parts of the CT domain also uncovered functional aspects of macrocyclization and ring size control.94 Thus, elongation and cyclization are competitive processes in CT domains, with macrocyclization being performed when the tail end of the linear depsipeptidyl chain is positioned in such a way that the hydroxy group is able to approach the thioester and the catalytic histidine of the CT domain. According to this model, product ring size is primarily determined by the size of the cyclization pocket, collectively formed by the CT-NTD (N-terminal subdomain) and the CT-CTD (C-terminal subdomain). Exploiting this “gauge” role of the CT domain, uNP cyclodepsipeptides with altered numbers of monomers were produced by swapping CT domains that favor different chain lengths (Fig. 8). The new tetrameric product FX1 (octa-beauvericin) was generated by substituting the CT of the beauvericin synthetase with the bassianolide CT (in the form of T2a–T2b–CT, T2b–CT or CT alone), and expressing the chimera in yeast. Utilization of A. niger as a robust heterologous host supported the production of hexa-bassianolide at a very high titer of 1.3 g L−1, and allowed the biosynthesis of new-to-nature octa-enniatin B (4 mg L−1) and octa-beauvericin (10.8 mg L−1) with chimeric synthetases (Fig. 8).94 While the selected CT domains firmly controlled the chain lengths of the products, these domains showed considerable promiscuity towards aliphatic as well as aromatic amino acid side chains (N-Me-L-Val/Leu/Ile/Phe). Importantly, the two hybrid cyclooctadepsipeptides showed up to 12-fold higher activity against the parasites Leishmania donovani and Trypanosoma cruzi compared to the reference drugs miltefosine and benznidazole, respectively. In addition, desmethyl congeners were also produced by deleting the M domains, demonstrating that the absence of structure-modulating N-methylations was tolerated by the assembly lines. From the twin T domains, the T2a domain alone was sufficient for complete precursor processing, though product yields were lower without the T2b domain partner.78,94 These studies provided highly important insights into the programming of the C2, the CT, the twin T2 and the M domains of cyclooligomer depsipeptide synthetases that may be exploited to design and generate novel uNPs. However, future work should define the key amino acid residues that influence chain length control in the CT domains, and those that modulate precursor permissiveness in the C2 and the CT domains.
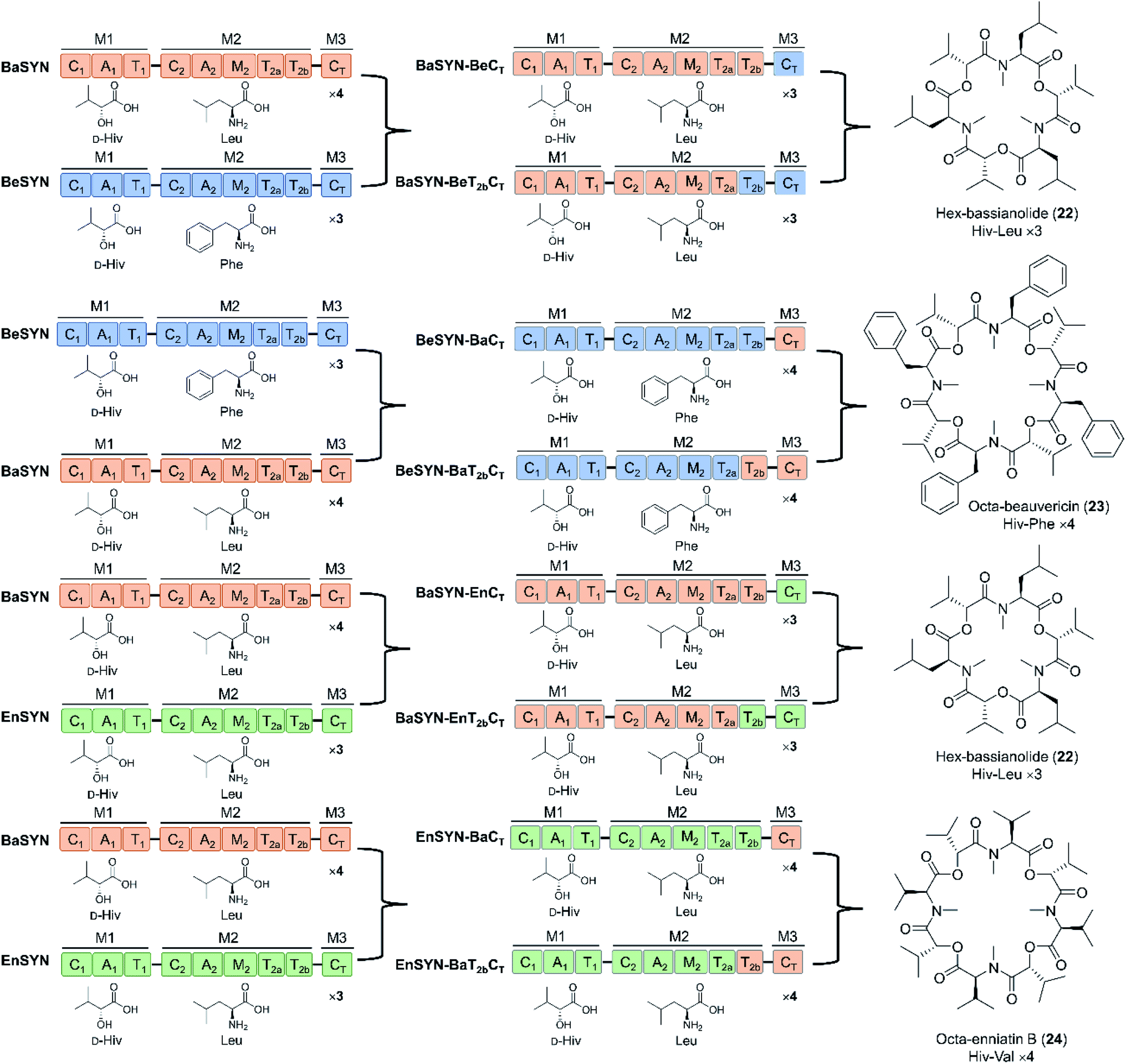 | ||
| Fig. 8 New trimeric and tetrameric uNP congeners of cyclooligomer depsipeptides generated by swapping the CT domains with or without the T2 domains.78,94 | ||
In another interesting attempt, CT domains were reassigned to fill the role of a canonical condensation domain at the junction of two consecutive modules. Thus, the beauvericin, bassianolide, and enniatin synthetases were fused head-to-tail in different combinations, with or without the T2b domain, but with the CT domain replacing the nonfunctional C1 domain of the downstream synthetase (Fig. 9).66 The C1 starter domain at the N-terminal end of the fused NRPS was retained, considering that deletion of such C1 domains was shown to have a deleterious effect on the product yield.78 Remarkably, the CT turned out to be at least partially competent to act in a linear assembly mode, catalyzing the condensation of the peptidyl intermediates to the precursor activated by the downstream A domain. This then led to the production of novel hybrid cyclooligomer depsipeptides with asymmetric structures instead of symmetric oligomers with tandem repeats of identical dipeptidols. The fusion of all three synthetases led to a cocktail of “rainbow” cyclooligomer depsipeptides (Fig. 9).66 Removal of T2b was shown to promote the linear mode of precursor incorporation: apparently, the presence of both T2 domains somehow facilitates loop-back elongation.92,94
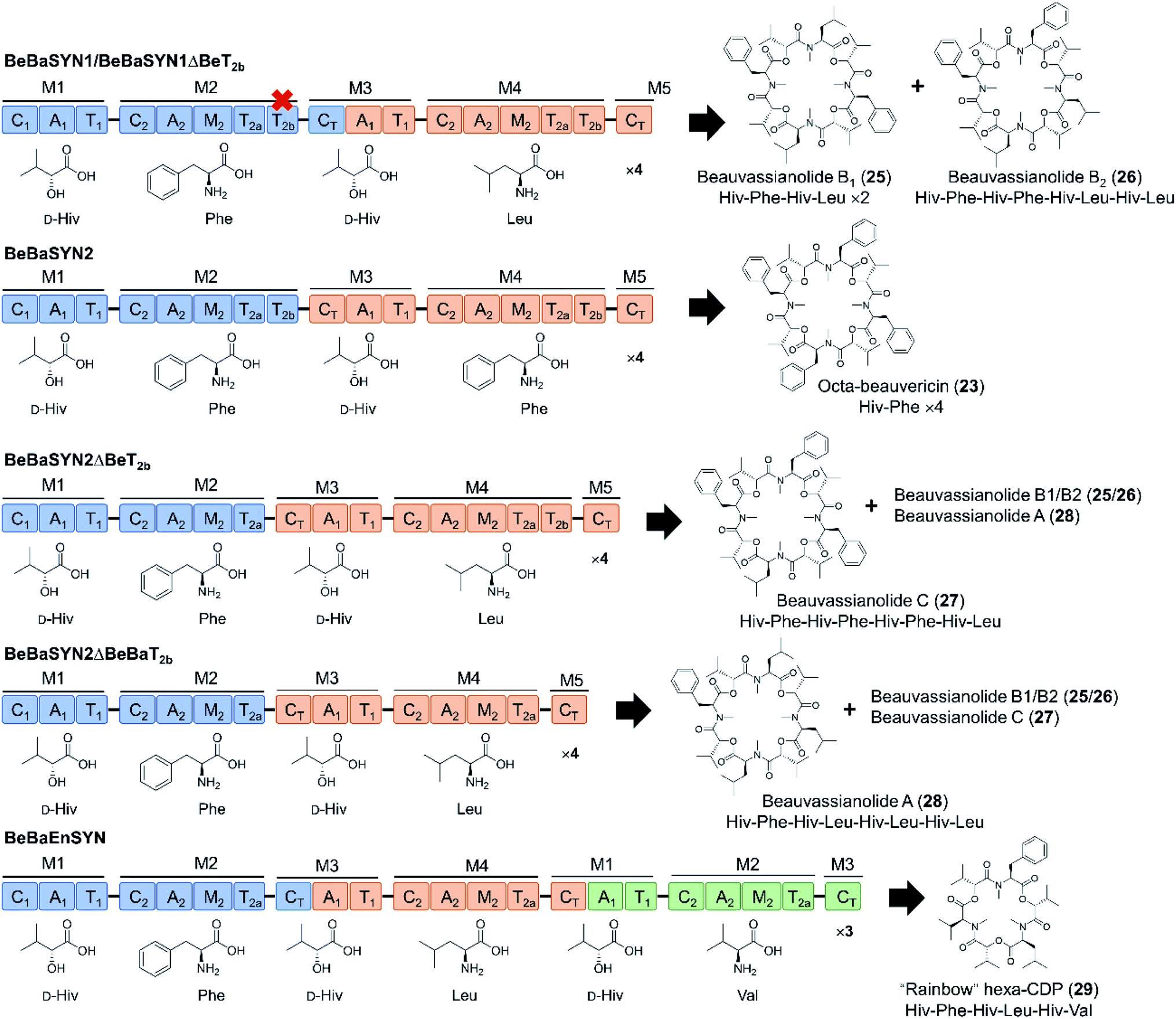 | ||
| Fig. 9 “Rainbow” cyclooligomer depsipeptide analogues generated in a mixed linear/iterative assembly mode by fusing two or three iterative NRPS.66 The assembly line was constructed with or without the T2b domains, while the C1 domains of the downstream synthetases were replaced by CT domains. Colors are as described for Fig. 3. | ||
Modules from NRPSs of both linear and iterative assembly modes were also successfully combined. Different swapping sites were found to be productive, while respecting the integrity and specificity of the C domains was necessary.82 Thus, modules 2 (processing N-methyl-L-leucine), 4 (processing N-methyl-L-valine), 7 (processing N-methyl-L-glycine) and 10 (processing N-methyl-L-leucine) of the cyclosporine synthetase (CySYN; linear assembly mode) from Tolypocladium inflatum82 could be integrated into cyclooligomer depsipeptide synthetases (iterative assembly mode). However, only those CySYN modules were accepted that displayed an identical substrate specificity with the native assembly lines. Thus, modules 2, 4 and 10 were compatible with the enniatin synthetase, while only modules 2 and 10 were functional as part of the bassianolide synthetase. These chimeric NRPSs showed higher product specificity compared to the native enzymes, but no new cyclooligomer depsipeptide was obtained. Nevertheless, with the growing number of characterized linear fungal NRPS systems and our expanding knowledge of building block recognition and processing, this approach should eventually result in the production of new-to-nature NRPs.
More generally, these experiments also helped to develop a novel strategy (the “two-face exchange system”) to fuse different NRPSs (Fig. 10).82 This strategy combines various exchange units (XU) in a way to maximize their fit with the substrate requirements of the C domains at both sides of the fusion. Thus, an appropriate A–T, C–A–T, C–A–T–C, or A–T–C domain assembly (plus any additional processing domains within these modules) is selected as an XU for a given C domain, based on the following generalized rules: (1) C domains should be presented by the upstream XU with a peptide chain that ends in a building block that is identical (or at least sterically similar) to the native donor-site substrate of the C domain; (2) the precursor offered by the downstream XU must strictly meet the C domain acceptor-site specificity requirements;84 (3) the crossover sites should be positioned right at the C-terminal end of the T domain for new T–C fusions,92,94 and at the C-terminus of the C domain66 or after the α-helical linker element84 for C–A fusions.
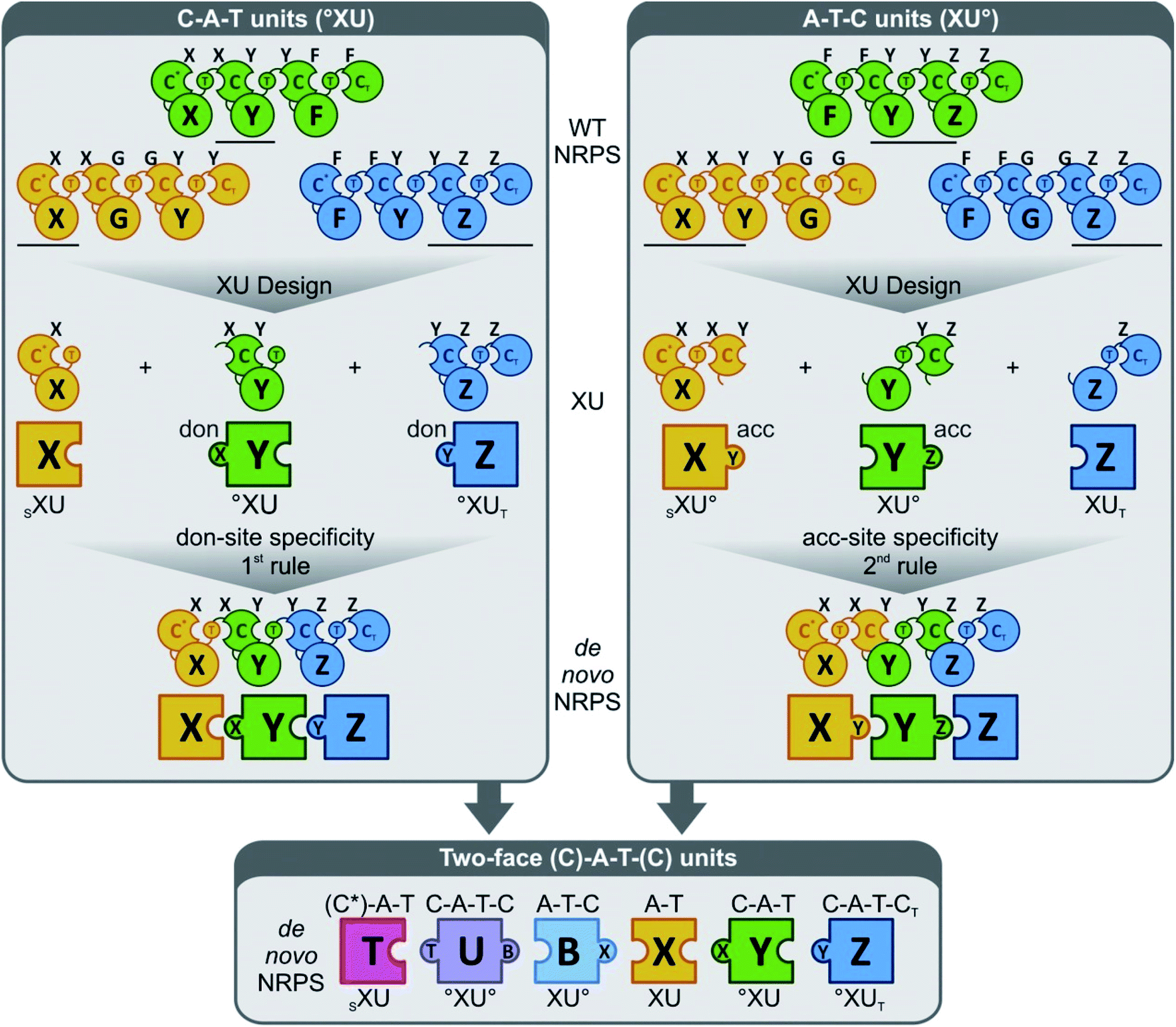 | ||
| Fig. 10 The “two-face” exchange concept for fungal NRPS engineering (reprinted with permission from Steiniger et al.82). The donor (don) and acceptor (acc) site specificities are indicated as a capital letter on top of each C domain. Combining exchange units (XU) of the C–A–T (upper left)22,96 and the A–T–C type84 (upper right) to the so-called two-face system (bottom)82 broadens combinatorial possibilities. °XU, selectivity requirement that must be met by the adjacent XU (upstream or downstream); SXU, starter exchange unit; XUT, termination exchange unit. | ||
3.2 NRPS for cyclic peptides
Cyclic peptides are one of the most abundant types of fungal NRPs as evidenced by genome sequencing-based secondary metabolome inventories. The predicted biosynthetic gene clusters for these metabolites have been extensively engineered in the native producer organisms to activate silent gene clusters, or to elucidate NRPS mechanisms. Precursor-directed biosynthesis was accomplished as early as in 1989 when three novel cyclosporine analogues, incorporating allylglycine, β-cyclohexylalanine or D-serine, respectively, were generated by feeding nonproteinogenic amino acids to cultures of Tolypocladium inflatum. Notably, cyclosporine A (3), with D-serine replacing the natural D-alanine at position 8, exhibited potent biological activity.97 However, the fungal cyclopeptide NRPSs themselves have not been engineered for uNP production, perhaps due to their large size. In contrast, manipulation of the tailoring enzymes turned out to be successful, as exemplified by the engineered production of the echinocandin/pneumocandin-type lipohexapeptides. These fungal NRPs inhibit the fungal 1,3-β-glucan synthase, and are used as first-line treatments against invasive mycosis as the active ingredients of the semisynthetic drugs caspofungin, micafungin, and anidulafungin. The structure complexity of these lipohexapeptides is due, at least in part, to several hydroxylated non-proteinogenic amino acids. While the majority of the functional groups in the side chains are not essential for bioactivity,6 they do affect potency, solubility, and other pharmacological properties.98 Previous reviews described the history of echinocandin research, and the diverse structural elements, biosynthetic background, and structure–activity relationships of these peptides.7,99–101 After early attempts by mutagenesis and medium manipulation,102–104 the identification of biosynthetic gene clusters for these NRPs from several fungi made it possible to engineer lipohexapeptide biosynthesis in a more targeted manner,105–107 substituting the native lipid side chain with similar ones, or editing the hydroxylation level of some nonproteinogenic amino acid constituents by targeting tailoring genes. Thus, the biosynthesis of the fatty acyl side chain and the lipo-initiation step of pneumocandin assembly was elucidated to be conducted by the highly reducing polyketide synthase GLPKS4, the type II thioesterase GLHYD, and the acyl-AMP ligase GLligase. Mutasynthesis following the disruption of the GLPKS4-encoding gene afforded pneumocandin analogues with altered side chains, such as acrophiarin and four new pneumocandin congeners with C14, C15, and C16 fatty acyl side chains (Fig. 11).108 A comprehensive biological evaluation showed that one compound, pneumocandin I, has elevated antifungal activity and similar hemolytic activity compared to pneumocandin B0, the precursor for the semisynthetic caspofungin. Future protein engineering of GLligase, the gatekeeper for the lipid starter unit, may allow the further diversification of the pneumocandin family of compounds.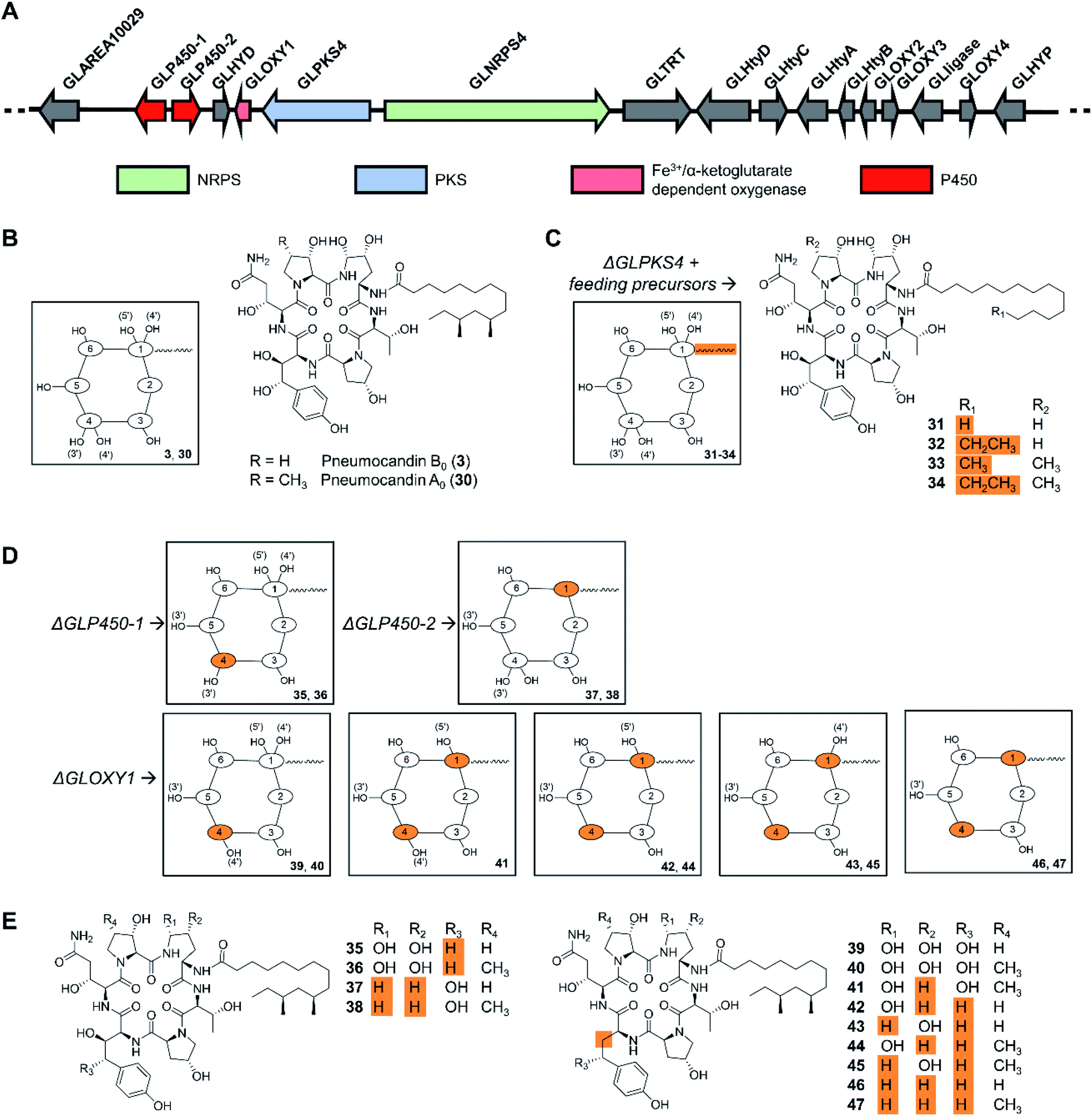 | ||
| Fig. 11 Inactivation of biosynthetic genes results in uNP pneumocandin analogues.98,108 (A) Genetic organization of the pneumocandin biosynthetic gene cluster. The core NRPS gene and the inactivated genes are color-coded as indicated. (B) Structures of pneumocandins B0 (3) and A0 (30). (C) Structures of mutasynthetic pneumocandin analogues resulting from the inactivation of the PKS-encoding gene GLPKS4 and feeding of various fatty acid precursors. (D) Schematic illustration of the structures of uNPs resulting from the inactivation of tailoring enzyme-encoding genes. (E) Structures of pneumocandin analogues. | ||
Insertional inactivation of two P450-type hemeprotein monooxygenase-encoding genes (GLP450-1 and GLP450-2) and one non-heme mononuclear iron oxygenase gene (GLOXY1) in Glarea lozoyensis generated 13 different pneumocandin analogues that lack one, two, three, or four hydroxyl groups from the 4R,5R-dihydroxy-ornithine and 3S,4S-dihydroxy-homotyrosine moieties of the parent hexapeptide (Fig. 11).98 Among them, pneumocandins F and G were more potent in vitro against Candida species and Aspergillus fumigatus than the principal native fermentation products, pneumocandins A0 and B0.
In a similar manner, deletion of the ecdH gene encoding a cytochrome P450 monooxygenase in Emericella rugulosa generated echinocandin analogues lacking both hydroxyl groups of the 4R,5R-dihydroxy-Orn1 moiety present in the parental molecule (Fig. 12).109 The deletion of the nonheme mononuclear iron oxygenase-encoding gene ecdG led to the production of echinocandin analogues with a non-hydroxylated homotyrosine residue. In vitro evaluation of the deshydroxy-echinocandin scaffolds in an anticandidal assay revealed up to a threefold loss of potency for the products from the ΔecdG strain. In contrast, a threefold gain of potency was seen for the ΔecdH-derived compounds, in line with prior results on deoxyechinocandin homologues.
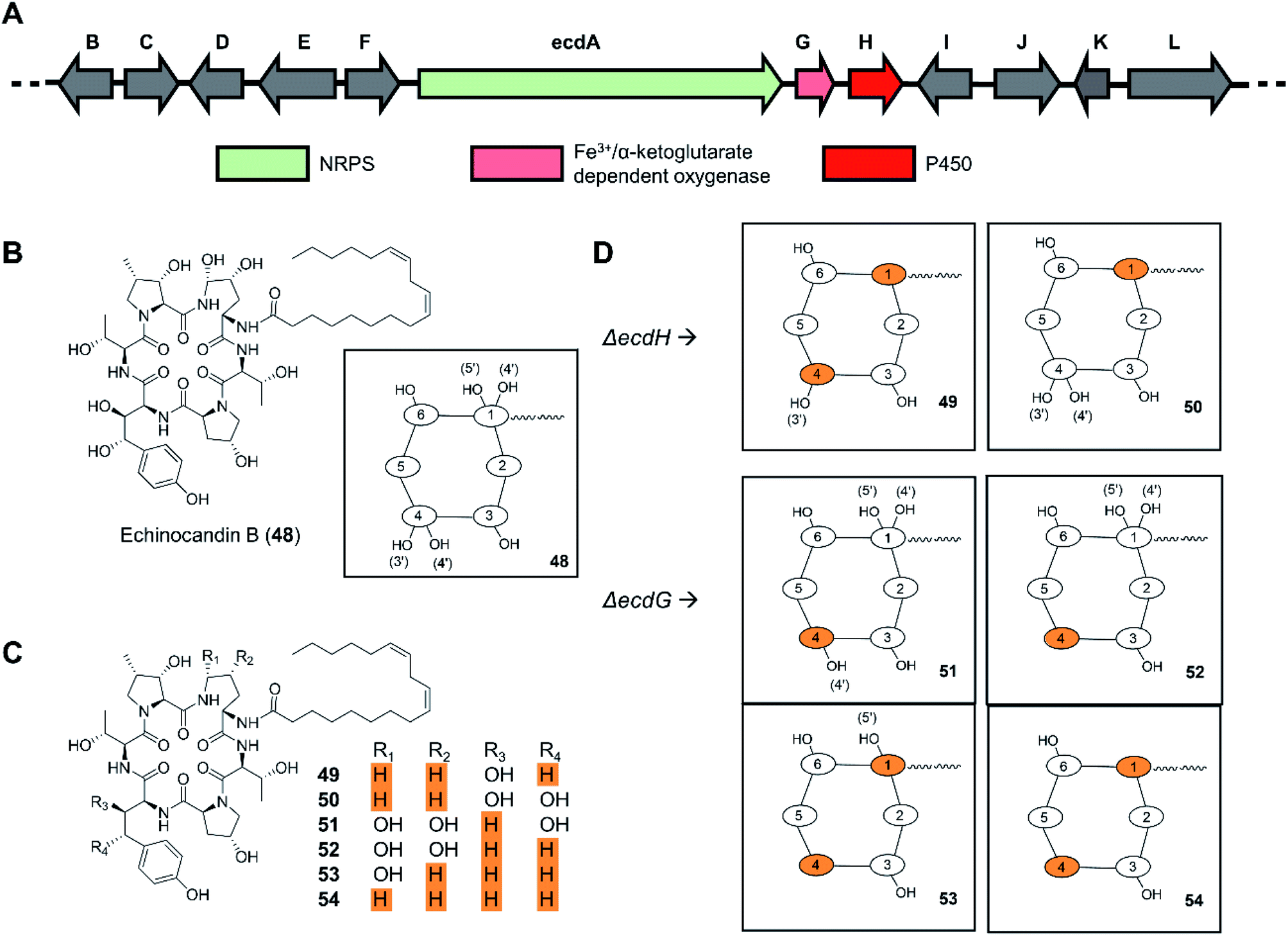 | ||
| Fig. 12 Inactivation of tailoring enzyme-encoding genes results in new uNP echinocandin analogues.109 (A) Genetic organization of the echinocandin B biosynthetic gene cluster. The core NRPS gene and the inactivated genes are color-coded as indicated. (B) Structures of echinocandin B (48). (C) Structures of echinocandin analogues. (D) Schematic illustration of the structures of echinocandin analogues. | ||
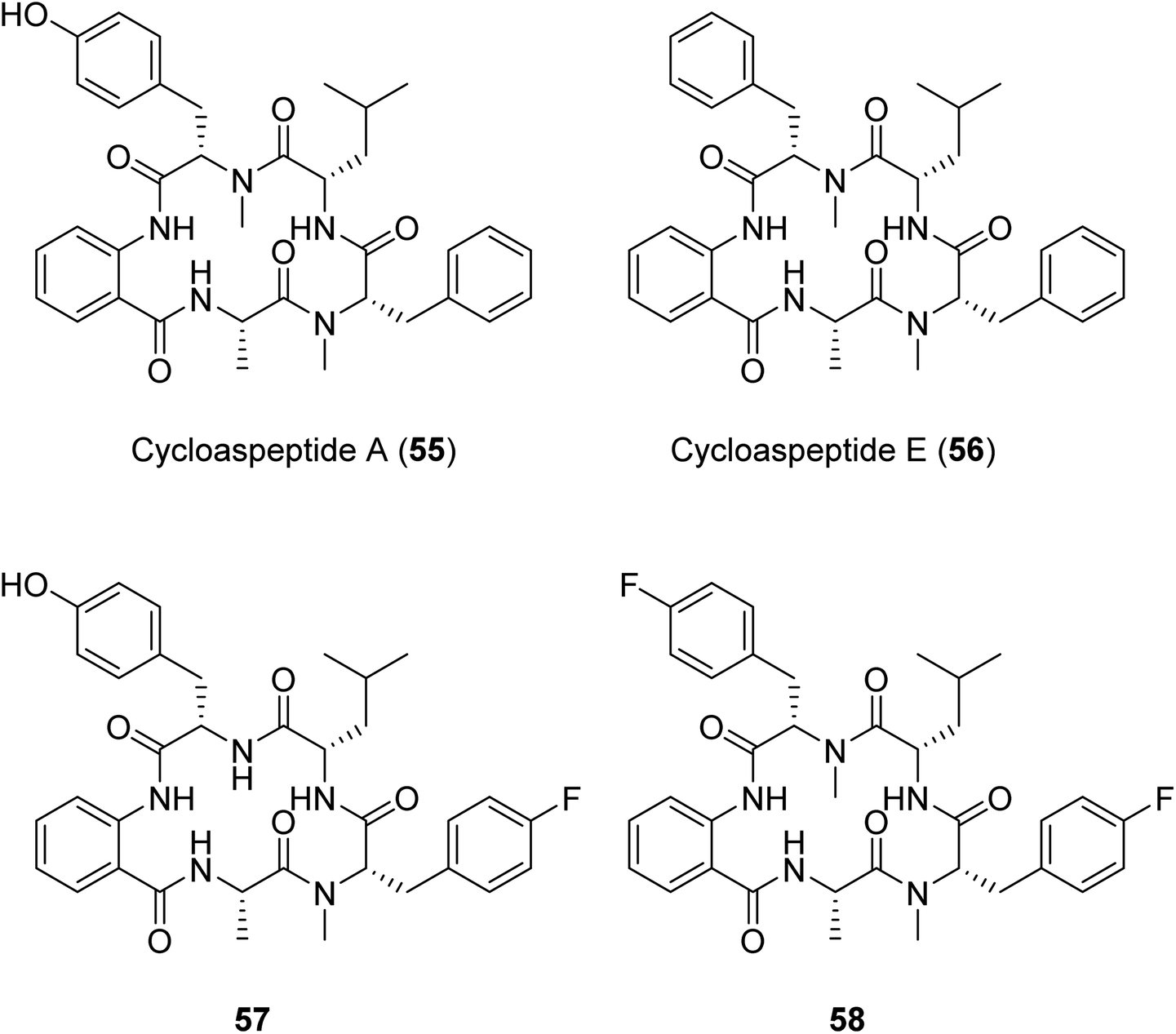
Another example of a successful mutasynthesis of a cyclic peptide-producing NRP was described for the cycloaspeptides, cyclic pentapeptides with various bioactivities obtained from Isaria farinosa and various Aspergillus and Penicillium species.110 The minor analogue, cycloaspeptide E, has shown potent insecticidal activity and drawn interest from the agricultural industry. The cycloaspeptides biosynthetic gene cluster contains a five-module NRPS where each module consists of C–A–T domains only. Two of the modules (modules 3 and 5) preferentially accept and incorporate N-methylated amino acids supplied by a trans-acting N-methyltransferase. Deletion of the N-methyltransferase-encoding gene and supplementation of the fermentation medium with N-methylated amino acids resulted in the production of analogues with fluorinated amino acids at the third and/or the fifth positions of the pentapeptide scaffold.110 A similar N-methyltransferase also participates in the ditryptophenaline biosynthetic pathway,110 suggesting the possibility for an analogous engineering approach to produce uNPs in a broader subclass of N-methylated cyclic peptides featuring trans-acting N-methyltransferases.
3.3 NRPS modules in hybrid megasynthetase systems
Fungal type I polyketide synthases (PKSs) are monomodular multidomain enzymes that use a single set of domains iteratively to synthesize structurally diverse natural products that exhibit a wide range of biological activities.5 In contrast to NRPSs, fungal polyketide synthases (PKSs) assemble malonyl-CoA-derived building blocks instead of amino acids, and form C–C bonds instead of amide bonds. Highly reducing PKSs (hrPKS; those that utilize a full set of reductive domains to synthesize variably reduced medium chain carboxylic acids following a cryptic metaprogram) may integrate with monomodular NRPSs resulting in a bimodular PKS–NRPS hybrid: these then generate structurally complex polyketide/amino acid hybrid molecules with various biological activities (Fig. 13). In addition to canonical PKS–NRPSs that feature full modules from each constituent, hybrid assembly lines with truncated modules, or those with a reversed module order (i.e., NRPS–PKS) have also been reported (Fig. 13C).111–115 Details on the domain architectures, catalytic functions, native product structures and bioactivities of these hybrid enzymes have been summarized in previous reviews.6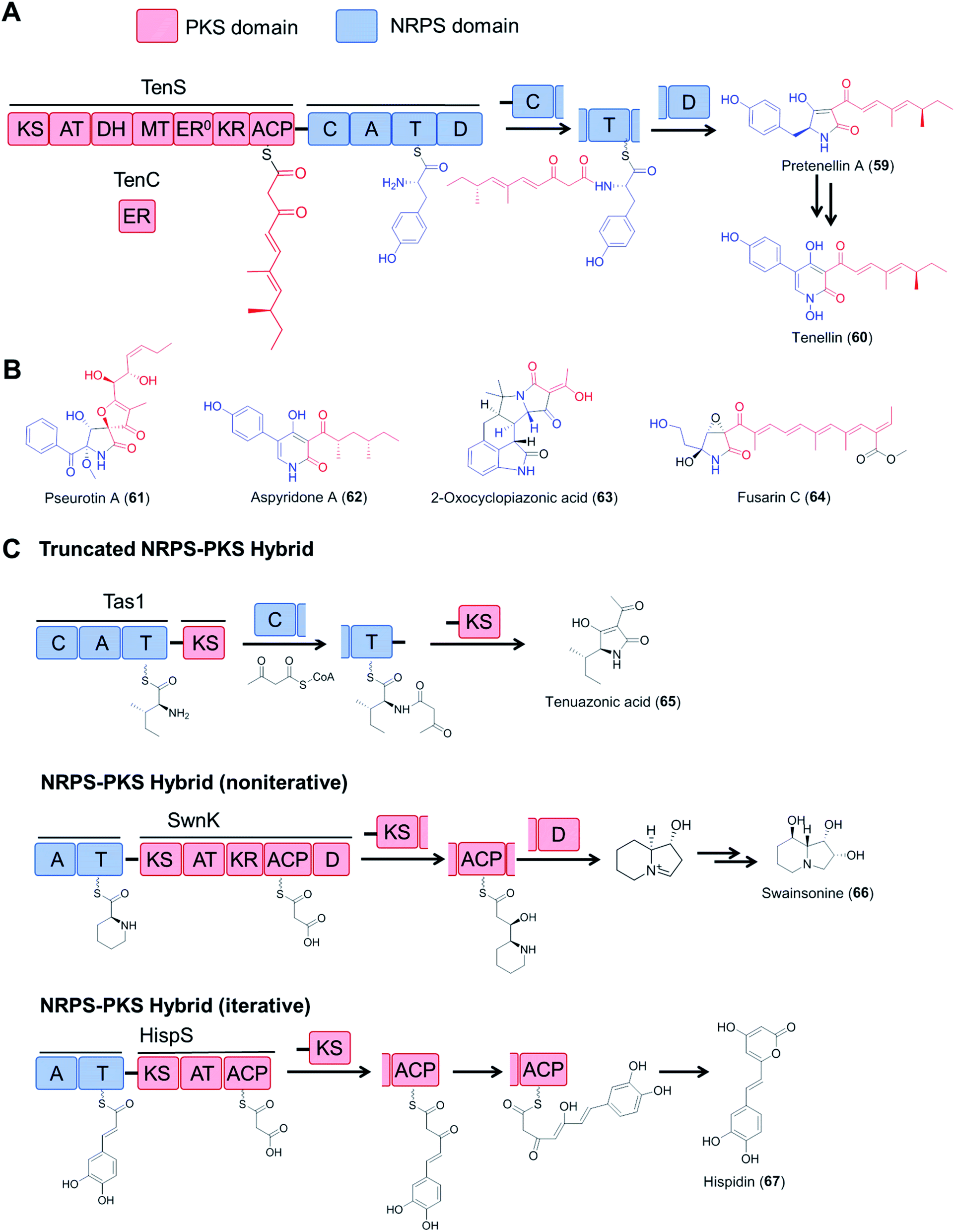 | ||
| Fig. 13 Fungal hybrid enzymes assembled from PKS and NRPS components. (A) Domain organization and reaction mechanisms of a canonical PKS–NRPS hybrid enzyme, represented by the tenellin synthetase TenS.116,117 The PKS module is composed of KS–AT–DH–MT–KR–ACP domains and uses the TenC ER domain as a trans-acting enzyme. The NRPS module has a typical domain organization of C–A–T–D. The product is released by the D domain (Dieckmann cyclase, also known as R, reductive release domain) from the NRPS T domain by forming a tetramate (or pyridone) moiety, which may be further modified by tailoring enzymes. (B) Structures of selected fungal polyketide–amino acid hybrids. The polyketide moieties and amino acid moieties are drawn in red and blue, respectively. The inactive ER domain is represented with ER0. (C) Examples of fungal NRPS–PKS hybrids with incomplete PKS or NRPS modules and iterative or noniterative assembly mechanisms. Tas1, tenuazonic acid synthetase;113 SwnK, swainsonine synthetase;114 HispS, hispidin synthetase.115 | ||
Fungal iterative PKS–NRPS hybrid systems have been successfully engineered to expand product structure diversity by combining non-cognate PKS and NRPS modules.6,77 These works demonstrated that the combination of heterologous PKS and NRPS modules, constructed by domain or module swapping116–118 or assembled as freestanding modules acting in trans,49 can generate chimeric new molecules as expected, albeit often in a reduced yield. Unfortunately, the condensation reaction between the polyketide chain and the incoming amino acid monomer failed in many cases in these unnatural assembly lines, especially when the similarity between the parent enzymes was not high enough.
In a pioneering study, the dissected ApdA PKS and NRPS modules of the aspyridone synthetase and the standalone ER ApdC were incubated in equimolar amounts in the presence of appropriate cofactors and building blocks to afford preaspyridone in a comparable yield to the intact ApdA.49 When the ApdA PKS module and the ApdC ER were coexpressed with the NRPS module of the cyclopiazonic acid PKS–NRPS CpaS in yeast, a new analogue was produced, but at a greatly reduced yield (2.5% relative to that of the intact ApdA and ApdC; Fig. 14).
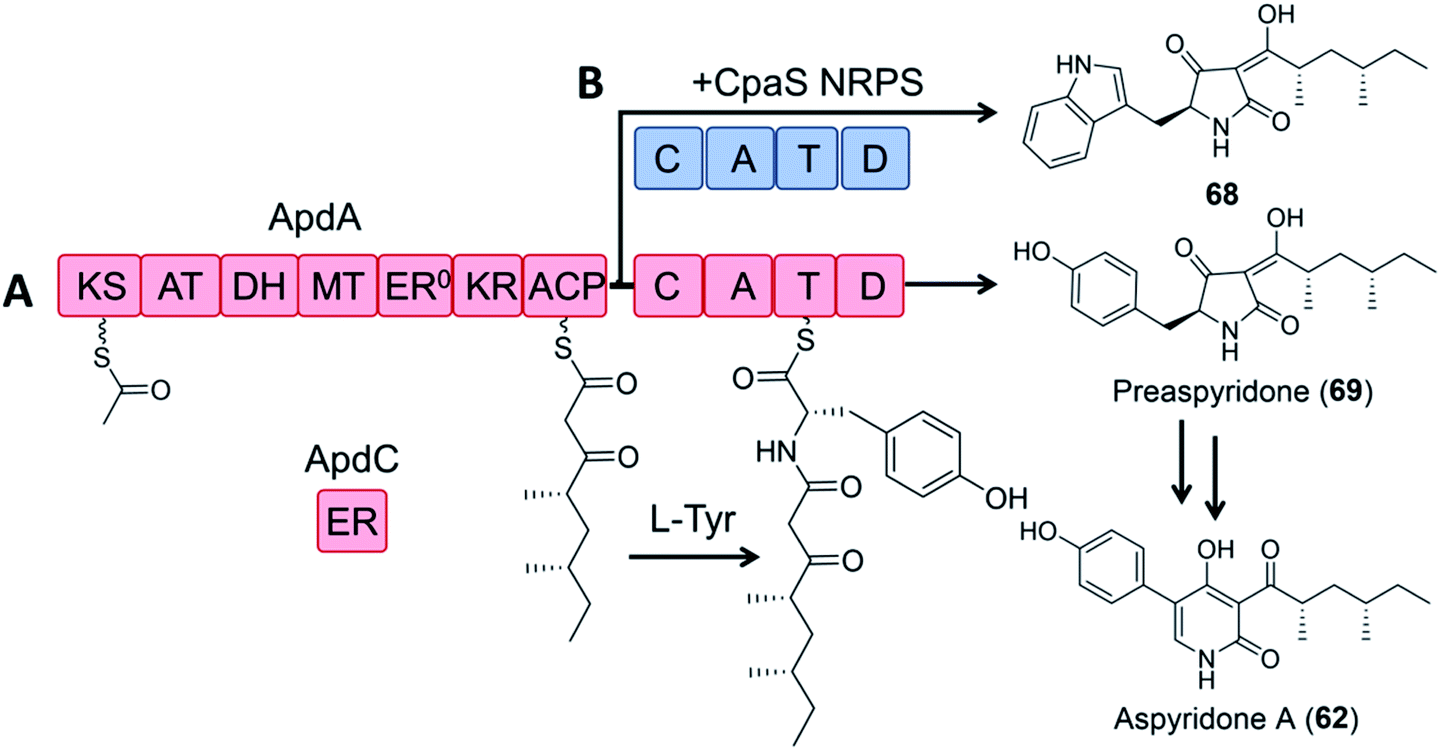 | ||
| Fig. 14 Hybrid PK–NRP production with dissected PKS–NRPS modules.49 (A) Biosynthesis of preaspyridone 69 by ApdA (PKS–NRPS) and ApdC (trans-acting ER). Preaspyridone is converted to the final natural product aspyridone A after enzymatic oxidation and ring expansion.30 (B) Dissected modules from ApdA (the PKS module) and CpaS (the NRPS module) were expressed as freestanding proteins in the same host cells of S. cerevisiae BJ5464-NpgA to afford the expected chimeric product 68. | ||
Domain swapping between the tenellin synthetase TenS and the desmethylbassianin synthetase DmbS, sharing 87% amino acid sequence identity and incorporating the same amino acid building block, was also successful: the chimeric enzyme as well as combinations of the trans-acting ER domains produced the expected tetramic acids (Fig. 15).116,117 What's more, no reductions in the titer were observed with these chimeric systems; the yield of the TenS PKS–DmbS NRPS hybrid enzyme was even higher than that of the native TenS.116
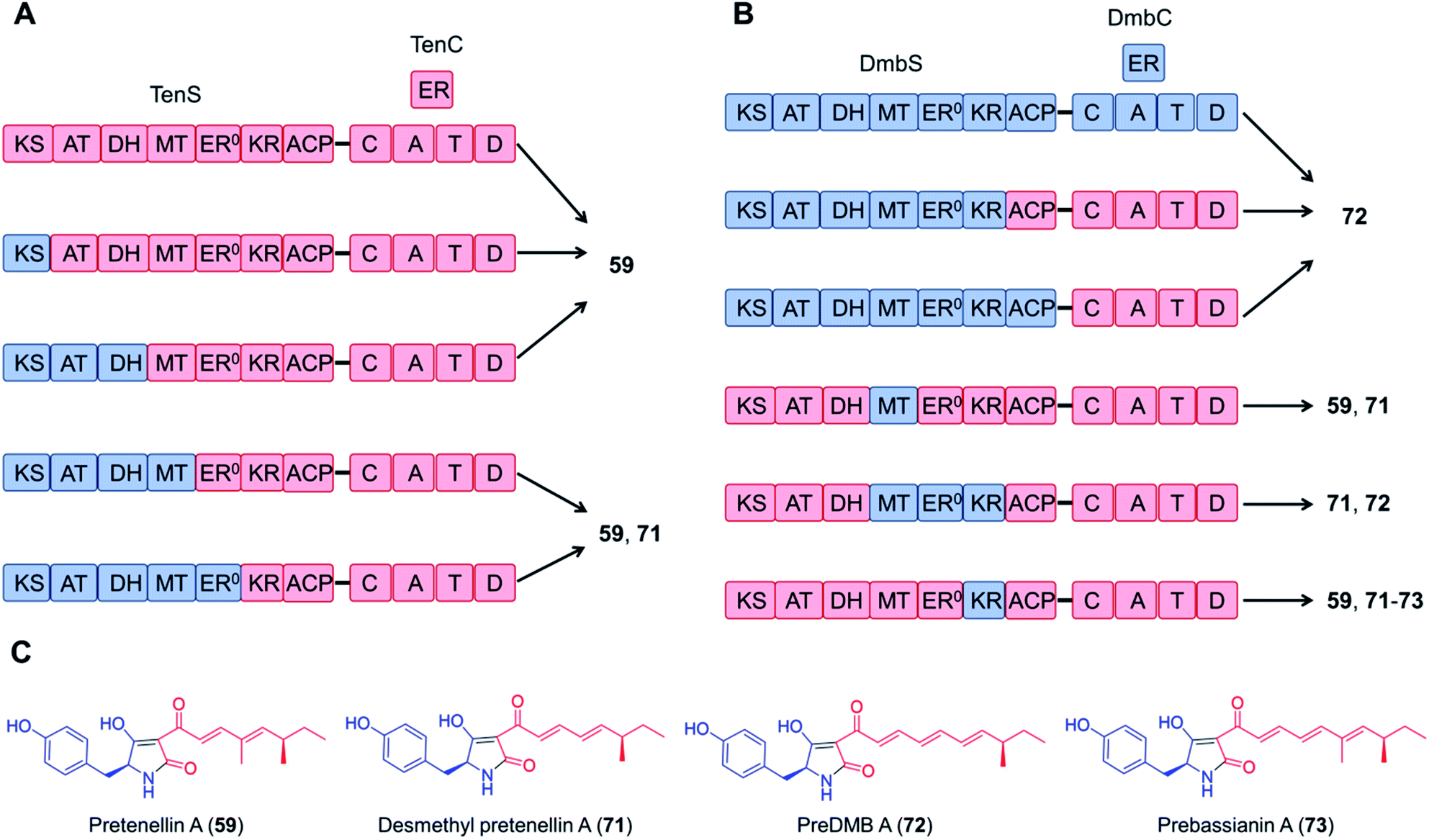 | ||
| Fig. 15 Chimeric PKS–NRPS enzymes constructed from the tenellin synthetase TenS and the desmethylbassianin synthetase DmbS biosynthesize the expected tetramic acid products.116 (A) and (B) Domain compositions of the chimeric synthetases. (C) Structures of products. The polyketide moieties and amino acid moieties are shown in red and blue, respectively. No significant variations in the total titer were observed between the native and the hybrid systems. | ||
Encouragingly, module swapping between the PKS–NRPS CcsA from Aspergillus clavatus involved in the biosynthesis of cytochalasin E, and Syn2 from Magnaporthe oryzae (52% amino acid sequence identity) led to the biosynthesis of the novel products niduchimaeralin A and B (Fig. 16).120
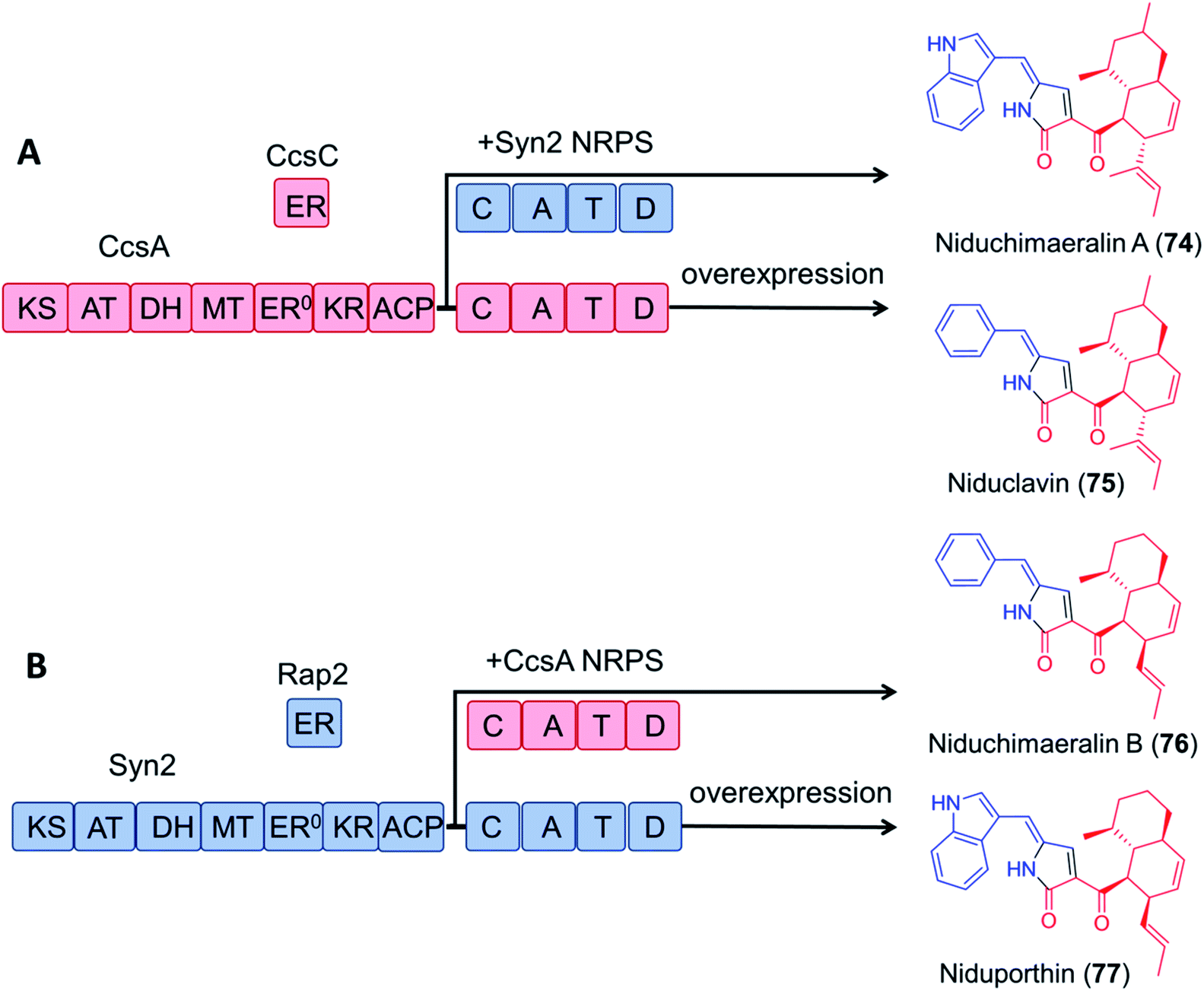 | ||
| Fig. 16 Chimeric PK–NRP production with hybrid enzymes constructed from the cytochalasin E synthetase CcsA119 and the related synthetase Syn2 from Magnaporthe oryzae.120 (A) Overexpression of CcsA and CcsC in A. nidulans led to the production of niduclavin. Expression of chimeric CcsA–Syn2 affords niduchimaeralin A. (B) Expression of chimeric Syn2–CcsA leads to production of niduchimaeralin B. Overexpression of Syn2 and Rap2 of M. oryzae in A. nidulans affords niduporthin. | ||
In an even more daring attempt, PKS and NRPS modules from five different PKS–NRPS enzymes (EqxS,121 FsdS,122 CpaS,123 PsoA,124 and LovB125) synthesizing five, chemically highly distinct fungal NPs (equisetin, fusaridione, cyclopiazonic acid, pseurotin, and lovastatin, respectively) were swapped in 34 combinations, utilizing several fusion sites. Unfortunately, only 16 fusion enzymes yielded any product at all, and most of the combinations only afforded the polyketide portions of the expected hybrid molecules. Only the equisetin PKS (EqxS) and the fusaridione NRPS (FsdS) proved compatible to produce a single hybrid PK–NRP product, presumably due to the relatively high similarity (47% amino acid sequence identity) between EqxS and FsdS (Fig. 17).112 The PKS module of the lovastatin synthetase LovB also failed to produce hybrid compounds when paired with other NRPS modules such as that of the chaetoglobosin A synthetase CheA.118
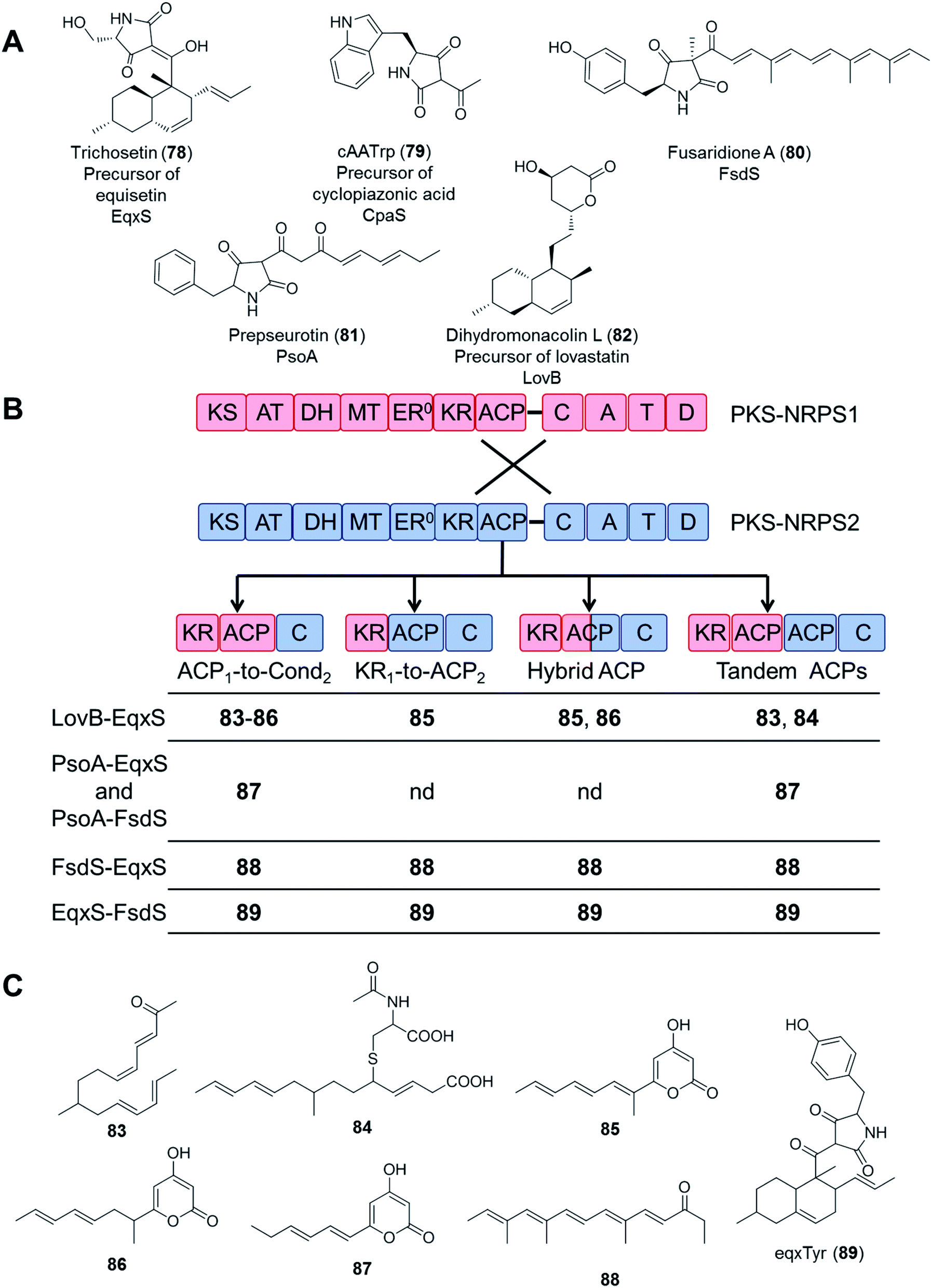 | ||
| Fig. 17 Compounds biosynthesized by chimeric PKS–NRPS enzymes, constructed by using different fusion sites.112 (A) Structures of the natural products of the five PKS–NRPS enzymes that were used for the construction of the chimeras. The names of the parental PKS–NRPS enzymes responsible for their synthesis are shown under the structures. Dihydromonacolin L is not an amidated product because its biosynthetic enzyme, LovB, possesses a truncated NRPS module. (B) Design of the fusion sites. PKS–NRPS1 (blue) represents PsoA, CpaS, LovB, EqxS or FsdS; and PKS–NRPS2 (red) represents EqxS or FsdS. (C) Compounds afforded by the different PKS/NRPS fusions. Compounds 83, 84, 87, 88 and 89 are novel (nd = no product was detected). Note: compound 83, all double bonds are trans. | ||
These attempts highlight the significance of considering the selectivity of the downstream NRPS module (with the C domain likely being the most demanding) for the successful biosynthesis of uNPs with PKS–NRPS chimeras. At this stage, the importance of appropriate protein–protein interactions between the PKS and the NRPS modules cannot be discounted either. The PKS and NRPS modules are connected by a relatively long inter-modular linker (70–150 amino acid residues), with no apparent sequence conservation among different synthetases. While PKS–NRPS hybrids were seen to tolerate the alteration of the sequence and the length of such linkers,120 the compatibility of the noncognate domains turned out to be far more important for a successful chimeric enzyme. The significance of selecting an appropriate ACP domain for the PKS module was demonstrated by the systematic study of fusion sites around the ACPs (Fig. 17B and C). All PKSs were active in making polyketides when fused to their own ACP domains, but only a subset of fusions with a non-cognate ACP led to polyketide products.112 Whether the polyketide products can be passed on to the downstream NRPS module largely depends on the selectivity of the C domains. For example, although the EqxS–FsdS fusion produced a chimeric product, the reciprocal fusion (FsdS–EqxS) yielded only the polyketide part assembled by the FsdS PKS module, without being fused to the serine moiety expected from the EqxS NRPS partner. The fusion product was absent even when the native EqxS ACP was present (Fig. 17C).112 Since both the FsdS and the EqxS ACPs could collaborate with the other domains of either PKSs, this incompatibility was unlikely to be caused by insufficient protein–protein interactions between the two modules. Instead, the failure must have been the consequence of the EqxS C domain rejecting the intermediate synthesized by the FsdS PKS partner.112
Successful biosynthesis of uNP analogues with PKS–NRPS megasynthetases may require the identification of promiscuous NRPS catalysts. Thus, the NRPS module (NRPS325) of the only PKS–NRPS (ATEG00325) of A. terreus produces thiol-substituted pyrazines in vitro. Substrate promiscuity of NRPS325 toward different amino acids and free thiols allowed the production of 63 different thiopyrazine compounds in good yields (Fig. 18).80
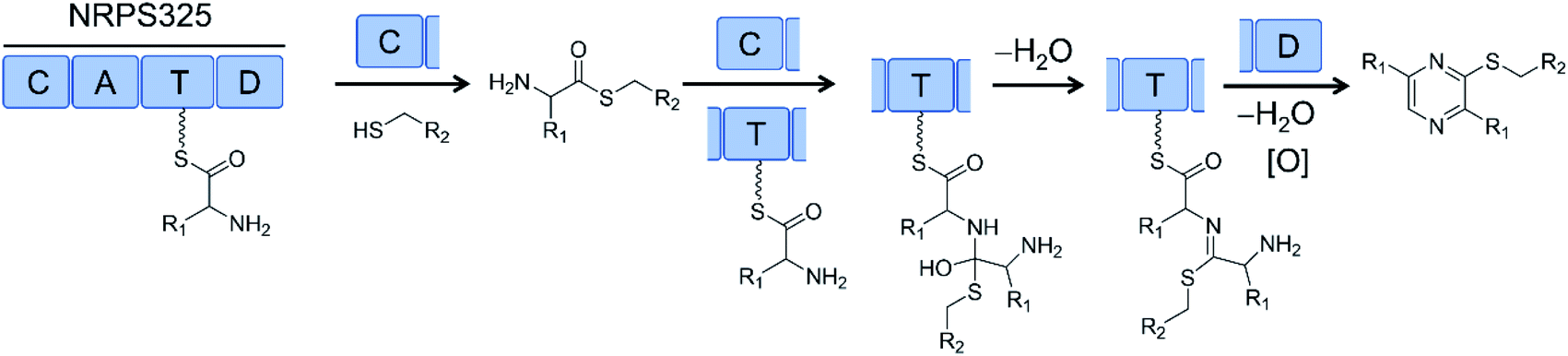 | ||
| Fig. 18 The NRPS module (NRPS325) of a PKS–NRPS from Aspergillus terreus was reconstituted in vitro and its substrate promiscuity towards different amino acids and free thiols was explored to produce >60 different thiopyrazine compounds.80 R1 and R2 represent various amino acid and thiol residues, respectively. | ||
Creation of chimeric products with NRPS–PKS enzymes does not need to consider the limitation of the stringent filtering function of C domains, and may take advantage of the inbuilt promiscuity of some KS and even A domains. For example, an NRPS–PKS hybrid enzyme (AnATPKS) from Aspergillus niger yields compounds where an amino acid starter is extended by a diketide, forming the α-pyrones pyrophen and campyrone B. Upon heterologous expression of the synthetase and precursor feeding, the promiscuous A domain loaded a variety of substituted phenylalanine analogues to the T domain, and these were then successfully extended by the PKS module to afford a library of substituted pyrophen analogues. These analogues may also be further processed by O-methylation and N-acetylation (Fig. 19).126
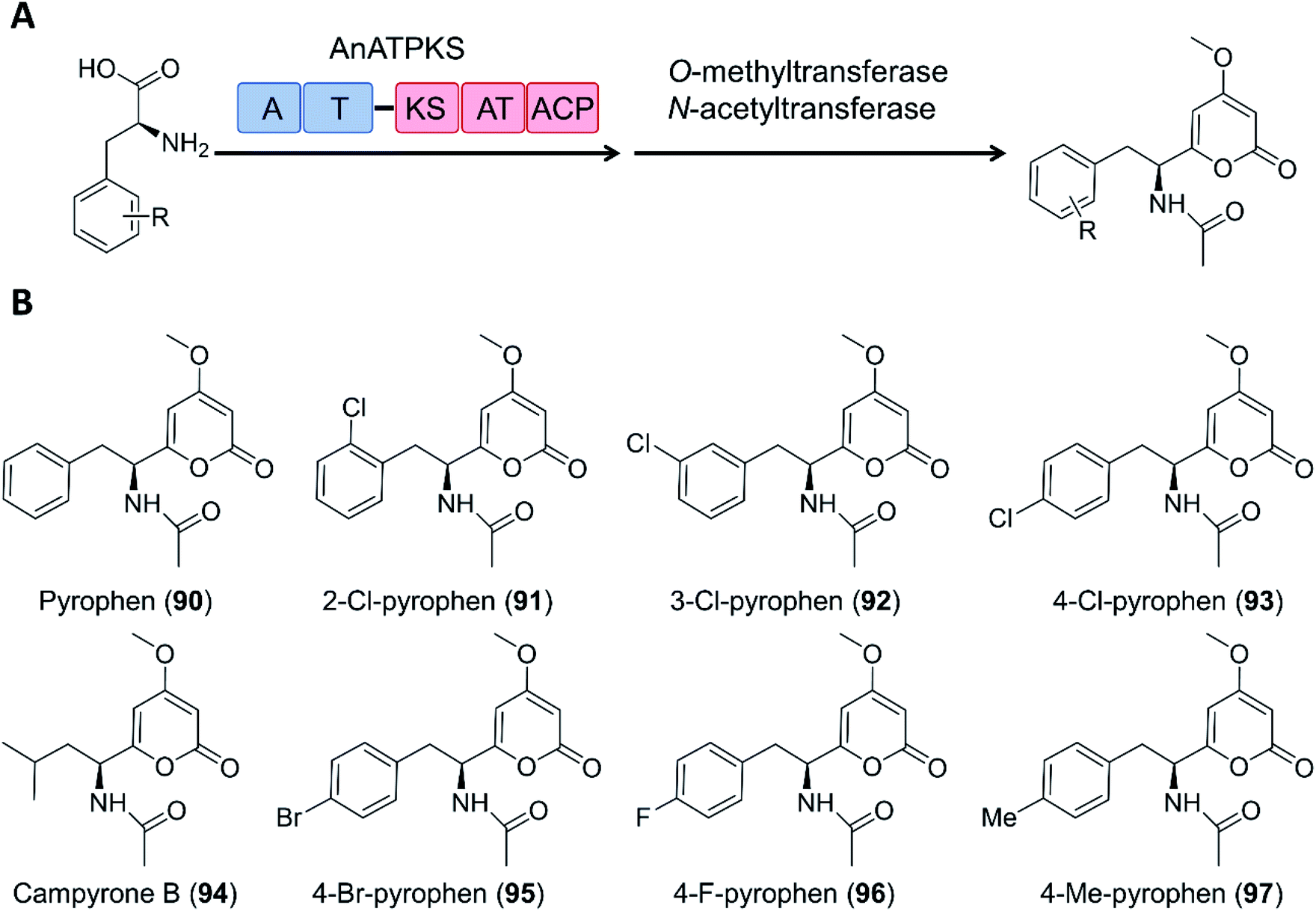 | ||
| Fig. 19 The promiscuity of the A domain and the PKS module of AnATPKS allows the production of various pyrophen analogues.126 (A) Domain organization of AnATPKS and illustration of the biosynthetic pathway. (B) The structure of pyrophen, campyrone B and the analogues obtained upon precursor feeding and heterologous expression of AnATPKS and an associated O-methyltransferase (AnOMT) in A. nidulans. | ||
3.4 NRPS-like enzymes
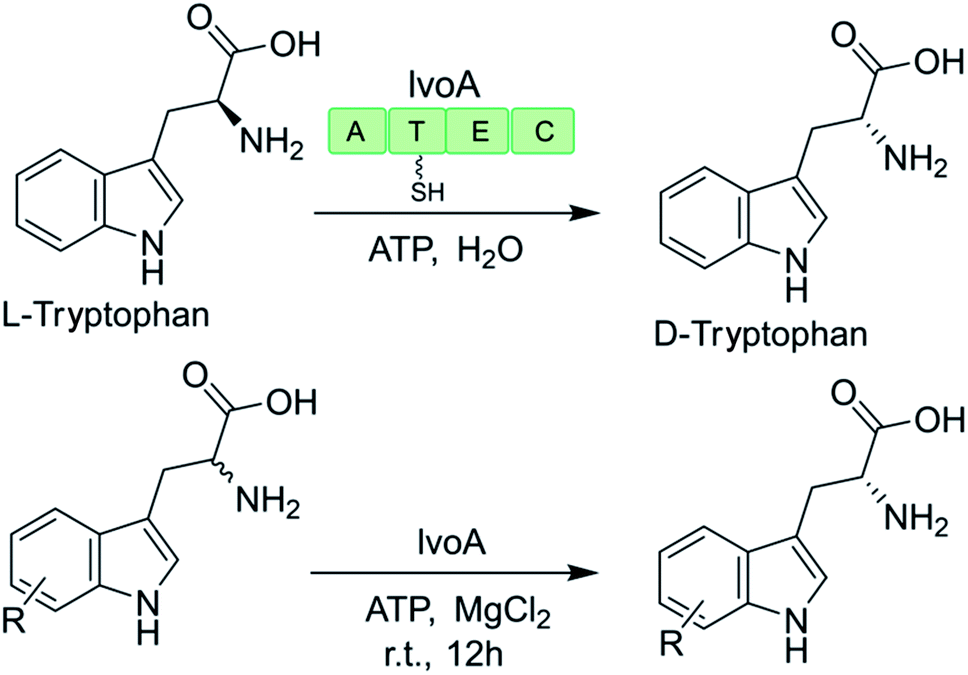
The promiscuity of some NRPS-like enzymes has also facilitated the production of uNPs. For example, the NRPS-like enzyme IvoA is essential for fungal pigment biosynthesis and catalyzes ATP-dependent unidirectional stereoinversion of L-tryptophan to D-tryptophan with near complete stereoselectivity (ee >99% when heterologously expressed in yeast).127,128 The change in the configuration of the chiral center is catalyzed by the IvoA epimerization (E) domain, while the terminal condensation (C) domain stereoselectively hydrolyzes the D-tryptophanyl-S-phosphopantetheine thioester. The purified enzyme also accepts a variety of tryptophan analogues with a substitution at almost any position on the indole ring (Table 1). However, substrates with larger substituents (e.g. 5-NO2, 5-CN, 6-Br, 7-Br) were generally poorly converted, reflecting the size limit of the active site of the IvoA A domain.128|
|
|||
|---|---|---|---|
| Substrate | Product | ee (%) | |
| Configuration | R | ||
| a Reaction conditions: 1.5 mM substrate, 5 μM IvoA, 5 mM ATP, 10 mM MgCl2, 100 mM K2HPO4, pH 7.5. | |||
| L | H | 98 | >99 |
| L | 5-OMe | 99 | >99 |
| rac | 5-CN | 100 | 63 |
| rac | 5-NO2 | 101 | 44 |
| rac | 4-F | 102 | >99 |
| rac | 5-F | 103 | >99 |
| rac | 6-F | 104 | >99 |
| rac | 5-CI | 105 | >99 |
| rac | 6-CI | 106 | 98 |
| rac | 5-Br | 107 | >99 |
| rac | 6-Br | 108 | 75 |
| rac | 7-Br | 109 | 87 |
| rac | 2-Me | 110 | 1.3 |
| rac | 4-Me | 111 | >99 |
| rac | 5-Me | 112 | >99 |
| rac | 6-Me | 113 | 97 |
| rac | 7-Me | 114 | >99 |
The hancockiamides are a family of N-cinnamoylated piperazines, obtained from the Australian soil fungus Aspergillus hancockii. An NRPS-like enzyme (Hkm11) activates and transfers trans-cinnamate to the piperazine scaffold (Fig. 20).129 The substrate flexibility of Hkm11 allowed the production of bioisosteric thienyl and furyl analogues 121–126 by expressing the truncated biosynthetic gene cluster (hkm4–12) lacking an acetyltransferase in A. nidulans (Fig. 20B). The structure diversity of novel hancockiamides was further expanded by supplementing the culture medium with unnatural cinnamic acid congeners. Notably, compounds 115 and 118 displayed potent cytotoxicity against murine myeloma NS-1 cells (MIC 1.6 and 3.1 μg mL−1, respectively), but were inactive against neonatal foreskin normal fibroblasts, suggesting potential applications in cancer chemotherapy. Compound 117, the likely end product of the hkm pathway, also showed potent anti-germination activity against Arabidopsis thaliana seeds (MIC of 6.3 μg mL−1), but was inactive against the monocot Eragrostis teff seeds, suggesting that this compound may be used as a monocots-targeting herbicide lead. The herbicidal activity of compound 117 was proposed to derive from its inhibition of plant lignin biosynthesis due to its phenylpropanoid-like structure.
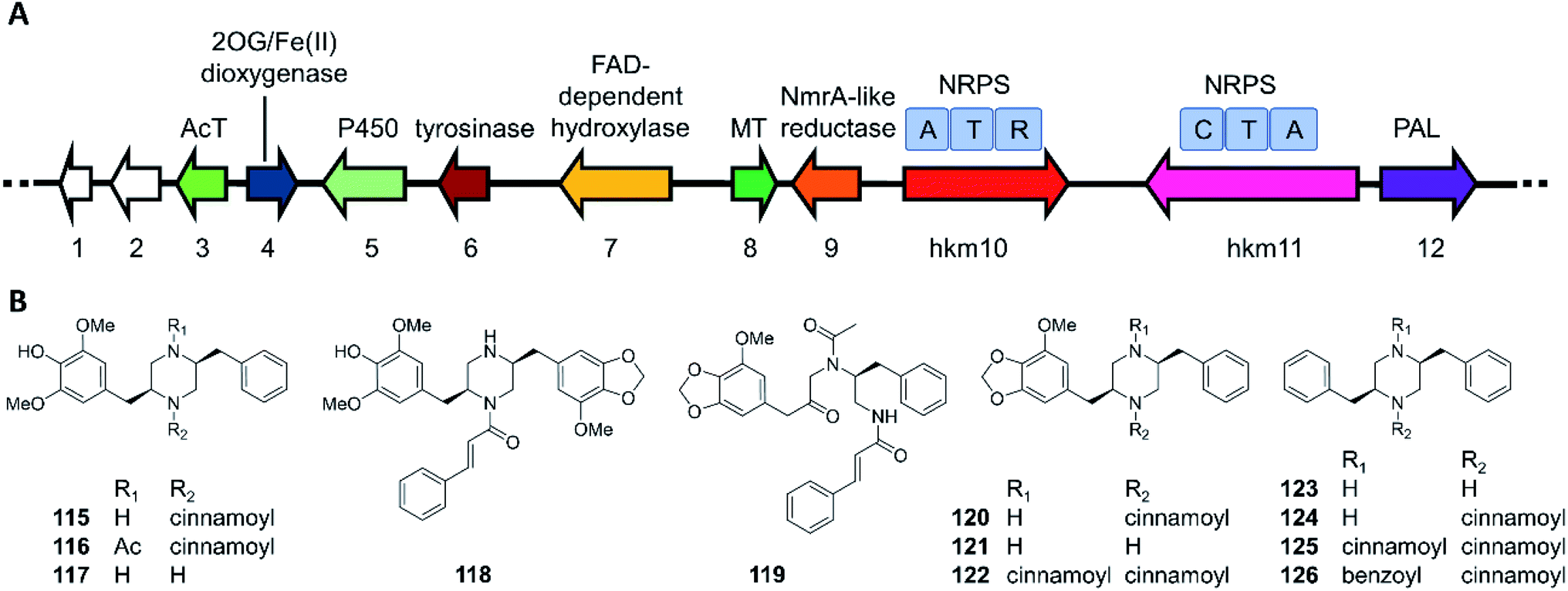 | ||
| Fig. 20 Biosynthesis of hancockiamide analogues.129 (A) The hkm gene cluster is responsible for the production of hancockiamides. AcT, acetyltransferase; MT, methyltransferase; PAL, phenylalanine ammonia lyase. (B) Structures of hancockiamides A–F (115–120) isolated from A. hancockii, and hancockiamides G–I (121–123) and xenocockaimides A–D (124–126) obtained by heterologous expression of the truncated hkm gene cluster in A. nidulans. | ||
As an example of domain swapping in NRPS-like enzymes, the A domain of the BtyA enzyme responsible for butyrolactone IIa (128) production in A. terreus was replaced by an equivalent A domain from the aspulvinone E synthase ApvA, reconstituting the biosynthesis of butyrolactone IIa (128). More interestingly, replacement of the A domain of BtyA with that of the phenguignardic acid (129) synthetase PgnA afforded the new compound phenylbutyrolactone IIa (130) in A. nidulans (Fig. 21).130
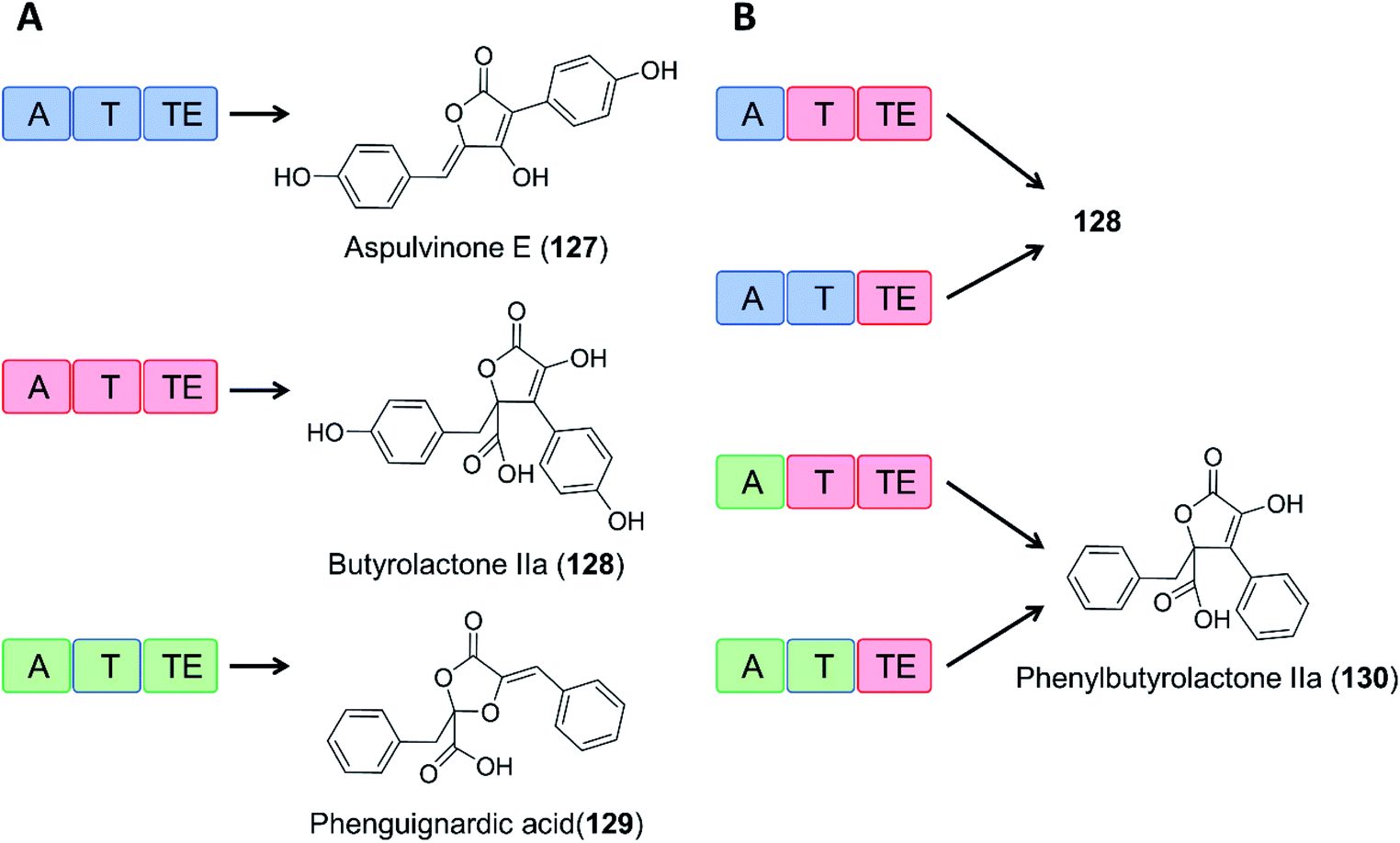 | ||
| Fig. 21 Domain swapping in related NRPS-like enzymes yields an uNP.130 (A) Three NRPS-like enzymes from Aspergillus terreus and their products, aspulvinone E (127), butyrolactone IIa (128), and phenguignardic acid (129).131 (B) Domain organization of chimeric NRPS-like enzymes and their products. The two ApvA/BtyA hybrids produce butyrolactone IIa (128), while both PgnA/BtyA hybrids yield the uNP phenylbutyrolactone IIa (130). | ||
4. Conclusions and discussions
Natural products (NPs) have long inspired drug discovery and continue to be an important source for developing new therapeutic agents.1,132 Amongst NPs, nonribosomal peptides (NRPs) represent an important class with impressive chemical diversity and wide variety of bioactivities. However, NPs often require structural modifications to limit toxicity, address resistance, and improve activity, stability and/or bioavailability. Engineering NRP biosynthetic pathways is a promising method to generate uNPs with improved properties, considering that in theory, the modular architecture of NRPSs lends itself to the modification, replacement, insertion or removal of domains, domain assemblies or even whole modules in a design-based, “plug and play” manner. Encouraged by this prospect, synthetic biology laboratories have been increasingly successful in the rational modification of NRP assembly lines from bacteria, and started to apply the lessons learned to fungal systems.However, it is also becoming increasingly clear that NRP assembly line engineering is a complex puzzle with multiple interrelated factors. Before we can harness the full potential of designer NRP biosynthesis, we will have to understand the reasons for drastic drops in titer, and often the complete loss of peptide production when modifying NRP assembly lines. An immediate need is to understand the intrinsic programming of A domain substrate selectivity: we need to decipher the specificity-conferring code of fungal A domains; and next, we need to derive facile engineering rules for the reprogramming of these domains in a keyhole surgery-like manner. A corollary of having a reliable fungal A domain specificity code, especially in conjunction with a more accurate prediction of C domain subtypes, would be a more precise prediction of the structures of the peptidyl backbones assembled by the huge number of NRPSs emerging from fungal genome sequencing projects.
The second factor is the significant influence of the C domain on the rate and specificity of amino acid incorporation, as well as its proofreading activity for peptide intermediates.133 The C domain has a pseudo-dimeric structure with a catalytic center at the interface of two sub-domains, occupied by the two T-domain-tethered amino acids during catalysis.83 Therefore, C domains have selectivity towards both the donor (the growing peptide) and the acceptor (the activated amino acid) substrates, and these domains enact a proofreading role to ensure the synthesis of the correct peptide sequence. This selectivity restricts the amino acids that may be incorporated by engineered NRPSs to only those building blocks that are structurally similar to the native substrate. NRPS engineering strategies such as the “two-face exchange system”82 try to overcome this bottleneck by combining appropriate domain assemblies with A–T, C–A–T, C–A–T–C, or A–T domain constituents to match, as best as possible, the substrate requirements of the C domains in hybrid assembly lines. Another attempt to solve the C domain bottleneck problem is to assemble chimeric C domains from two noncognate subdomains, each with the desired specificity, to create an “exchange unit condensation domain”.83 Either way, more C domains with characterized selectivity for both of their substrates are required to be catalogued to (1) provide a sufficient toolbox for sub-domain selection; and (2) further elucidate the “specificity code” of C domains that is apparently more complex than that of the A domains.
The C domain bottleneck, and the attempts to generate chimeric C domains or fused assembly lines also emphasizes the need for a better understanding of the protein–protein interactions within the subdomains of the C domain, and those with the other domains of the NRPS assembly line.12 Compared to bacterial NRPSs, much less structural information is available for fungal synthetases.134 However, advances in AI-based computational biology tools such as AlphaFold2 (ref. 135) and RosettaFold136 will facilitate the understanding of the structural basis for the dynamic intra- and inter-domain interactions in fungal NRPSs.
Another barrier, the enormous size of the fungal megaenzymes and their encoding genes, continues to pose a challenge for NRPS engineering in both the endogenous producer and in heterologous hosts. In prokaryotes, NRPS assembly lines are usually encoded by several genes, while in fungi, the megaenzyme is typically the product of a single gene. Therefore, multiple bacterial NRPS subunits may be independently manipulated and then expressed in heterologous hosts such as E. coli. These subunits then may efficiently cooperate through N- and C-terminal docking domains (originally referred to as communication domains) composed of 15–25 amino acids, and synthesize the targeted product.137 Artificial docking domains138 and synthetic zippers139 have also been developed to engineer bacterial NRPS exchange units without natural docking domains. In contrast, fungal multimodule NRPSs are too large to be routinely transferred to model synthetic biology chassis such as S. cerevisiae or E. coli, limiting the development of fungal NRPS engineering. Currently, the successfully engineered fungal NRPSs or PKS–NRPS hybrids contain no more than three modules.
Fungal NRP assembly lines represent a promising yet underexplored target for biosynthetic engineering. While many efforts have been focused on their bacterial counterparts, it is clear that harnessing the exceptional versatility of fungal NRPS assembly lines by advanced synthetic biology tools will offer tremendous opportunities for expanding the chemical diversity of NRP-based uNPs for medical and veterinary drug discovery and crop protection applications.
5. Author contributions
LZ and CW conceptualized the review process, wrote the manuscript and drew the figures. KC drew the figures. WZ reviewed and edited the manuscript. YX and IM conceptualized and supervised the review process; reviewed and edited the manuscript.6. Conflicts of interest
I. M. has declared financial interests in TEVA Pharmaceutical Works Hungary that are unrelated to the subject of the research described here. All other authors declare no competing financial interests.7. Acknowledgements
Research in the laboratories of the authors were supported by the USDA-NIFA (Grant #2017-68005-26867, and Hatch project ARZT-1361640-H12-224 to I. M.), VTT Research Centre of Finland (to I. M.), the National Natural Science Foundation of China (32070053 and 31870076 to Y. X., 32070064 to L. Z., and 32170070 to C. W.); the Youth Innovation Program of Chinese Academy of Agricultural Sciences (Y2022QC13 to C. W.); and the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP to Y. X. and L. Z., and Y2020XK20 to L. Z.).8. References
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS.
- G. Bills, Y. Li, L. Chen, Q. Yue, X. M. Niu and Z. Q. An, Nat. Prod. Rep., 2014, 31, 1348–1375 RSC.
- E. Marschall, M. J. Cryle and J. Tailhades, J. Biol. Chem., 2019, 294, 18769–18783 CrossRef CAS.
- D. A. Alonzo, C. Chiche-Lapierre, M. J. Tarry, J. Wang and T. M. Schmeing, Nat. Chem. Biol., 2020, 16, 493–496 CrossRef CAS PubMed.
- D. A. Alonzo and T. M. Schmeing, Protein Sci., 2020, 29, 2316–2347 CrossRef CAS PubMed.
- A. Miyanaga, F. Kudo and T. Eguchi, Nat. Prod. Rep., 2018, 35, 1185–1209 RSC.
- W. Hüttel, Appl. Microbiol. Biotechnol., 2021, 105, 55–66 CrossRef.
- X. Yang, P. Feng, Y. Yin, K. Bushley, J. W. Spatafora and C. Wang, mBio, 2018, 9, e01211–01218 CAS.
- L. Zhang, Z. Zhou, Q. Guo, L. Fokkens, M. Miskei, I. Pócsi, W. Zhang, M. Chen, L. Wang, Y. Sun, B. G. G. Donzelli, D. M. Gibson, D. R. Nelson, J. Luo, M. Rep, H. Liu, S. Yang, J. Wang, S. B. Krasnoff, Y. Xu, I. Molnár and M. Lin, Sci. Rep., 2016, 6, srep23122 CrossRef.
- J. M. Palmer and N. P. Keller, Curr. Opin. Microbiol., 2010, 13, 431–436 CrossRef CAS.
- T. E. Witte, N. Villeneuve, C. N. Boddy and D. P. Overy, Front. Microbiol., 2021, 12, 664276 CrossRef PubMed.
- R. Maini, S. Umemoto and H. Suga, Curr. Opin. Chem. Biol., 2016, 34, 44–52 CrossRef CAS.
- J. M. Reimer, M. Eivaskhani, I. Harb, A. Guarné, M. Weigt and T. M. Schmeing, Science, 2019, 366, eaaw4388 CrossRef CAS.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS.
- L. Zhang, O. E. Fasoyin, I. Molnár and Y. Xu, Nat. Prod. Rep., 2020, 37, 1181–1206 RSC.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, ChemBioChem, 2002, 3, 490–504 CrossRef CAS.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef.
- D. Liu, G. M. Rubin, D. Dhakal, M. Chen and Y. Ding, iScience, 2021, 24, 102512 CrossRef CAS.
- K. Takesako, S. Mizutani, H. Sakakibara, M. Endo, Y. Yoshikawa, T. Masuda, E. Sono-Koyama and I. Kato, J. Antibiot., 1996, 49, 676–681 CrossRef CAS PubMed.
- D. Matthes, L. Richter, J. Müller, A. Denisiuk, S. C. Feifel, Y. Xu, P. Espinosa-Artiles, R. D. Süssmuth and I. Molnár, Chem. Commun., 2012, 48, 5674–5676 RSC.
- J. Thirlway, R. Lewis, L. Nunns, M. Al Nakeeb, M. Styles, A.-W. Struck, C. P. Smith and J. Micklefield, Angew. Chem., Int. Ed., 2012, 51, 7181–7184 CrossRef CAS PubMed.
- K. T. Nguyen, D. Ritz, J. Q. Gu, D. Alexander, M. Chu, V. Miao, P. Brian and R. H. Baltz, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17462–17467 CrossRef CAS PubMed.
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509–523 CrossRef CAS.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- H.-N. Lyu, H.-W. Liu, N. P. Keller and W.-B. Yin, Nat. Prod. Rep., 2020, 37, 6–16 RSC.
- H. Mohimani and P. A. Pevzner, Nat. Prod. Rep., 2016, 33, 73–86 RSC.
- A. Vassaux, L. Meunier, M. Vandenbol, D. Baurain, P. Fickers, P. Jacques and V. Leclère, Biotechnol. Adv., 2019, 37, 107449 CrossRef CAS PubMed.
- N. Sbaraini, R. L. M. Guedes, F. C. Andreis, A. Junges, G. L. de Morais, M. H. Vainstein, A. T. R. de Vasconcelos and A. Schrank, BMC Genomics, 2016, 17, 736 CrossRef.
- N. P. Keller, Nat. Rev. Microbiol., 2019, 17, 167–180 CrossRef CAS PubMed.
- S. Bergmann, J. Schümann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS.
- E. Skellam, Trends Biotechnol., 2019, 37, 416–427 CrossRef CAS.
- J. A. van Santen, S. A. Kautsar, M. H. Medema and R. G. Linington, Nat. Prod. Rep., 2021, 38, 264–278 RSC.
- M. A. Skinnider, C. W. Johnston, M. Gunabalasingam, N. J. Merwin, A. M. Kieliszek, R. J. MacLellan, H. X. Li, M. R. M. Ranieri, A. L. H. Webster, M. P. T. Cao, A. Pfeifle, N. Spencer, Q. H. To, D. P. Wallace, C. A. Dejong and N. A. Magarvey, Nat. Commun., 2020, 11, 9 CrossRef.
- E. Stylos, M. V. Chatziathanasiadou, A. Syriopoulou and A. G. Tzakos, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1047, 15–38 CrossRef CAS.
- R. M. Gathungu, R. Kautz, B. S. Kristal, S. S. Bird and P. Vouros, Mass Spectrom. Rev., 2020, 39, 35–54 CrossRef CAS.
- M. X. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W. T. Liu, M. Criisemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderon, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C. C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C. C. Liaw, Y. L. Yang, H. U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Q. Dai, R. Neupane, J. Gurr, A. M. C. Rodriguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P. M. Allard, P. Phapale, L. F. Nothias, T. Alexandrovr, M. Litaudon, J. L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D. T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Muller, K. M. Waters, W. Y. Shi, X. T. Liu, L. X. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutierrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS.
- R. Reher, H. W. Kim, C. Zhang, H. H. Mao, M. Wang, L. F. Nothias, A. M. Caraballo-Rodriguez, E. Glukhov, B. Teke, T. Leao, K. L. Alexander, B. M. Duggan, E. L. Van Everbroeck, P. C. Dorrestein, G. W. Cottrell and W. H. Gerwick, J. Am. Chem. Soc., 2020, 142, 4114–4120 CrossRef CAS PubMed.
- T. Q. Shi, G. N. Liu, R. Y. Ji, K. Shi, P. Song, L. J. Ren, H. Huang and X. J. Ji, Appl. Microbiol. Biotechnol., 2017, 101, 7435–7443 CrossRef CAS PubMed.
- X. M. Mao, W. Xu, D. Li, W. B. Yin, Y. H. Chooi, Y. Q. Li, Y. Tang and Y. Hu, Angew. Chem., Int. Ed., 2015, 54, 7592–7596 CrossRef CAS PubMed.
- C. Greco, B. T. Pfannenstiel, J. C. Liu and N. P. Keller, ACS Chem. Biol., 2019, 14, 1121–1128 CrossRef CAS PubMed.
- N. P. Keller, Nat. Chem. Biol., 2015, 11, 671–677 CrossRef CAS.
- B. T. Pfannenstiel and N. P. Keller, Biotechnol. Adv., 2019, 37, 14 CrossRef PubMed.
- C. J. B. Harvey, M. Tang, U. Schlecht, J. Horecka, C. R. Fischer, H. C. Lin, J. Li, B. Naughton, J. Cherry, M. Miranda, Y. F. Li, A. M. Chu, J. R. Hennessy, G. A. Vandova, D. Inglis, R. S. Aiyar, L. M. Steinmetz, R. W. Davis, M. H. Medema, E. Sattely, C. Khosla, R. P. St Onge, Y. Tang and M. E. Hillenmeyer, Sci. Adv., 2018, 4, eaar5459 CrossRef.
- S. E. Unkles, V. Valiante, D. J. Mattern and A. A. Brakhage, Chem. Biol., 2014, 21, 502–508 CrossRef CAS PubMed.
- Y. He, B. Wang, W. P. Chen, R. J. Cox, J. R. He and F. S. Chen, Biotechnol. Adv., 2018, 36, 739–783 CrossRef CAS.
- F. Alberti, G. D. Foster and A. M. Bailey, Appl. Microbiol. Biotechnol., 2017, 101, 493–500 CrossRef CAS PubMed.
- C. M. Lazarus, K. Williams and A. M. Bailey, Nat. Prod. Rep., 2014, 31, 1339–1347 RSC.
- R. A. Cacho and Y. Tang, Methods Mol. Biol., 2016, 1401, 103–119 CrossRef CAS.
- Y. Tsunematsu, K. Ishiuchi, K. Hotta and K. Watanabe, Nat. Prod. Rep., 2013, 30, 1139–1149 RSC.
- W. Xu, X. L. Cai, M. E. Jung and Y. Tang, J. Am. Chem. Soc., 2010, 132, 13604–13607 CrossRef CAS PubMed.
- H. D. Mootz, K. Schörgendorfer and M. A. Marahiel, FEMS Microbiol. Lett., 2002, 213, 51–57 CAS.
- D. C. Anyaogu and U. H. Mortensen, Front. Microbiol., 2015, 6, 1–6 Search PubMed.
- A. Yoshimi, S. Yamaguchi, T. Fujioka, K. Kawai, K. Gomi, M. Machida and K. Abe, Front. Microbiol., 2018, 9, 690 CrossRef.
- K. C. Van de Bittner, M. J. Nicholson, L. Y. Bustamante, S. A. Kessans, A. Ram, C. J. van Dolleweerd, B. Scott and E. J. Parker, J. Am. Chem. Soc., 2018, 140, 582–585 CrossRef CAS.
- F. Guzmán-Chávez, R. D. Zwahlen, R. A. L. Bovenberg and A. J. M. Driessen, Front. Microbiol., 2018, 9, 2768 CrossRef.
- R. Süssmuth, J. Müller, H. von Döhren and I. Molnár, Nat. Prod. Rep., 2011, 28, 99–124 RSC.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS.
- C. Rausch, T. Weber, O. Kohlbacher, W. Wohlleben and D. H. Huson, Nucleic Acids Res., 2005, 33, 5799–5808 CrossRef CAS.
- M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Commun., 2020, 11, 4554 CrossRef CAS PubMed.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 CrossRef CAS.
- B. S. Evans, Y. Chen, W. W. Metcalf, H. Zhao and N. L. Kelleher, Chem. Biol., 2011, 18, 601–607 CrossRef CAS PubMed.
- D. L. Niquille, D. A. Hansen, T. Mori, D. Fercher, H. Kries and D. Hilvert, Nat. Chem., 2018, 10, 282–287 CrossRef CAS.
- F. Ishikawa, A. Miyanaga, H. Kitayama, S. Nakamura, I. Nakanishi, F. Kudo, T. Eguchi and G. Tanabe, Angew. Chem., Int. Ed., 2019, 58, 6906–6910 CrossRef CAS PubMed.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Prod. Rep., 2018, 35, 1210–1228 RSC.
- K. Blin, S. Shaw, A. M. Kloosterman, Z. Charlop-Powers, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2021, 49(W1), W29–W35 CrossRef CAS PubMed.
- C. Steiniger, S. Hoffmann and R. D. Süssmuth, ACS Synth. Biol., 2019, 8, 661–667 CrossRef CAS.
- S. Dekimpe and J. Masschelein, Nat. Prod. Rep., 2021, 38, 1910–1937 RSC.
- T. Caradec, M. Pupin, A. Vanvlassenbroeck, M. D. Devignes, M. Smail-Tabbone, P. Jacques and V. Leclere, PLoS One, 2014, 9, e85667 CrossRef PubMed.
- X. Zhang, E. King-Smith, L. B. Dong, L. C. Yang, J. D. Rudolf, B. Shen and H. Renata, Science, 2020, 369, 799–806 CrossRef CAS PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- J. Grünewald, S. A. Sieber, C. Mahlert, U. Linne and M. A. Marahiel, J. Am. Chem. Soc., 2004, 126, 17025–17031 CrossRef.
- J. W. Trauger, R. M. Kohli, H. D. Mootz, M. A. Marahiel and C. T. Walsh, Nature, 2000, 407, 215–218 CrossRef CAS.
- R. M. Kohli, C. T. Walsh and M. D. Burkart, Nature, 2002, 418, 658–661 CrossRef CAS.
- Y. Ding, C. M. Rath, K. L. Bolduc, K. Håkansson and D. H. Sherman, J. Am. Chem. Soc., 2011, 133, 14492–14495 CrossRef CAS PubMed.
- J. W. Bogart, M. D. Cabezas, B. Vögeli, D. A. Wong, A. S. Karim and M. C. Jewett, ChemBioChem, 2021, 22, 84–91 CrossRef CAS.
- J. Li, L. K. Zhang and W. Q. Liu, Synth. Syst. Biotechnol., 2018, 3, 83–89 CrossRef.
- J. A. Baccile, H. H. Le, B. T. Pfannenstiel, J. W. Bok, C. Gomez, E. Brandenburger, D. Hoffmeister, N. P. Keller and F. C. Schroeder, Angew. Chem., Int. Ed., 2019, 58, 14589–14593 CrossRef CAS PubMed.
- D. Yu, F. Xu, S. Zhang and J. Zhan, Nat. Commun., 2017, 8, 15349 CrossRef CAS PubMed.
- Y. Hai, M. Jenner and Y. Tang, Chem. Sci., 2020, 11, 11525–11530 RSC.
- K. Qiao, H. Zhou, W. Xu, W. Zhang, N. Garg and Y. Tang, Org. Lett., 2011, 13, 1758–1761 CrossRef CAS.
- R. Süssmuth, J. Müller, H. von Döhren and I. Molnár, Nat. Prod. Rep., 2011, 28, 99–124 RSC.
- C. Steiniger, S. Hoffmann and R. D. Sussmuth, Cell Chem. Biol., 2019, 26, 1526–1534 CrossRef CAS.
- K. A. J. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y. N. Shi, P. Grün and H. B. Bode, Nat. Chem., 2019, 11, 653–661 CrossRef.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C. P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275–281 CrossRef.
- W. Weckwerth, K. Miyamoto, K. Iinuma, M. Krause, M. Glinski, T. Storm, G. Bonse, H. Kleinkauf and R. Zocher, J. Biol. Chem., 2000, 275, 17909–17915 CrossRef CAS.
- M. Krause, A. Lindemann, M. Glinski, T. Hornbogen, G. Bonse, P. Jeschke, G. Thielking, W. Gau, H. Kleinkauf and R. Zocher, J. Antibiot., 2001, 54, 797–804 CrossRef CAS PubMed.
- S. C. Feifel, T. Schmiederer, T. Hornbogen, H. Berg, R. D. Süssmuth and R. Zocher, ChemBioChem, 2007, 8, 1767–1770 CrossRef CAS.
- J. Müller, S. C. Feifel, T. Schmiederer, R. Zocher and R. D. Süssmuth, ChemBioChem, 2009, 10, 323–328 CrossRef PubMed.
- Y. Xu, J. Zhan, E. M. Wijeratne, A. M. Burns, A. A. L. Gunatilaka and I. Molnár, J. Nat. Prod., 2007, 70, 1467–1471 CrossRef CAS.
- K. Yanai, N. Sumida, K. Okakura, T. Moriya, M. Watanabe and T. Murakami, Nat. Biotechnol., 2004, 22, 848–855 CrossRef CAS.
- Y. Xu, E. M. K. Wijeratne, P. Espinosa-Artiles, A. A. L. Gunatilaka and I. Molnár, ChemBioChem, 2009, 10, 345–354 CrossRef CAS PubMed.
- D. Yu, F. Xu, D. Gage and J. Zhan, Chem. Commun., 2013, 49, 6176–6178 RSC.
- S. Zobel, S. Boecker, D. Kulke, D. Heimbach, V. Meyer and R. D. Süssmuth, ChemBioChem, 2016, 17, 283–287 CrossRef CAS.
- C. Steiniger, S. Hoffmann, A. Mainz, M. Kaiser, K. Voigt, V. Meyer and R. D. S. Süssmuth, Chem. Sci., 2017, 8, 7834–7843 RSC.
- K. M. Hoyer, C. Mahlert and M. A. Marahiel, Chem. Biol., 2007, 14, 13–22 CrossRef CAS PubMed.
- T. Stachelhaus, A. Schneider and M. A. Marahiel, Science, 1995, 269, 69–72 CrossRef CAS PubMed.
- R. Traber, H. Hofmann and H. Kobel, J. Antibiot., 1989, 42, 591–597 CrossRef CAS PubMed.
- Y. Li, L. Chen, Q. Yue, X. Z. Liu, Z. Q. An and G. F. Bills, ACS Chem. Biol., 2015, 10, 1702–1710 CrossRef CAS PubMed.
- G. Bills, Y. Li, L. Chen, Q. Yue, X. M. Niu and Z. An, Nat. Prod. Rep., 2014, 31, 1348–1375 RSC.
- W. Hüttel, Z. Naturforsch., C: J. Biosci., 2017, 72, 1–20 CrossRef.
- Y. Li, N. Lan, L. Xu and Q. Yue, Appl. Microbiol. Biotechnol., 2018, 102, 9881–9891 CrossRef CAS PubMed.
- L. A. Petersen, D. L. Hughes, R. Hughes, L. DiMichele, P. Salmon and N. Connors, J. Ind. Microbiol. Biotechnol., 2001, 26, 216–221 CrossRef CAS.
- P. S. Masurekar, J. M. Fountoulakis, T. C. Hallada, M. S. Sosa and L. Kaplan, J. Antibiot., 1992, 45, 1867–1874 CrossRef CAS PubMed.
- S.-P. Zou, W. Zhong, C.-J. Xia, Y.-N. Gu, K. Niu, Y.-G. Zheng and Y.-C. Shen, Bioprocess Biosyst. Eng., 2015, 38, 1845–1854 CrossRef CAS.
- R. A. Cacho, W. Jiang, Y. H. Chooi, C. T. Walsh and Y. Tang, J. Am. Chem. Soc., 2012, 134, 16781–16790 CrossRef CAS PubMed.
- L. Chen, Q. Yue, X. Zhang, M. Xiang, C. Wang, S. Li, Y. Che, F. J. Ortiz-Lopez, G. F. Bills, X. Liu and Z. An, BMC Genomics, 2013, 14, 339 CrossRef CAS.
- W. Hüttel, L. Youssar, B. A. Grüning, S. Günther and K. G. Hugentobler, BMC Genomics, 2016, 17, 570 CrossRef PubMed.
- L. Chen, Y. Li, Q. Yue, A. Loksztejn, K. Yokoyama, E. A. Felix, X. Liu, N. Zhang, Z. An and G. F. Bills, ACS Chem. Biol., 2016, 11, 2724–2733 CrossRef CAS PubMed.
- W. Jiang, R. A. Cacho, G. Chiou, N. K. Garg, Y. Tang and C. T. Walsh, J. Am. Chem. Soc., 2013, 135, 4457–4466 CrossRef CAS.
- K. M. J. de Mattos-Shipley, C. Greco, D. M. Heard, G. Hough, N. P. Mulholland, J. L. Vincent, J. Micklefield, T. J. Simpson, C. L. Willis, R. J. Cox and A. M. Bailey, Chem. Sci., 2018, 9, 4109–4117 RSC.
- Y. Hai and Y. Tang, J. Am. Chem. Soc., 2018, 140, 1271–1274 CrossRef CAS.
- T. B. Kakule, Z. Lin and E. W. Schmidt, J. Am. Chem. Soc., 2014, 136, 17882–17890 CrossRef CAS PubMed.
- C. S. Yun, T. Motoyama and H. Osada, Nat. Commun., 2015, 6, 8758 CrossRef CAS PubMed.
- D. Cook, B. G. G. Donzelli, R. Creamer, D. L. Baucom, D. R. Gardner, J. Pan, N. Moore, S. B. Krasnoff, J. W. Jaromczyk and C. L. Schardl, G3: Genes, Genomes, Genet., 2017, 7, 1791–1797 CrossRef CAS.
- A. A. Kotlobay, K. S. Sarkisyan, Y. A. Mokrushina, M. Marcet-Houben, E. O. Serebrovskaya, N. M. Markina, L. Gonzalez Somermeyer, A. Y. Gorokhovatsky, A. Vvedensky, K. V. Purtov, V. N. Petushkov, N. S. Rodionova, T. V. Chepurnyh, L. I. Fakhranurova, E. B. Guglya, R. Ziganshin, A. S. Tsarkova, Z. M. Kaskova, V. Shender, M. Abakumov, T. O. Abakumova, I. S. Povolotskaya, F. M. Eroshkin, A. G. Zaraisky, A. S. Mishin, S. V. Dolgov, T. Y. Mitiouchkina, E. P. Kopantzev, H. E. Waldenmaier, A. G. Oliveira, Y. Oba, E. Barsova, E. A. Bogdanova, T. Gabaldón, C. V. Stevani, S. Lukyanov, I. V. Smirnov, J. I. Gitelson, F. A. Kondrashov and I. V. Yampolsky, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12728–12732 CrossRef CAS PubMed.
- K. M. Fisch, W. Bakeer, A. A. Yakasai, Z. S. Song, J. Pedrick, Z. Wasil, A. M. Bailey, C. M. Lazarus, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 16635–16641 CrossRef CAS.
- M. N. Heneghan, A. A. Yakasai, K. Williams, K. A. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2011, 2, 972–979 RSC.
- D. Boettger, H. Bergmann, B. Kuehn, E. Shelest and C. Hertweck, ChemBioChem, 2012, 13, 2363–2373 CrossRef CAS.
- K. Qiao, Y. H. Chooi and Y. Tang, Metab. Eng., 2011, 13, 723–732 CrossRef CAS PubMed.
- M. L. Nielsen, T. Isbrandt, L. M. Petersen, U. H. Mortensen, M. R. Andersen, J. B. Hoof and T. O. Larsen, PLoS One, 2016, 11, 18 Search PubMed.
- T. B. Kakule, D. Sardar, Z. Lin and E. W. Schmidt, ACS Chem. Biol., 2013, 8, 1549–1557 CrossRef CAS.
- T. B. Kakule, R. C. Jadulco, M. Koch, J. E. Janso, L. R. Barrows and E. W. Schmidt, ACS Synth. Biol., 2015, 4, 625–633 CrossRef CAS.
- M. Tokuoka, Y. Seshime, I. Fujii, K. Kitamoto, T. Takahashi and Y. Koyama, Fungal Genet. Biol., 2008, 45, 1608–1615 CrossRef CAS PubMed.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS PubMed.
- S. Maiya, A. Grundmann, X. Li, S. M. Li and G. Turner, ChemBioChem, 2007, 8, 1736–1743 CrossRef CAS PubMed.
- Y. Hai, A. Huang and Y. Tang, J. Nat. Prod., 2020, 83, 593–600 CrossRef CAS.
- C. T. Sung, S. L. Chang, R. Entwistle, G. Ahn, T. S. Lin, V. Petrova, H. H. Yeh, M. B. Praseuth, Y. M. Chiang, B. R. Oakley and C. C. C. Wang, Fungal Genet. Biol., 2017, 101, 1–6 CrossRef CAS.
- Y. Hai, M. Jenner and Y. Tang, J. Am. Chem. Soc., 2019, 141, 16222–16226 CrossRef CAS PubMed.
- H. Li, A. E. Lacey, S. Shu, J. A. Kalaitzis, D. Vuong, A. Crombie, J. Hu, C. L. M. Gilchrist, E. Lacey, A. M. Piggott and Y. H. Chooi, Org. Biomol. Chem., 2021, 19, 587–595 RSC.
- J. W. A. van Dijk, C.-J. Guo and C. C. C. Wang, Org. Lett., 2016, 18, 6236–6239 CrossRef CAS.
- C. J. Balibar, A. R. Howard-Jones and C. T. Walsh, Nat. Chem. Biol., 2007, 3, 584–592 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- M. Kaniusaite, J. Tailhades, E. A. Marschall, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, Chem. Sci., 2019, 10, 9466–9482 RSC.
- T. V. Lee, L. J. Johnson, R. D. Johnson, A. Koulman, G. A. Lane, J. S. Lott and V. L. Arcus, J. Biol. Chem., 2010, 285, 2415–2427 CrossRef CAS PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Nature, 2020, 577, 706–710 CrossRef CAS.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS.
- M. Hahn and T. Stachelhaus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15585–15590 CrossRef CAS PubMed.
- X. Cai, L. Zhao and H. B. Bode, Org. Lett., 2019, 21, 2116–2120 CrossRef CAS.
- K. A. J. Bozhueyuek, J. Watzel, N. Abbood and H. B. Bode, Angew. Chem., Int. Ed., 2021, 60, 17531–17538 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this review. |
| This journal is © The Royal Society of Chemistry 2023 |