
Structure-based engineering of α-ketoglutarate dependent oxygenases in fungal meroterpenoid biosynthesis
Takayoshi
Awakawa
 *ab,
Takahiro
Mori
abc,
Richiro
Ushimaru
abd and
Ikuro
Abe
*ab
*ab,
Takahiro
Mori
abc,
Richiro
Ushimaru
abd and
Ikuro
Abe
*ab
aGraduate School of Pharmaceutical Sciences, the University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: awakawa@mol.f.u-tokyo.ac.jp; abei@mol.f.u-tokyo.ac.jp
bCollaborative Research Institute for Innovative Microbiology, the University of Tokyo, Yayoi 1-1-1, Bunkyo-ku, Tokyo 113-8657, Japan
cPRESTO, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan
dACT-X, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan
First published on 1st June 2022
Abstract
Non-heme iron- and α-ketoglutarate-dependent oxygenases (αKG OXs) are key enzymes that play a major role in diversifying the structure of fungal meroterpenoids. They activate a specific C–H bond of the substrate to first generate radical species, which is usually followed by oxygen rebound to produce cannonical hydroxylated products. However, in some cases remarkable chemistry induces dramatic structural changes in the molecular scaffolds, depending on the stereoelectronic characters of the substrate/intermediates and the resulting conformational changes/movements of the active site of the enzyme. Their molecular bases have been extensively investigated by crystallographic structural analyses and structure-based mutagenesis, which revealed intimate structural details of the enzyme reactions. This information facilitates the manipulation of the enzyme reactions to create unnatural, novel molecules for drug discovery. This review summarizes recent progress in the structure-based engineering of αKG OX enzymes, involved in the biosynthesis of polyketide-derived fungal meroterpenoids. The literature published from 2016 through February 2022 is reviewed.
 Takayoshi Awakawa | Takayoshi Awakawa received his BS from the Agricultural Department, the University of Tokyo in 2006; and PhD from the Agricultural and Life Sciences, the University of Tokyo, in 2011. After obtaining his PhD, he worked as an assistant professor (2011–2017), lecturer (2018–2019), and associate professor (2019–now). He also carried out postdoctoral research in the laboratory of Prof. Bradley S. Moore at UC San Diego, Scripps Institute of Oceanography (2014–2016) as a visiting scholar. His research interests focus on the engineered biosynthesis and chemical biology of natural products. |
 Takahiro Mori | Takahiro Mori received his BS from the School of Pharmaceutical Sciences, the University of Shizuoka in 2010; and his MS and PhD from the Graduate School of Pharmaceutical Sciences, the University of Tokyo, in 2012 and 2016, respectively. After obtaining his PhD he carried out postdoctoral research in the laboratory of Prof. Donald Hilvert at ETH (2016–2017). He was appointed assistant professor at the Graduate School of Pharmaceutical Sciences, the University of Tokyo in 2018. His research interests are in the structural analysis and engineering of natural product biosynthetic enzymes. |
 Richiro Ushimaru | Richiro Ushimaru is an assistant professor in the Graduate School of Pharmaceutical Sciences at the University of Tokyo. He received BS and MS degree from the Department of Chemistry at Nagoya University. He then joined the research group of Prof. Hung-wen Liu at the University of Texas at Austin, where he received his PhD in chemistry in 2019. His current research focuses on natural product biosynthesis and mechanistic enzymology. |
 Ikuro Abe | Ikuro Abe received his BS (1984) and PhD (1989) from the University of Tokyo under the direction of Prof. Ushio Sankawa and Prof. Yutaka Ebizuka, studying chemistry and biochemistry of natural products biosynthesis. After postdoctoral research with Prof. Guy Ourisson at the CNRS Institut de Chimie des Substances Naturelles, and with Prof. Michel Rohmer at the Ecole Nationale Supérieure de Chimie de Mulhouse (1989–1991), he stayed in the United States to work with Prof. Glenn D. Prestwich at the State University of New York at Stony Brook (1991–1996) and then at the University of Utah (1996–1998). In 1998, he moved back to Japan to join the faculty at the University of Shizuoka (1998–2009), and was then appointed Professor of Natural Products Chemistry at the Graduate School of Pharmaceutical Sciences, the University of Tokyo (2009-). His research interests mostly focus on exploring and engineering natural products biosynthesis. He received the Japanese Society of Pharmacognosy Award (2017), Sumiki Umezawa Memorial Award (2017), the Pharmaceutical Society of Japan Award (2019), and Prizes for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology, Japan (2019). He is a Fellow of the Royal Society of Chemistry (FRSC) and a former President of the Japanese Society of Pharmacognosy. |
1 Introduction
Meroterpenoids are defined as a group of hybrid natural products that consist of non-terpenoid and terpenoid moieties. The fungal meroterpenoids mainly possess polyketide-derived skeletons, although they occasionally include molecular scaffolds derived from other biosynthetic origins, as in the indole and shikimate-derived meroterpenoids.1 The enzymes responsible for fungal meroterpenoid biosynthesis have been well characterized,2 and the information regarding their structure and function has been utilized to create structurally diverse, unnatural novel meroterpenoids through engineered biosynthesis.3–5 This review mainly focuses on work toward the discovery, structural analysis, and protein engineering of the enzymes involved in the biosynthesis of polyketide-derived fungal meroterpenoids.The biosynthesis of polyketide-derived meroterpenoids is achieved by polyketide synthases (PKSs), prenyltransferases (PTs), flavin-dependent monooxygenases (FMOs), terpene cyclases (CYCs),6 and tailoring enzymes, including isomerases, transferases, and oxidases, such as cytochrome P450 oxygenases (P450s)7 or non-heme iron- and α-ketoglutarate-dependent oxygenases (αKG OXs).2,8–11 PKSs construct polyketide cores from acetate-derived building blocks, PTs install prenyl side-chains, FMOs oxidize the olefin of the prenyl group to form epoxides, and CYC catalyzes protonation-initiated cyclization of the terpene moiety, and a tailoring enzyme decorates the resultant scaffold to generate complex meroterpenoid structures. Among the tailoring enzymes, αKG OXs often play a crucial role in amplifying the structural diversity and complexity of the molecules.12–17 They employ mononuclear non-heme Fe(II) and the co-substrate αKG, and generate a highly reactive ferryl–oxo intermediate (Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) with the concomitant oxidative decarboxylation of αKG to succinate.18,19 The Fe(IV)O first abstracts a hydrogen atom from a specific unactivated C–H bond in the substrate (Fig. 1). The generated intermediate radical species facilitate a wide range of reactions, including hydroxylation, desaturation, epoxidation, C–X bond formation, and C–C bond reconstruction.2,3,5,8–10,12–21 As a result, diverse meroterpenoid structures are produced, as exemplified by the biosyntheses of farnesyl-3,5-dimethylorsellinic acid (DMOA) 1 derived terretonin 2, austinol 3, emeridone F 4, anditomin 5, paraherquonin 6, and novofumigatonin 7 (Fig. 2).2 The structural basis of the αKG OX enzyme reactions has recently been extensively studied by X-ray crystallography and site-directed mutagenesis experiments. In this review, recent progress in the structure-based engineering of αKG OX enzymes to create unnatural novel molecules for drug discovery is summarized.
O) with the concomitant oxidative decarboxylation of αKG to succinate.18,19 The Fe(IV)O first abstracts a hydrogen atom from a specific unactivated C–H bond in the substrate (Fig. 1). The generated intermediate radical species facilitate a wide range of reactions, including hydroxylation, desaturation, epoxidation, C–X bond formation, and C–C bond reconstruction.2,3,5,8–10,12–21 As a result, diverse meroterpenoid structures are produced, as exemplified by the biosyntheses of farnesyl-3,5-dimethylorsellinic acid (DMOA) 1 derived terretonin 2, austinol 3, emeridone F 4, anditomin 5, paraherquonin 6, and novofumigatonin 7 (Fig. 2).2 The structural basis of the αKG OX enzyme reactions has recently been extensively studied by X-ray crystallography and site-directed mutagenesis experiments. In this review, recent progress in the structure-based engineering of αKG OX enzymes to create unnatural novel molecules for drug discovery is summarized.
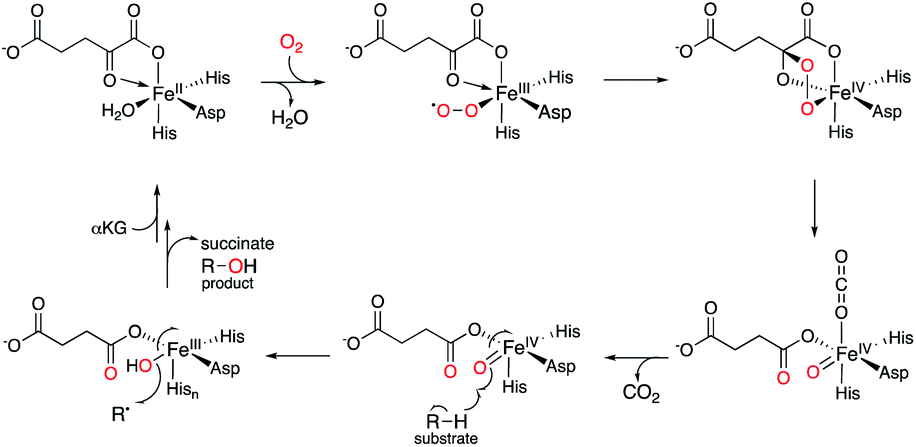 | ||
| Fig. 1 The general catalytic cycle for non-heme iron- and α-ketoglutarate-dependent oxygenase. | ||
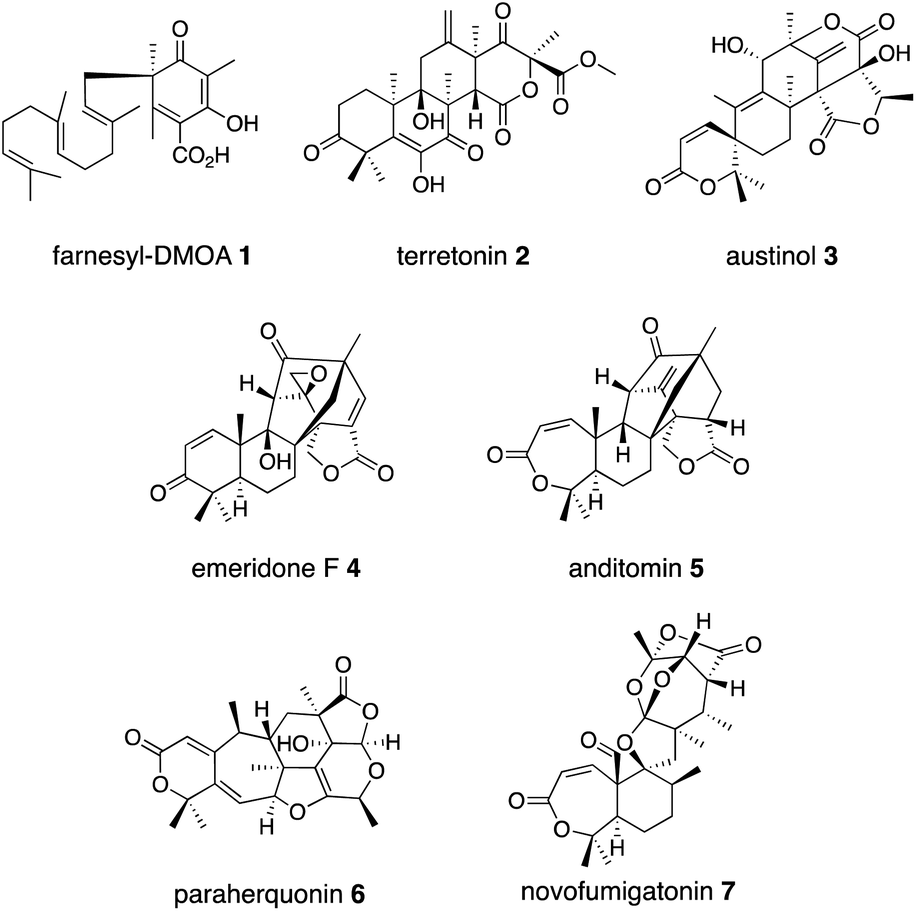 | ||
| Fig. 2 Fungal meroterpenoids derived from farnesyl-DMOA 1. | ||
2 AusE/PrhA in austinol/paraherquonin biosynthesis
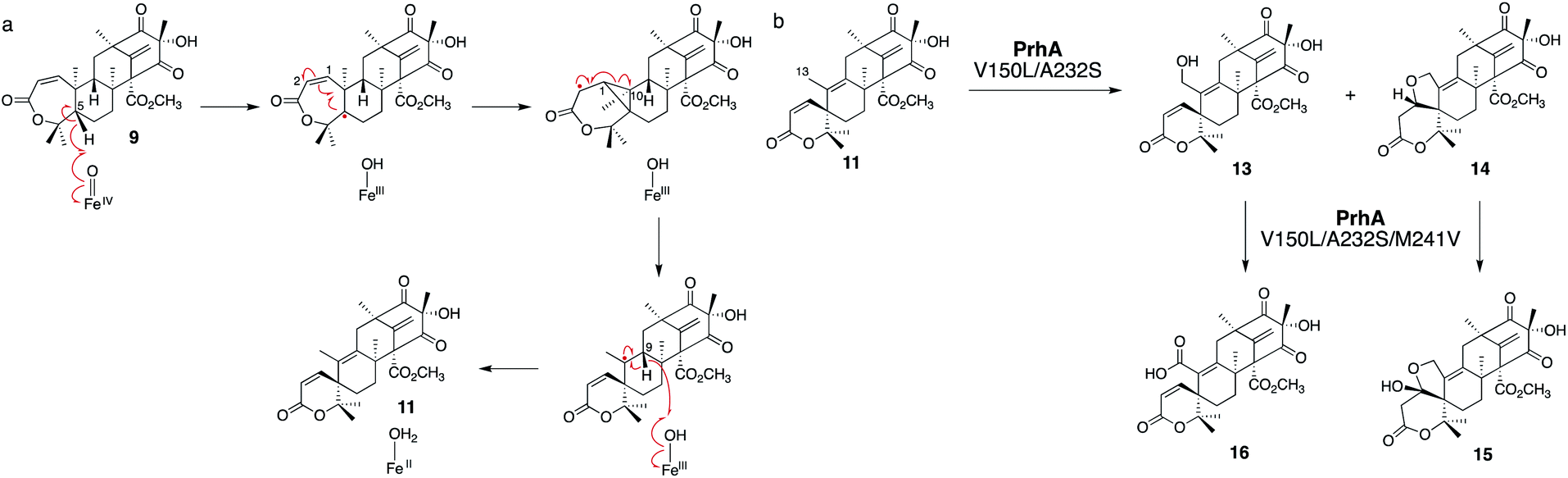
AusE and PrhA are multifunctional αKG OX enzymes that accept the common substrate preaustinoid A1 8, but catalyze different regioselective dehydrogenations to produce preaustinoid A2 9 in austinol 3via biosynthesis in Aspergillus nidulans; and berkeleyone B 10 in paraherquonin 6via biosynthesis in Penicillium brasilianum, respectively (Fig. 3).22,23 AusE further catalyzes spirolactone ring formation to produce preaustinoid A3 11, while PrhA catalyzes cycloheptadiene formation to produce berkeleydione 12. Because AusE and PrhA share high amino acid sequence identity (78%), the two homologous enzymes are excellent models for a comparative analysis to investigate the underlying differences in the catalytic mechanisms of meroterpenoid diversification. The X-ray crystal structures of AusE/PrhA were reported as the first examples of the αKG OXs in fungal meroterpenoid biosynthesis.24 While AusE was crystallized in complex with Mn(II), which does not serve as a functional cofactor for the oxygenation, PrhA was crystallized with Fe(II) in an active site under anaerobic conditions. The overall structure of AusE/PrhA complexes are symmetric homodimers consisting of a double-stranded β-helix (DSBH) core, a.k.a. jelly-roll, cupin, or jumonji C fold, which includes the residues for substrate binding and the 2-H–1-D (H130–D132–H214) triad for metal-binding (Fig. 4a).12 The analysis of PrhA complexed with Fe, αKG, and 8 (PrhA-Fe/αKG/8) provided important information regarding the manner of substrate binding (Fig. 4b). Remarkably, the structural analyses revealed significant conformational changes upon substrate binding. The Cα of the S66 loop A in chain B moves 5.4 Å toward the active site, to interact with D276′ and the 5′-OH and 6′-carbonyl of 8. The newly generated hydrogen bonds fix hairpin B, which is only detected when the protein is complexed with 8. Upon acceptance by the enzyme, the conformation of the A-ring of 8 is altered from a chair to boat conformation. The A/B-rings of 8 are surrounded by three hydrophobic residues V150, A232, and M241, and one hydrophilic residue N152 (Fig. 4b). In contrast, the D-ring of 8 interacts with several hydrophilic residues, including R72 at the β-sheet, N127 and R128 at loop C, and D276′ in hairpin B, as well as loop A 57–69 and hairpin 267′–280′ (from another monomer). The region including these residues was proposed to function as a lid for substrate binding. The 6′-carbonyl of 8 is hydrogen bonded with the C-1 carboxylate of αKG, suggesting the requirement of αKG for substrate binding. | ||
| Fig. 3 Syntheses of preaustinoid A3 11 and berkeleydione 12 from the common substrate preaustinoid A1 8 by AusE and PrhA. PrhA-V150L/A232S catalyzes the AusE reaction to produce 11, and AusE-L150V/S232A catalyzes the PrhA reaction to produce 12. | ||
 | ||
| Fig. 4 Structure of AusE and PrhA. (a) The overall structure of PrhA (PDBID: 5YBN). αKG and catalytic triad residues are shown with blue and magenta sticks, respectively. (b) Active site structure of PrhA-Fe/αKG/8 (PDBID: 5YBO). Loop A and loop C in chain B are represented with the turquoise and salmon illustrations, respectively. The hairpin loop from the adjoining monomer is depicted with the yellow illustration. 8 and αKG are shown with the blue and grey sticks, respectively. (c) Comparison of the active site residues between the AusE and PrhA structures (PDBID: 5YBL and 5YBO). PrhA and AusE are represented with the salmon- and green-colored residues, respectively. 8 and αKG are shown with the white and grey sticks, respectively. (d) Comparison of binding modes of 8 in PrhA wild type (salmon sticks, PDBID: 5YBO) and PrhA-V150L/A232S (yellow sticks, PDBID: 5YBP). 8 in PrhA wild type, 8 in PrhA-V150L/A232S, and αKG are shown with the blue, yellow, and grey sticks, respectively. Overall: Yellow dashed lines show hydrogen bond interactions. Blue dashed lines represent coordination of metal atom. Orange and purple spheres show iron and manganese atoms, respectively. The key distance between the iron and carbon atoms are shown with arrows. The units of distance are Å. The oxygen, nitrogen, and sulfur atoms are colored red, blue, and yellow, respectively. | ||
The comparison of the AusE and PrhA structures revealed that V150, A232, and M241 in PrhA are replaced with L150, S232, and V241 in AusE (Fig. 4c). To evaluate the effects of these substitutions, these three amino acid pairs were replaced with their counterpart residues. Remarkably, the AusE S232A variant produced 12 and 11 in comparable amounts, and AusE L150V/S232A produced 12 as a major product. As anticipated, the PrhA V150L/A232S variant produced 11 as a major product. The kcat and Km values of AusE L150V/S232A and PrhA V150L/A232S were very similar to those of PrhA and AusE, respectively, indicating that they efficiently catalyze the non-native reactions. Thus, the structure-based engineering successfully achieved functional interconversion of the two enzymes. The crystal structures of PrhA-V150L/A232S and PrhA-V150L/A232S/M241V complexed with each substrate revealed that the position of the D-ring of the substrate does not change, because it is tightly fixed by loop A and hairpin B, while significant conformational changes shift the orientation of the A/B rings toward the iron center (Fig. 4d). The wild-type PrhA abstracts H-5 of 8, leading to the production of 10, while PrhA V150L/A232S abstracts H-2 to produce 9, as in the case of wild-type AusE (Fig. 3). Based on the altered regioselectivities, it is likely that the A-ring of 8 undergoes a boat-to-chair flip and the C-2 of the substrate becomes closer to the iron in PrhA V150L/A232S than that in the wild-type enzyme (4.2 Å vs. 4.4 Å). This example shows how the enzyme active site dictates the conformation of the terpene moiety of the substrate, leading to the two different regioselectivities of the reactions. In PrhA V150L/A232S complexed with 9, the distance between H-5 and Fe(II) is the shortest, which is consistent with the proposed mechanism of the reaction initiation by the abstraction of H-5. After the formation of 9, the reconstruction of the spirocycle ring system is proposed as follows. The C-5 radical first reacts with the double bond between C-1 and C-2, resulting in the cyclopropylcarbinyl radical intermediate, and the C-2 radical then reacts with an electron of the bond between C-1 and C-10, followed by the dehydrogenation of H-9 and the formation of the C-9/C-10 double bond, to produce 11 (Fig. 5a).
 | ||
| Fig. 5 Proposed reaction mechanisms of AusE and PrhA V150L/A232S to synthesize 11 from 9 (a), and the reactions of PrhA V150L/A232S and V150L/A232S/M241V to synthesize 13, 14, 15 and 16, respectively (b). | ||
Interestingly, the PrhA V150L/A232S variant oxidizes C-13 on 11 to produce unnatural novel products, 13 and 14, and PrhA V150L/A232S/M241V further oxidizes 13 into 16, and 14 into 15 (Fig. 5b). The crystal structure of PrhA V150L/A232A with 11 revealed that the C-10/C-13 bond is rotated clockwise by 30°, thus reducing the distance from C-13 to Fe(II).
It is remarkable that the double amino acid substitutions V150L and A232S in PrhA increase the number of oxidations from two (8 to 11) to five times (8 to 16). Because the structure of the D-ring of the substrate does not change during the reaction, the enzyme can recognize and accommodate the oxidized products 9, 11, 13, and 14, even though its A/B ring structures are altered significantly. The prediction and evaluation of the conformational changes of substrates in the active site of the enzyme are crucial to rationally design the structures of molecules, as illustrated in the in silico-designed targeted protein engineering of class I terpene cyclases.25,26
3 AndA in anditomin biosynthesis
AndA, another multifunctional αKG OX active during anditomin 5 biosynthesis, desaturates the bond between the C-1 and C-2 of preandiloid B 17 to produce preandiloid C 18, and also catalyzes the subsequent isomerization (not oxidation!) to yield andiconin 19 through a dynamic skeletal rearrangement (Fig. 6).27 The X-ray crystallographic analysis and density functional theory (DFT) calculations of the reaction intermediates were conducted to clarify its catalytic mechanism.28 The apo structure of AndA exhibits the conserved DSBH fold with a funnel-like reaction chamber (Fig. 7a),12,24 and the complex structures with 17 or 18 revealed that the substrates are bound to the interface of the two monomers, in a similar manner to AusE and PrhA (Fig. 7b and c). Loop A (loop 58–76, between α2 and β3) from one monomer, and loop B′ (loop 261′–281′, between α7′ and α8′) from the other monomer, and R239 located at the loop structure, constitute a lid for the active site. Loop A is not visible in the apo-structure, and in the complex structure it is bound to the D/E rings of the substrate. The two carbonyl oxygen atoms O-3 and O-5 of 17 are anchored to a water molecule (w1) supported by the side chain of Y272′ and the main chains of N67 and I70, and O-4 is bound to two water molecules (w2 and w3) coordinated by the side chain of E66 and the main chains of K63 and N67 (Fig. 7b). The terpenoid moiety (the A/B/C rings) of 17 is mainly supported by hydrophobic interactions, except for one hydrogen bond between O-1 and N121. Since a water molecule (w4) and the carboxylic oxygen of αKG (O-2/αKG) are considered to occupy identical positions to the oxo species of Fe(IV)O, the distances from each carbon atom of the substrate to w4 or O-2/αKG were compared. As a result, the calculated distances were 4.4 Å (C-1 to w4), 4.0 Å (C-2 to w4), 3.3 Å (C-1 to O-2/αKG), and 3.7 Å (C-2 to O-2/αKG), indicating that both H-1 and H-2 are sufficiently close for the hydrogen atom abstraction (Fig. 7b). The exact mechanism of the desaturation remains unclear even with the crystal structures, although three pathways can be proposed as shown in the mechanistic study of desaturation by AsqJ αKG OX in the viridicatin-type alkaloid biosynthetic pathway.29
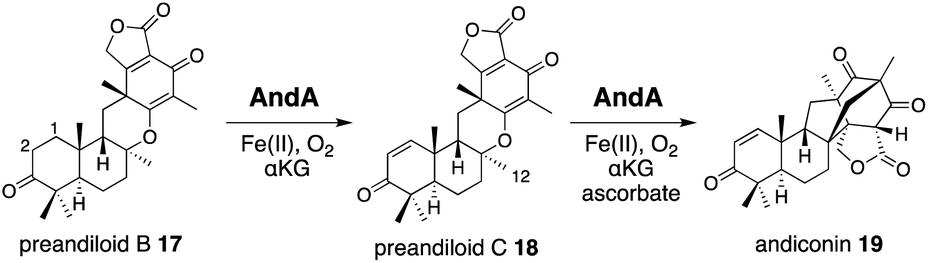 | ||
| Fig. 6 Andiconin 19 synthesis from preandiloid B 17 by AndA. | ||
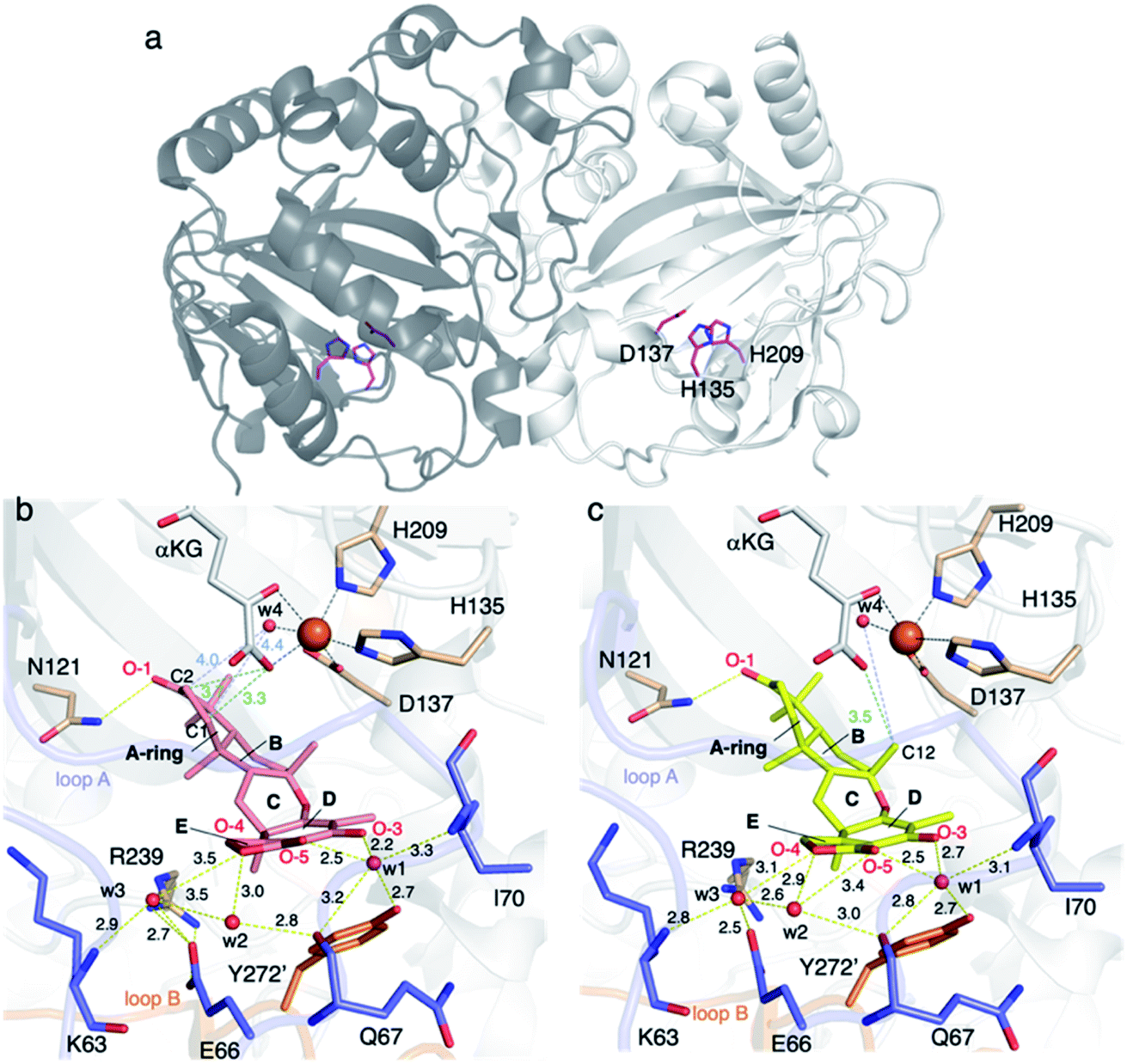 | ||
| Fig. 7 Structure of AndA. (a) The overall structure of AndA (PDBID: 5ZM2). The catalytic triad residues are shown in magenta sticks. (b) The active site architecture of AndA-Fe/αKG/17 (PDBID: 5ZM3). 17 and αKG are represented as salmon and white sticks, respectively. (c) The active site architecture of AndA-Fe/αKG/18 (PDBID: 5ZM4). 18 and αKG are represented as yellow and white sticks, respectively. Overall: Loop A and loop B are colored blue and orange, respectively. Yellow dashed lines show hydrogen bond interactions. Black dashed lines represent coordination of metal atoms. The orange spheres show iron atoms. The units of distance are Å. The oxygen and nitrogen atoms are colored red and blue, respectively. Red spheres show water molecules. | ||
In the isomerization reaction, which starts with the abstraction of H-12 of 18 which is bound to an enzyme via O-1, -3, -4, and O-5 like 17, the two distances from H-12 to O-2/αKG (3.5 Å) and to w4 (4.9 Å) support that the structure is reasonable for H-12 abstraction (Fig. 6 and 7c). To further investigate the dynamics of the isomerization, the DFT calculation was performed.28 After the H-12 abstraction of 18 to generate 18a, four reaction steps can be proposed (Fig. 8): (i) C8–O2 bond cleavage (18a to 18b), (ii) C12–C5′ bond formation (18b to 18c), (iii) C8–C2′ bond formation (18c to 18d), and (iv) quench of the C-7′ radical by ascorbic acid (18d to 19). In accordance with the prediction that the energetically stable tertiary carbon radical 18b is preferred, the calculated energy for the C–O bond cleavage is only 6.6 kcal mol−1. In another pathway, anti-Baldwin 5-end-trig cyclization proceeds to yield the α-oxy carbon radical intermediate 18b′ with an activation energy of 23.4 kcal mol−1, indicating that the pathway to 18b is more likely than this pathway. During the conversion from 18c to 18d, several conformational changes with minute activation barriers are expected to occur, and the C-8 radical approaches to the Michael acceptor carbon center C-2′. The radical at C-7′ in 18d is finally quenched by a reducing agent, resulting in the generation of 19.
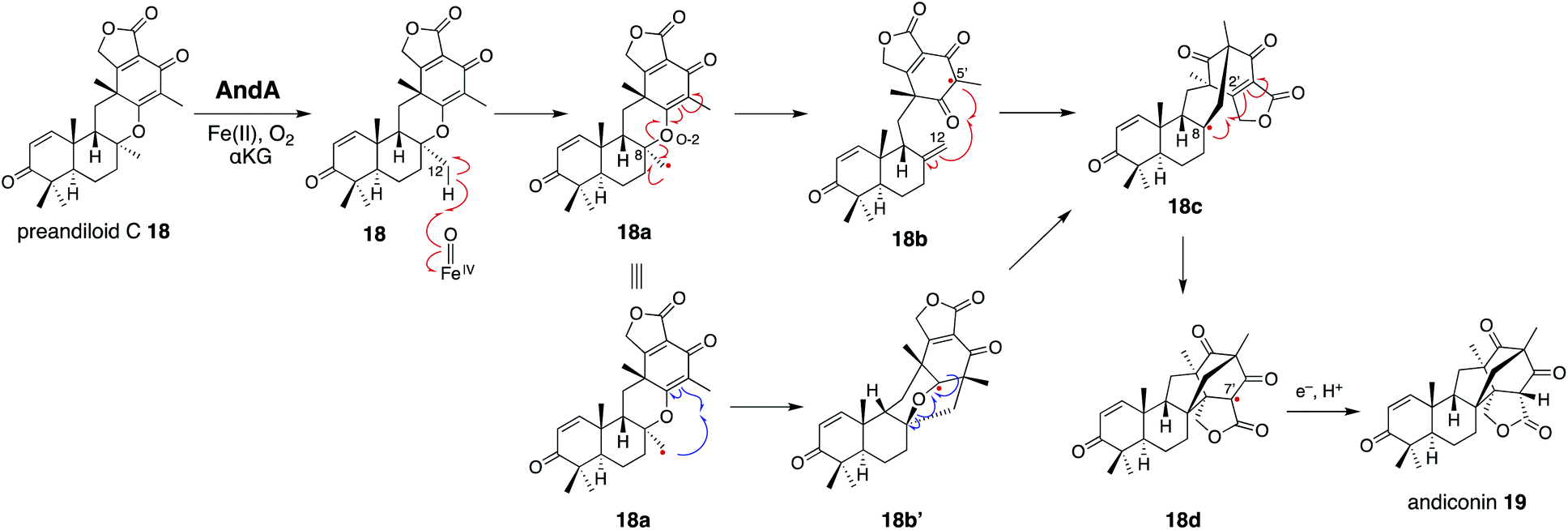 | ||
| Fig. 8 Proposed reaction mechanism of AndA to synthesize andiconin 19 from preandiloid C 18. | ||
To examine the roles of the amino acids in the active site of AndA, N121, R239, and Y272′ were substituted with alanine or apolar residues of similar bulkiness. The AndA N121A variant exhibited reduced activity, and the AndA R239A, R239M, R239V, and Y272A variants only yielded 18 and failed to produce 19. Because R239 and Y272′ interact with the lid-like loop region, these substitutions likely interrupted the sequential conformational changes required for the isomerization reaction. These observations indicated the importance of the lid region to maintain the reaction.
The combination of structural and computational studies nicely illustrates the mechanism of the isomerization enzyme reaction to produce the unique bicyclo[2.2.2]octane system. Like AusE and PrhA,24 AndA recognizes the A/B rings of 17 and 18 by hydrogen bonding with N121 and hydrophobic interactions, but not tight interactions as seen in those with the D ring, thus allowing the dynamic conformational changes of the intermediates during the enzyme reaction. Computational analyses of the enzyme-bound reaction intermediates will provide important information regarding how the enzyme manages the conformational changes of the substrate/intermediates. For example, the underlying reasons for the requirement of ascorbate in the in vitro enzyme reaction, the native reductant, and the mechanism of the reduction from 18d to 19 remain unknown. Similar reductive radical quenching is also observed in several unique αKG OX enzyme reactions, including FtmOx1 (endoperoxidase),30–32 CarC (epimerase),33 and SnoN (epimerase),34 but the mechanisms of their reductions remain obscure, even though they are essential for the functional conversions of αKG OXs from oxygenases to isomerases.
4 NvfI in novofumigatonin biosynthesis
Another remarkable αKG OX is NvfI, which installs the peroxide bridge on asnovolin A 20 to produce fumigatonoid A 21 in novofumigatonin 7 biosynthesis (Fig. 9).35 The quantification of αKG and oxygen consumption during the reaction suggested that one turnover of the NvfI reaction requires one equivalent of αKG and two equivalents of molecular oxygen.36 Labeling experiments using 18O2 and H218O indicated that two oxygen atoms at the endoperoxide moiety are derived from one molecular oxygen, and the one oxygen atom at the 3′-OH is derived from H218O (Fig. 9). The gel filtration analysis revealed that NvfI is the first monomeric αKG OX found in fungal meroterpenoid biosynthesis, similar to the αKG OXs in the biosyntheses of other secondary metabolites, such as deacetoxycephalosporin C synthase.37,38 Given that the previously described AusE/PrhA and AndA utilize the interface of the two monomers for substrate accommodation, the substrate binding manner of NvfI was expected to be different. The X-ray crystal structural analysis revealed that NvfI also adopts the DSBH fold, and its β1 and β3 strands unusually form a small β-barrel-like structure with three anti-parallel β-sheets (β4′, β5′, and β7′) (Fig. 10a). The comparison of the apo- and complex-structures revealed that S122–G128 on loop I and W199-P209 on loop II undergo large conformational changes, when substrate 20 is bound (Fig. 10b). As loop I alters its orientation toward the active site, F127 approaches the catalytic center along with the movement of W199. The side chain of R201 also changes its orientation, and newly interacts with Y116 and D206 via water molecules. E208 also moves and forms a new hydrogen bond with K205, thus increasing the cavity volume of the DSBH fold. | ||
| Fig. 9 Synthesis of fumigatonin A 21 from asnovolin A 20 by NvfI. | ||
 | ||
| Fig. 10 Structure of NvfI. (a) The overall structure of NvfI. The catalytic triad residues and αKG are shown with green sticks. The DSBH fold and the smaller β-barrel fold are colored gray and turquoise, respectively. (b) Comparison of the active site structures of NvfI apo (PDBID: 7ENB) and NvfI-Fe/αKG/20 (PDBID: 7DE2). 20 and αKG are shown with magenta and turquoise sticks, respectively. Loop regions in apo and complex structures are colored green and turquoise, respectively. Direction of the conformational changes of the loop regions are indicated by the blue arrows. (c) Close-up view of the active site of NvfI-Fe/αKG/20 (PDBID: 7DE2). 20 and αKG are shown as magenta and white sticks, respectively. Yellow dashed lines show the hydrogen bond interactions. Black dashed lines represent the coordination of the metal atom. The key distances between the iron atom and carbon atoms are shown with arrows. The units of distance are in Å. The oxygen and nitrogen atoms are colored red and blue, respectively. Red spheres show the water molecules. | ||
Notably, the distance from C-7′ of 20 to Fe(II) (4.2 Å) is shorter than the distance from C-13 (6.5 Å) in the complex structure (Fig. 10c), suggesting that this binding model is an artifact or represents the reaction of a C-7′ oxidation reaction to produce the derailment product 22 (Fig. 11). The docking simulation of the NvfI-αKG binary complex with 20 indicated that the enzyme accommodates 20 by adjusting its conformation so that C-13 is located closer to Fe(II) than C-7′. Furthermore, the comparison of the NvfI-αKG and NvfI-αKG/20 complex structures suggested that the movement of the loop around E208 increases the volume of the enzyme active site around the A-ring of 20, and the radical formation at C-13 and the following conformational changes move the substrate location deeper inside the tunnel (Fig. 10b).
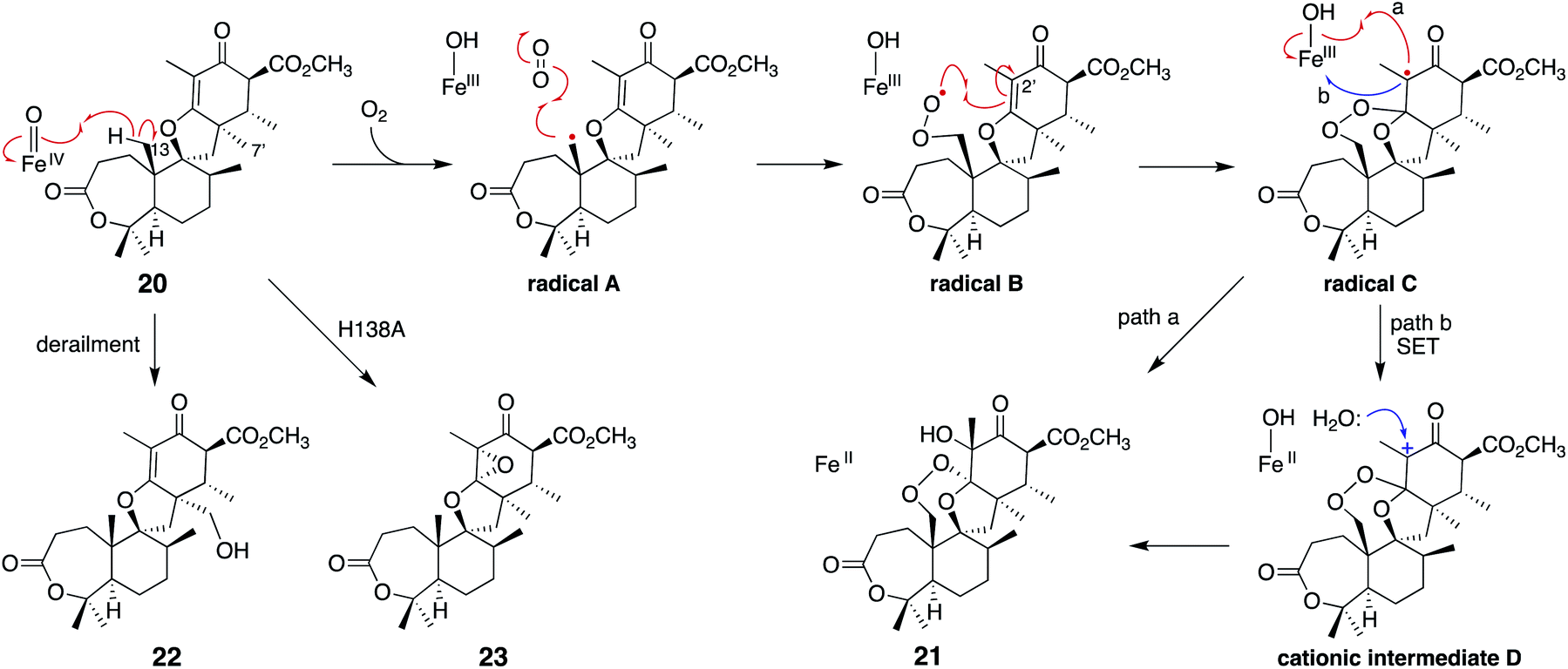 | ||
| Fig. 11 Proposed reaction mechanism of NvfI to synthesize 21 from 20. The structures of the derailment products 22 and the product 23 from the H138A variant reaction are also depicted. | ||
The substitution of the F127 on loop A and W199 on loop B lead to a loss of activity, which shows that these residues are essential for the endoperoxide forming activity. Their roles were proposed as gate-keepers that retain the correct conformation of 20. In other endoperoxide-forming enzymes; i.e., heme-dependent prostaglandin H synthase (PGHS, also known as cyclooxygenase, COX)39 and αKG OX fumitremorgin B endoperoxidase (FtmOx1),30–32 the active site tyrosine residue close to the Fe(IV)O was proposed to play a critical role to relay a radical from the reaction intermediate (Fig. 12). However, the substitution of the Y residues in the NvfI active site to F did not affect the reactivity. These data indicated that NvfI employs a distinct mechanism from those of PGHS and FtmOx1. Interestingly, NvfI H138A reduced the endoperoxide-forming activity by 20%, and formed newly generated 23 (Fig. 11), which indicated that the hydrogen bonding between H138 and the C-3 ester group is important to adjust the location of the substrate (Fig. 10c). Consequently, the reaction mechanism from 20 to 21 was proposed, as shown in Fig. 11. The Fe(IV)O species abstracts the hydrogen atom from C-13 to generate the primary radical A, which reacts with O2 to form the peroxide radical B. The generated radical B reacts with C-2′ to yield radical C, which then reacts with the Fe(III)-hydroxyl species to produce 21 (path a). It is also possible that electron transfer from the radical C to the ferric iron species generates the cationic intermediate D, and the successive attack by water yields 21 (path b). In path a, the oxygen ligands in the Fe(IV) and Fe(III) states should be exchanged with the solvent water during the catalysis as reported in the reaction of other αKG OXs.40 NvfI thus consumes only one molecule of αKG to produce one molecule of 21, as hypothesized in the reaction mechanism described above. The absolute configuration of OH-3′ is restricted to the R-configuration, indicating that the enzyme controls the orientation of the hydroxyl-rebound or the addition of water.
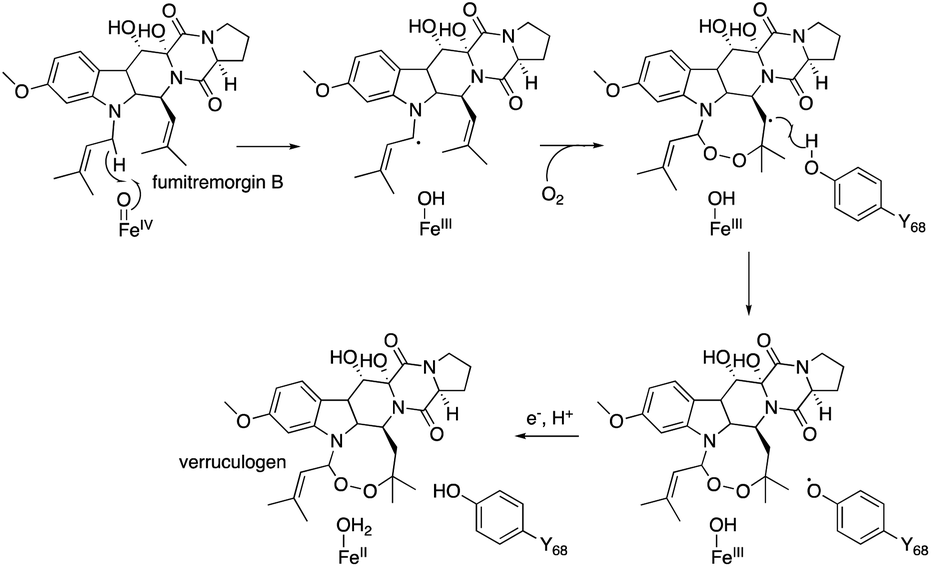 | ||
| Fig. 12 The reaction mechanism of FtmOx1 to synthesize verruculogen from fumitremorgin B. | ||
The structural analysis of NvfI revealed how the monomeric αKG OX accepts the substrate with the conformational changes of loop I and II. More importantly, this study proposed a new mechanism of the rare enzymatic endoperoxide formation reaction without the help of the tyrosine residue for the radical relay, although the possibility that other residues may work in the relay cannot be excluded. NvfI may suppress the direct oxygen rebound between the radical A and Fe(III)–OH by facilitating the large conformational changes of the radical A. The prolonged lifetime of the radical intermediates may lead to the reaction with molecular oxygen, although the mechanism of molecular oxygen retention in the enzyme remains enigmatic. This hypothesis of oxygen rebound prevention could be applicable to other non-hydroxylating αKG OX enzyme reactions.12–16 This study provided useful information for future enzyme engineering to introduce unique chemical scaffolds into the products.
5 TlxI-J in talaromyolides biosynthesis
Four αKG OXs – TlxA, C, I, and J – were identified in the biosynthesis of 6/6/6/6/6/6 hexacyclic meroterpenoids, talaromyolides 24a–f and 25 (Fig. 13).41,42 Their functions were analyzed by in vitro assays with 26 as a substrate, which showed that TlxJ oxidizes the C-9α of 26 to yield 27, but TlxA, C, I, and J were all inactive toward 27 when assayed individually.43 The bioinformatic analysis indicated that TlxI lacks an arginine residue for αKG binding,12 and the loops of TlxI and TlxC are shorter than those of other fungal αKG OXs. These analyses suggest that TlxI and TlxC lack oxygenase functions and play auxiliary roles. In fact, the reaction with TlxI-J consumed 27 to produce talaromyolide G 24a, talaromyolide C 24d, talaromyolide K 25, novel compounds 28, and 29 (Fig. 13 and 14). The 4,5-secodriman products 24a, 24d, 28, and 29 were generated by a retro-aldol reaction from the trans-drimane intermediate, and the hemiketal product 25 was produced in the reaction between the 9α-OH and 3-keto groups in the cis-drimane intermediate (Fig. 14a). Similarly, TlxA-C work together to hydroxylate 24a and 24d to produce 24b and 24e, respectively.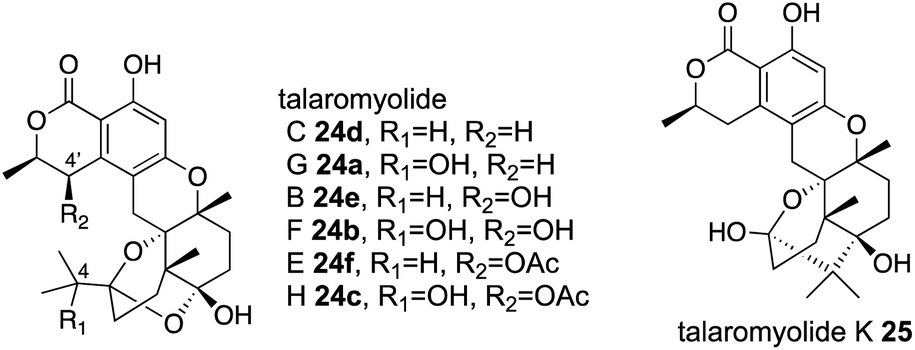 | ||
| Fig. 13 Chemical structures of talaromyolides 24a–f, 25. | ||
 | ||
| Fig. 14 Reactions of monomer TlxJ and heterodimer TlxI-J to synthesize talaromyolides 24a, 24b, 24d, 24e, and 25, and unnatural analogs 28 and 29. | ||
The reaction pathway from 27 to 24a, 24d, and 25 was proposed, as described below. First, TlxI-J abstracts H-5 to give the C-5a hydroxylated trans-drimane intermediate (Fig. 14a). The deprotonation of the OH-5a leads to the retro-aldol reaction which cleaves the C4–C5 bond, as proposed in yaminterritrem B biosynthesis,44 and the resultant intermediate undergoes successive ketal formation to produce 24d. The hydroxylation at C-4 of the retro-aldol reaction product generates 24a. It is intriguing that 25 is derived from the cis-drimane intermediate, while its yield is lower than those of the other products. After radical formation at C-5, the trans-drimane is likely transformed into cis-drimane before the oxygen rebound.
Interestingly, the unnatural meroterpenoids 27 and 28 were also observed in the TlxI-J reaction (Fig. 14b). They are expected to be generated via repeat demethylations of C-14 and C-15 through sequential oxygenation from the methyl group to the carboxylic acid followed by decarboxylation, as in cholesterol biosynthesis45 and terpenoid secondary metabolite biosynthesis.46,47 The successive C-3 demethylation possibly proceeds via oxidative decarboxylation, as in αKG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18,19 and 4-hydroxyphenylpyruvate decarboxylation.48,49
18,19 and 4-hydroxyphenylpyruvate decarboxylation.48,49
The gel filtration analyses of TlxI-J and TlxA-C revealed that they form unprecedented αKG OX enzyme heterodimers.12,13 The amounts of TlxJ and TlxA purified from an Escherichia coli expression system are low when they are solely expressed, but they are efficiently produced as a monomer or heterodimer when co-expressed with TlxI or TlxC. Together, these data indicate that TlxI and TlxC serve as chaperone-like proteins to increase the stability of TlxJ and TlxA, respectively. Interestingly, TlxC cannot be substituted for TlxI, and vice versa, indicating the rigid specificity for the heterodimer formation.
To investigate the molecular basis of the unprecedented αKG OX heterodimer, the X-ray crystal structure of TlxI-J complexed with Fe/N-oxalylglycine (NOG)50 was analyzed.43 TlxJ in the heterodimer adopts the conserved DSBH fold, while TlxI does not possess a complete DSBH fold. In fact, loop A′ in TlxI is much shorter than the corresponding region of TlxJ (Fig. 15a). These observations suggest that the reaction chamber cannot be constructed in the TlxI monomer. Thus, in analogy with the other homodimeric αKG OXs such as PrhA and AndA,11,16 a funnel-like substrate binding site is expected to be constructed with the DSBH fold (TlxJ) as a chamber, and loop A (TlxJ) and loop B′ (TlxI) as a lid. The metal binding site (H136, D138, and H213) and αKG binding site (R224, N133, and T173) are conserved in the TlxJ structure, but the residue corresponding to R224 of TlxJ is substituted with S211 in TlxI (Fig. 15b and c), indicating that TlxI cannot bind αKG.
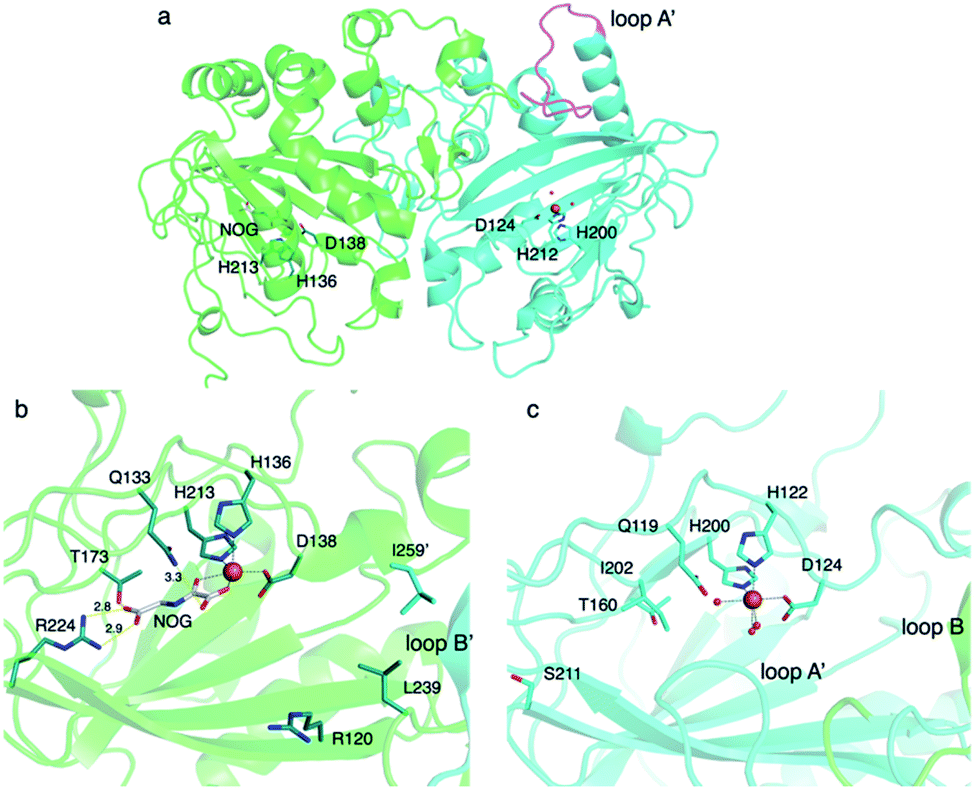 | ||
| Fig. 15 Structure of TlxI-J. (a) The heterodimer structure of TlxI-J-Fe/NOG (PDBID: 7VBQ). The structures of TlxI and TlxJ are colored turquoise and green, respectively. The catalytic triad residues and NOG in TlxJ are shown as green and white sticks, respectively. The catalytic triad residues in TlxI are shown as dark turquoise sticks. Loop A′ is highlighted in magenta. The corresponding loop region in TlxJ is disordered (b and c). Active site structures of TlxJ (green) and TlxI (turquoise). Active site residues in TlxJ and TlxI are represented in green and turquoise sticks. NOG is shown as a white stick. Yellow dashed lines show hydrogen bond interactions. Black dashed lines represent the coordination of the metal atom. The orange spheres show iron atoms. The units of distance are Å. The oxygen and nitrogen atoms are colored red and blue, respectively. Red spheres show water molecules. | ||
The substitution of the amino acid residues oriented toward the active site of TlxJ, including R120 and L239 located on the DSBH core and I259′ located on loop B′, significantly reduced the reactivity, while these variants maintained the heterodimer structure as confirmed by gel filtration, suggesting that these residues are required for substrate binding. Interestingly, TlxJL239A-I newly accumulated the C-1 hydroxylated 27, indicating that the substitutions in the hydrophobic reaction chamber are effective to alter the reaction profile, as observed in PrhA/AusE and AndA.24,28
To investigate the differences between the heterodimer and homodimer structures, the amino acid residues located at the interface of TlxI-J were substituted. There are several amino acid pairs responsible for the hydrogen bonding, including R63′ and S276, T224′ and F236 (main chain), R135′ and D152, and W262′ and P238 (Fig. 16a). As expected, the TlxI R63′A, T224′A, R135′A, and W262′A variants no longer form a heterodimer with TlxJ, as evidenced by the pull-down assays, indicating that they are important for the interaction between monomers. Comparison of the structure of TlxI-J with those of PrhA and AndA demonstrated that the method of heterodimer formation is quite different from that of homodimer formation (Fig. 16b and c).
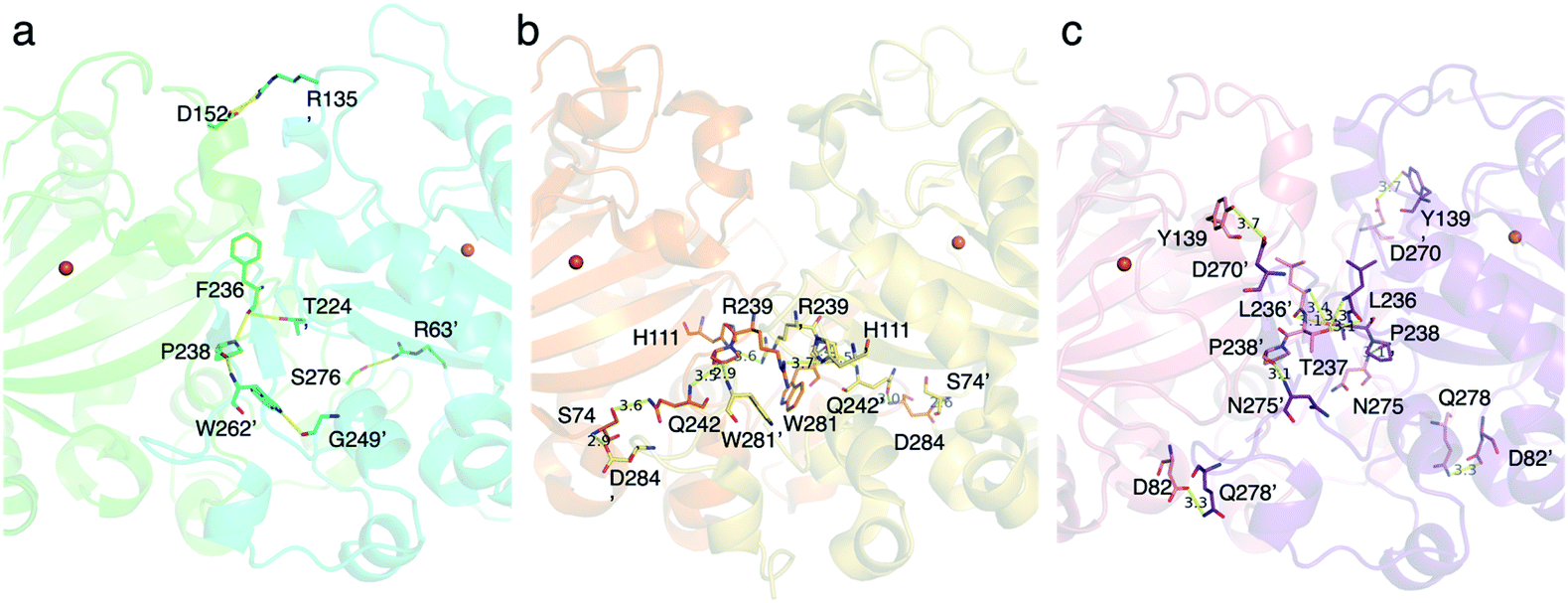 | ||
| Fig. 16 The amino acid residues located at the interface between two monomers of the dimers (a–c), close-up view of the interface between (a) TlxI and TlxJ heterodimer (PDBID: 7VBQ) colored turquoise and green, respectively, (b) the homodimer PrhA (PDBID: 5YBO) colored orange and yellow, and (c) the homodimer AndA (PDBID: 5ZM3) colored salmon and violet. Overall: the amino acid residues which form hydrogen bond interaction are represented as sticks. Hydrogen bond interactions are shown as yellow dashed lines. The orange spheres show iron atoms. The units of distances are Å. The oxygen and nitrogen atoms are colored red and blue, respectively. | ||
The reason why TlxJ requires TlxI to form a functional heterodimer remains to be elucidated. Since TlxJ exists as a monomer when it is produced in E. coli, it is likely that TlxJ cannot form a homodimer by itself. The TlxJ monomer can oxidize 26, although this reaction is much slower than that of TlxI-J. These results indicate that the TlxJ monomer can also form a premature reaction cavity, but it requires TlxI to form a reaction cavity that can accept 27. The CRISPR-based gene inactivation of TlxI prevented the production of talaromyolides and accumulated 27, implying that TlxI-J also forms a heterodimer in vivo. This study presented the characterization of the first heterodimeric αKG OXs. The interaction of each monomer in the heterodimer is quite different from that in the homodimer. Given that TlxI-J can catalyze a high number of oxygenations (possibly 11 times to synthesize 28 and 29), the heterodimer structure might decrease the rigidity of substrate recognition. If two αKG OXs are artificially complexed as a heterodimer, a multifunctional heterodimer like TlxI-J could be created. The simulation showing the creation of the cis-drimane intermediate in the enzyme after the C-5 radical intermediate would also be intriguing.
6 SptF in emeridons biosynthesis
SptF is an αKG OX that reportedly catalyzes two oxidation steps from andiconin D 31 to produce 32 in the biosynthesis of emeridone F 4.51 SptF hydroxylates C-11, followed by water elimination, carbon skeletal rearrangement, and deprotonation to yield 33, and epoxidizes the C-3′-9′ double bond of 33 to yield 32 (Fig. 17). Recent studies revealed that SptF further oxidizes 32 to give two products: 34 with an additional C-9 hydroxyl group and C1–C2 epoxide, and 35 with a new ether bridge between C-4′ and C-2′, a hydroxyl group at C-5′, and a double bond between C-7 and C-8.51 In accordance with the high sequence similarity between SptF and AndF (94%), which is involved in the formation of anditomin 5 from andilesin C 36,22 SptF also accepts andilesin C 36 to produce two products 37 and 38, which possess an epoxide group between C-3′ and C-9′ with two different absolute configurations (Fig. 18a).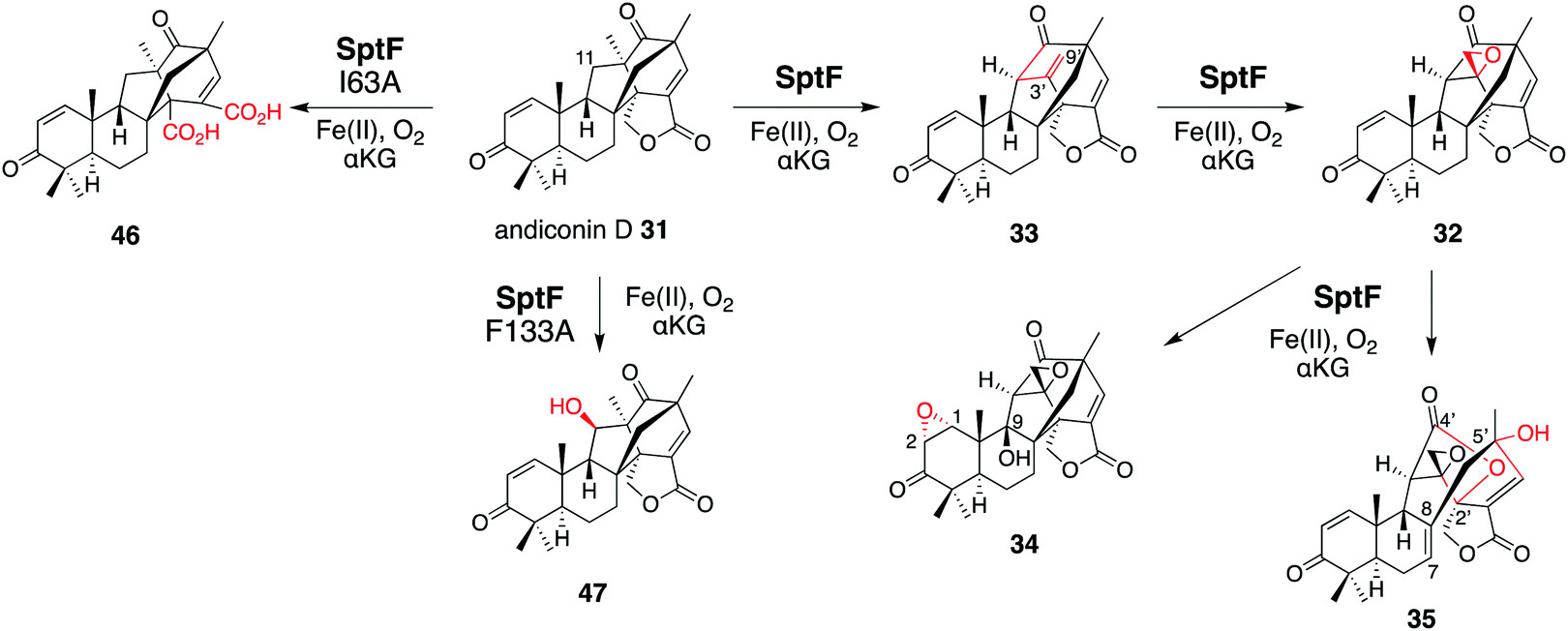 | ||
| Fig. 17 Synthetic reactions from andiconin D 31. The syntheses of 34 and 35 by SptF WT, the synthesis of 46 by SptF I63A, and the synthesis of 47 by SptF F133A. | ||
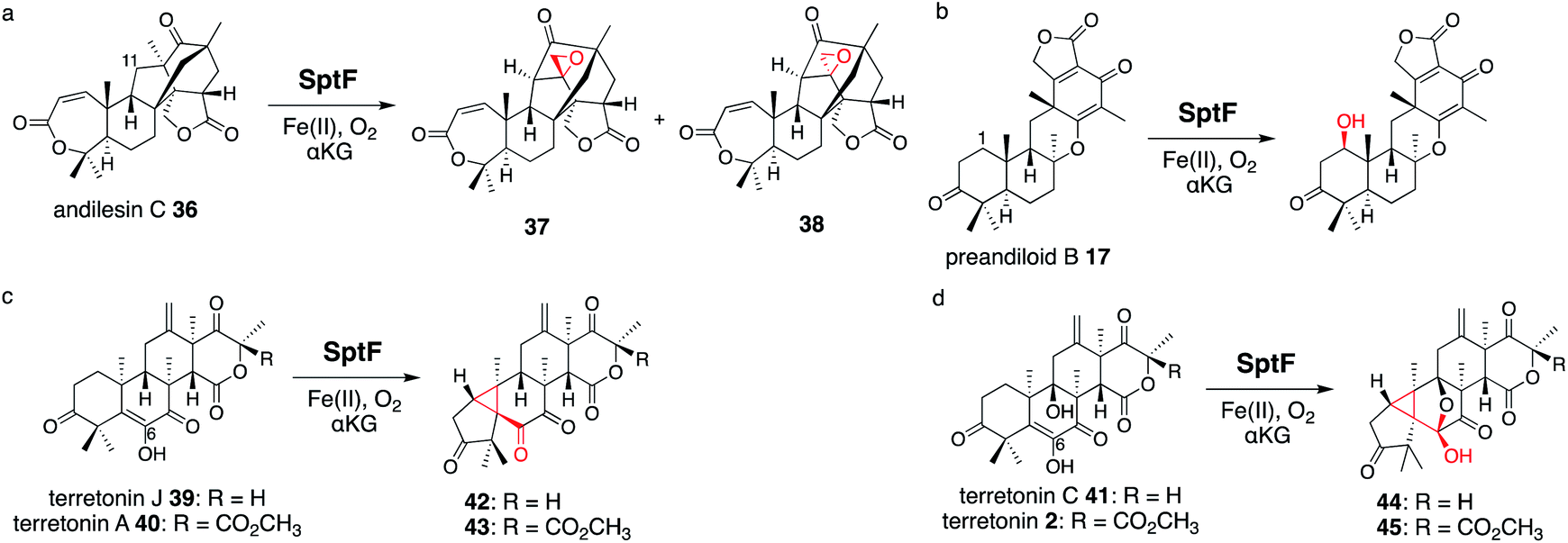 | ||
| Fig. 18 Synthesis of unnatural products by SptF from andilesin C 36 (a), preandiloid B 17 (b), terretonin J 39 and A 40 (c), and terretonin C 41 and terretonin 2 (d). | ||
Inspired by its catalytic versatility, the substrate scope of SptF was further investigated using six fungal meroterpenoids as unnatural substrates, including preandiloid B 17, preandiloid C 18, terretonin J 39, terretonin A 40, terretonin C 41, and terretonin 2 (Fig. 18b–d). Except for 18, all of the tested substrates were accepted by SptF, indicating that SptF has relaxed substrate specificity. SptF catalyzed the C-1 hydroxylation of 17 (Fig. 18b), and reacted with 39 and 40 to generate 42 and 43 possessing the 5/3/6/6/6 scaffold, and the reaction with 41 and 2 to yield 44 and 45 possessing the 5/3/5/5/6/6 scaffold, respectively (Fig. 18c and d). The consumption rates of 40 and 2 by SptF were lower than 10%, indicating that the bulky functional group at the D-ring hampers the reaction. SptF also oxidizes various kinds of steroids, including androsterone, testosterone, and progesterone, with moderate catalytic efficiencies (34–53%).
To determine the molecular basis of the exceptional promiscuity of SptF,51,52 its X-ray crystallographic structure was analyzed. SptF alone exhibits the conserved DSBH fold (Fig. 19a). The complex structures with 31 and 36 showed that these two substrates are located at almost identical positions, indicating that the A-ring of the substrates does not alter the binding manner. The distances from the iron to the initial reaction site C-11 of 31 and 36 are 4.2 and 4.3 Å, respectively (Fig. 19b and c), and are reasonable distances for the hydrogen abstraction. The hydrophobic surface, consisting of I63, F133, and I231, mainly supports the substrate in the enzyme cavity. The lid-like region interacts with the A-rings of 31 and 36via hydrogen bonding with only N65, while their E rings are supported by S114, the main chain of L199, and T148. The loose interaction with the lid-like loop explains the broad substrate specificity toward structurally diverse compounds. Interestingly, the unnatural substrate terretonin C 41 was also accepted by SptF in a different ligand binding mode, with the direct hydrogen bond interaction between the D-ring and N150 and the indirect hydrogen bond network via water molecules supported by T148, D130, and L199 (Fig. 19d). Remarkably, the lid-like loop region was not observed in the complex structure with 41, while the conformation of the other active site regions are almost identical. In this structure, the substrate is mainly supported by hydrophobic interactions with F133 and I231, which are close to the C-8, C-10, and C-13 methyl groups. The C-6 enol of 41 is closest to the iron (3.7 Å), suggesting that the formation of the cyclopropane rings of 42, 43, 44, and 45 are triggered by the hydrogen bond abstraction from this enol.
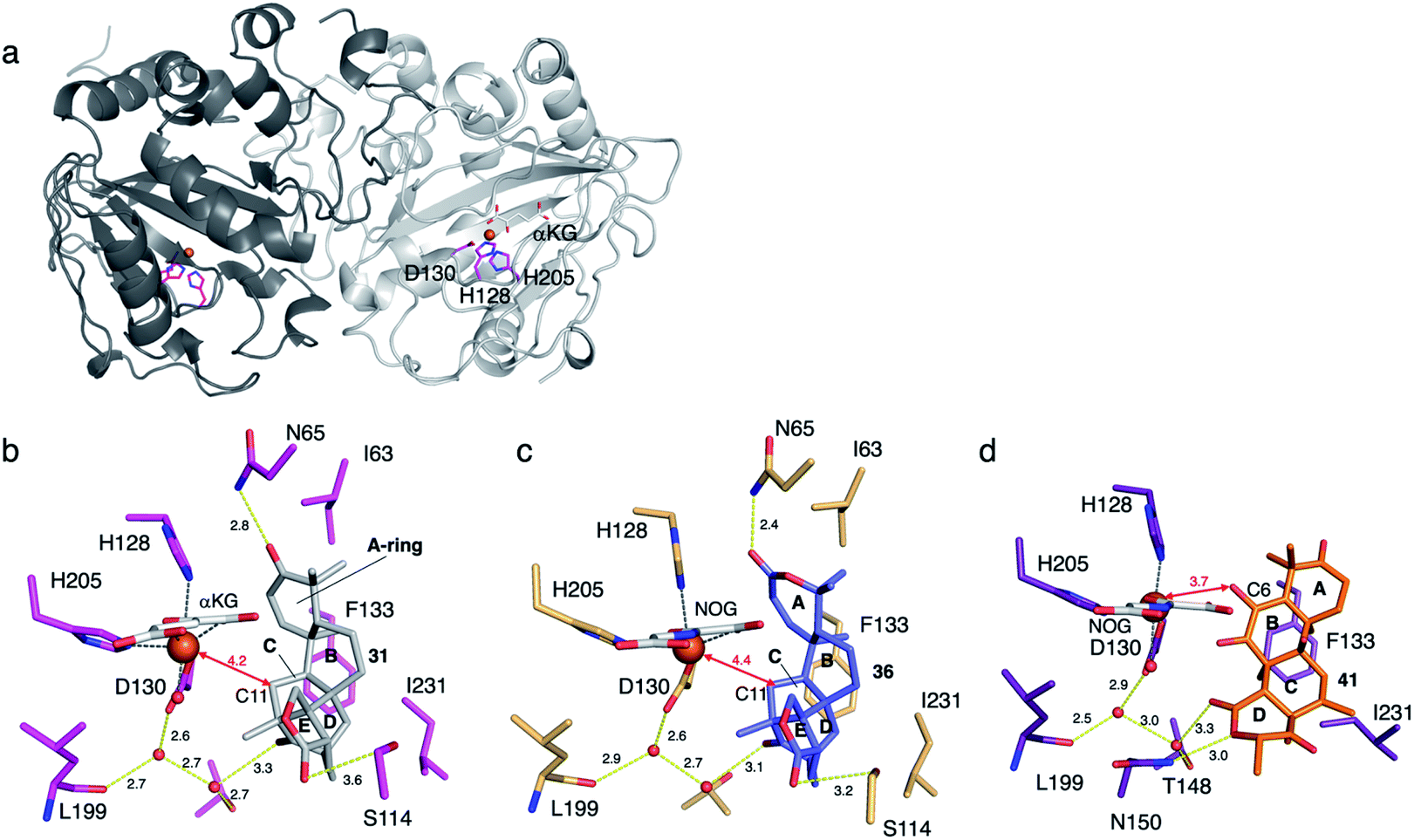 | ||
| Fig. 19 Structure of SptF. (a) The overall structure of SptF (PDBID: 7EYS). The catalytic triad residues are shown with magenta sticks. (b) Active site structures of SptF-Fe/αKG/31 (PDBID: 7EYS). 31 and αKG are shown with grey and white sticks, respectively. (c) Active site structures of SptF-Fe/NOG/36 (PDBID: 7EYT). 36 and NOG are shown as blue and white sticks, respectively. (d) Active site structures of SptF-Fe/NOG/41 (PDBID: 7EYW). 41 and NOG are shown as orange and white sticks, respectively. Yellow dashed lines show hydrogen bond interactions. Black dashed lines represent the metal atom coordination. The iron atom is represented by an orange sphere. The key distances between the iron atom and carbon atoms are shown as arrows. The units of distance are Å. The oxygen and nitrogen atoms are colored red and blue, respectively. | ||
To investigate the role of I63, F133, and I231, each was substituted with alanine. As a result, SptF I63A prevented the production of 34 and 35, but produced the novel product 46 (Fig. 17). The structure of 46 was determined to be an E-ring opened dicarboxylic acid, which is generated through two rounds of oxidation at the C-1′ of 31. In contrast, the SptF I63A and F133A variants almost completely lost their ability to produce 34 and 35, and accumulated 32 and 33, respectively. The SptF F133A variant also converted 31 into the C-11 hydroxylated 47 (Fig. 17). These data indicated the crucial role of hydrophobic interactions for substrate binding and the potential to engineer the enzyme reaction by site-directed substitutions of the hydrophobic residues.
The importance of the hydrophilic residues, including N65, S114, T148, and N150, was also investigated in detail. N65A significantly reduced the activity by 80% and accumulated 33. The SptF T148A, S114A, and N150A variants decreased the production efficiencies of 34 and 35 by 7, 17, and 48%, and changed the production ratios of 34:35 to 7:3 for the S114A and T148S variants and 1:9 for the N150A variant, as compared with 4:6 for SptF WT. These observations indicate that the three hydrophilic residues are important to define the orientation of 33 within the reaction chamber. When N65 was substituted with threonine, which is the corresponding residue in the closely related AndF, the variant produced 35 as a major product from 31, with a smaller amount of 34. In addition to the hydrophobic and hydrophilic interactions, the functions of I63 and N65 in the lid-like loop region were investigated. Remarkably, the SptF I63A and N65A variants accepted 41 with similar efficiencies to the wild-type enzyme, while the reactivity on 31 was 14 times lower, suggesting the different substrate binding modes between the two substrates. To evaluate the importance of the flexible lid-like loop region, the loop was truncated by 6 residues (Δ6, H61-V66), 9 residues (Δ9, K58-V66), 16 residues (Δ16, N54-K69), and 19 residues (W53-K71). The Δ6, Δ9, and Δ16 truncated SptFs were reacted with 31, 32, and 33. Interestingly, these variants lost the activities of 31 and 32, and generated several minor products, but no longer produced 33–35. In contrast, these variants showed 26–52% activity toward 33, and generated small amounts of 34 and 35. This observation suggested the importance of the lid-like region to control the reaction. While the unnatural meroterpenoid substrates were accepted by the variants to a lower extent, the steroidal substrates were no longer accepted.
Based on the investigations of the structure–function relationship, the reaction mechanisms for the SptF-catalyzed oxidation reactions were proposed (Fig. 20). Hydrogen abstraction at C-11 of 31 by the Fe(IV)O species firstly generates a radical, which undergoes the cleavage of the C-4′–C-3′ bond and the construction of the C-4′–C-11 bond. The following H-9′ abstraction initiates the formation of the exo-methylene 33, and the successive epoxidation produces 32. In pathway a, the second epoxidation of C-1–C-2 and the H-9 abstraction, followed by the rebound of the hydroxyl group, produced 34 as a product. In pathway b, H-7′ is abstracted to induce the cleavage of the C-8–C-2′ bond and generate the double bond between C-7 and C-8. The hydroxyl rebound between the radical at C-2′ and the Fe(III)-hydroxyl species yields IM2. The 2′-OH reacts with the 4′-carbonyl and the subsequent electron migration results in the protonation of C-7′ to generate IM3. The H-7′ in IM3 is abstracted by the Fe(IV)O species in the next round of oxidation, and the generated allylic radical receives a hydroxyl group at C-5′ from the Fe(III)-OH species to produce 35.
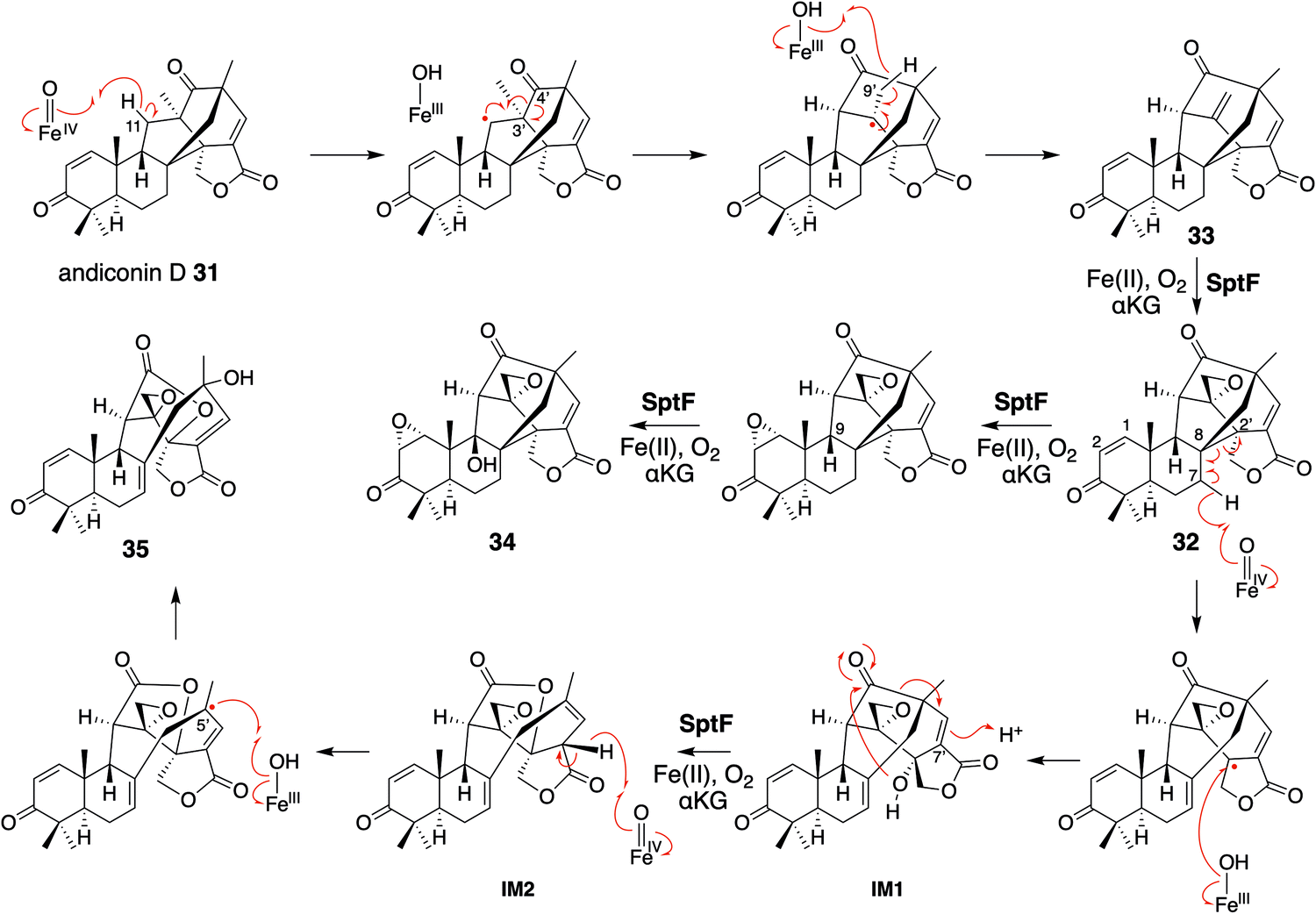 | ||
| Fig. 20 Proposed reaction mechanisms of SptF in the syntheses of 34 and 35 from andiconin D 31. | ||
SptF accepts a remarkably broad range of meroterpenoid and steroid substrates. The solved complex structures revealed the different substrate binding mode for each substrate. Compound 41 can bind to the enzyme in the inverse manner, as compared with those for 31 and 36. SptF also utilizes the loop region as a lid for the active site cavity, but it does not appear to bind the substrate tightly. The relaxed binding mode of the loop region and the hydrophobic reaction chamber would allow the promiscuous substrate binding.
7 Summary and future perspectives
In summary, the multifunctional fungal αKG OX enzymes catalyze remarkable chemistries, and often induce dramatic changes in molecular scaffolds, depending on the stereoelectronic character of the substrate/intermediates and the resulting conformational changes/movements of the active site of the enzyme. The X-ray structural analyses of the enzymes revealed which amino acid residues are responsible for the substrate binding as well as the active site architecture to accommodate the bulky terpenoid and polar polyketide moieties. The reaction chambers of αKG OXs are mainly composed of hydrophobic amino acids, which enable the accommodation of the terpene moiety and multiple oxidation reactions (Fig. 21). The structure of the flexible lid-like loop regions of the active site are significantly different in each enzyme. By manipulating the structure of the reaction chamber and the lid-like loop regions, their reactions could be modified.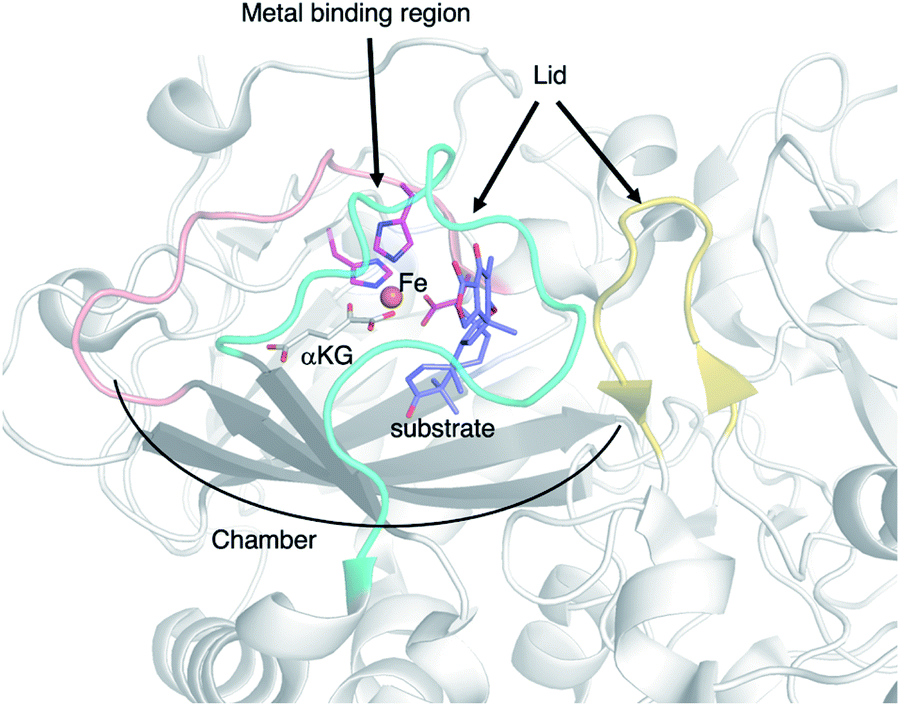 | ||
| Fig. 21 The important structural features of the αKG OX enzyme. | ||
To obtain more versatile and useful αKG OX enzymes, further search for new enzymes that catalyze novel reactions or exhibit unique substrate specificities is still needed. The genome mining method would be helpful in searching for such enzymes.53,54 In the study of the fungal αKG OX AsqJ in the quinolone alkaloid biosynthesis, various research techniques have been applied to elucidate its stepwise desaturation and epoxidation reactions (e.g. X-ray crystallography, pre-steady-state enzyme kinetics, Mössbauer spectroscopy, assays with unnatural substrates, DFT calculations, and molecular dynamic simulations).29,55–60 Likewise, studies on structural biology, bioorganic chemistry, and computational chemistry will provide important information for enzyme engineering of αKG OXs. Future development of computational calculations of catalytic reactions is expected to enable highly accurate simulations of enzyme dynamics, which will provide useful insight for more rigorous control of enzyme reactions and the creation of biocatalysts by de novo design. In addition, a recent engineering study to convert an αKG-dependent amino acid hydroxylase into a halogenase suggested some strategies to evolve the αKG OXs towards enzymes with different catalytic functions.61 During the writing of this manuscript, two studies based on the saturated random mutagenesis to evolve αKG OXs in meroterpenoid biosynthesis were published.62,63 Such random mutagenesis-based studies complement the rational enzyme engineering. Finally, the engineered enzymes can be utilized in combinatorial biosyntheses in heterologous expression hosts such as Aspergillus oryzae, to produce pharmaceutically useful molecules, as demonstrated by the recent successful productions of unnatural novel diterpene fungal meroterpenoids.64–66 Therefore, the discovery, engineering, and reconstitution of fungal meroterpenoid oxidation pathway will continue to provide important insight into natural product chemistry and enzymology, and will lead to the production of beneficial drugs.
8 Conflicts of interest
There are no conflicts to declare.9 Acknowledgements
The authors would like to express sincere appreciation to an excellent group of coworkers whose contributions are cited in the text, in particular to Dr Yudai Matsuda (City University of Hong Kong). This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI grant number JP16H06443, JP17H04763, JP18K19139, JP19H04641, JP19K15703, JP20H00490, JP20KK0173, JP20K22700, JP21H02636, JP21K14744, and JP21K18246), the New Energy and Industrial Technology Development Organization (NEDO, grant number JPNP20011), AMED (grant number JP21ak0101164), UTEC-UTokyo FSI Research grant program, the PRESTO and ACT-X Programs from Japan Science and Technology Agency, Fuji Foundation for Protein Research, Japan Foundation for Applied Enzymology, Kato Memorial Bioscience Foundation, The Asahi Glass Foundation, and Naito foundation.10 Notes and references
- R. Geris and T. J. Simpson, Nat. Prod. Rep., 2009, 26, 1063–1094 RSC
.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC
- Y. Matsuda, T. Awakawa, T. Mori and I. Abe, Curr. Opin. Chem. Biol., 2016, 31, 1–7 CrossRef CAS
- T. Awakawa and I. Abe, J. Fungi, 2021, 7, 486 CrossRef CAS
- H. Tao and I. Abe, Curr. Opin. Biotechnol., 2021, 69, 52–59 CrossRef CAS PubMed
- L. Barra and I. Abe, Nat. Prod. Rep., 2021, 38, 566–585 RSC
- X. Zhang, J. Guo, F. Chenga and S. Li, Nat. Prod. Rep., 2021, 38, 1072–1099 RSC
- H. Nakamura, Y. Matsuda and I. Abe, Nat. Prod. Rep., 2018, 35, 633–645 RSC
- M. C. Tang, Y. Zou, K. Watanabe, C. T. Walsh and Y. Tang, Chem. Rev., 2017, 117, 5226–5333 CrossRef CAS
- R. Cox, Nat. Prod. Rep., 2014, 31, 1405–1424 RSC
- T. Mori, T. Iwabuchi, S. Hoshino, H. Wang, Y. Matsuda and I. Abe, Nat. Chem. Biol., 2017, 13, 1066–1073 CrossRef CAS
- W. Aik, M. A. McDonough, A. Thalhammer, R. Chowdhury and C. J. Schofield, Curr. Opin. Struct. Biol., 2012, 22, 691–700 CrossRef CAS PubMed
- S. Islam, T. M. Leissing, R. Chowdhury, R. J. Hopkinson and C. J. Schofield, Annu. Rev. Biochem., 2018, 87, 585–620 CrossRef
- S. S. Gao, N. Naowarojna, R. Cheng, X. Liu and P. Liu, Nat. Prod. Rep., 2018, 35, 792–837 RSC
- C. R. Zwick and H. Renata, Nat. Prod. Rep., 2020, 37, 1065–1079 RSC
- I. Abe, Chem. Pharm. Bull., 2020, 68, 823–831 CrossRef CAS
- L. F. Wu, S. Meng and G. L. Tang, Biochim. Biophys. Acta, Proteins Proteomics, 2016, 1864, 453–470 CrossRef CAS PubMed
- X. Huang and J. T. Groves, J. Biol. Inorg Chem., 2017, 22, 185–207 CrossRef CAS PubMed
- C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. B. Jr, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS
- J. Li, H. J. Liao, Y. Tang, J. L. Huang, L. Cha, T. S. Lin, J. L. Lee, I. V. Kurnikov, M. G. Kurnikova, W. C. Chang, N. L. Chan and Y. Guo, J. Am. Chem. Soc., 2020, 142, 6268–6284 CrossRef CAS
- R. Bunno, T. Awakawa, T. Mori and I. Abe, Angew. Chem., Int. Ed., 2021, 60, 15827–15831 CrossRef CAS
- Y. Matsuda, T. Awakawa, T. Wakimoto and I. Abe, J. Am. Chem. Soc., 2013, 135, 10962–10965 CrossRef CAS PubMed
- Y. Matsuda, T. Iwabuchi, T. Fujimoto, T. Awakawa, Y. Nakashima, T. Mori, H. Zhang, F. Hayashi and I. Abe, J. Am. Chem. Soc., 2016, 138, 12671–12677 CrossRef CAS PubMed
- Y. Nakashima, T. Mori, H. Nakamura, T. Awakawa, S. Hoshino, M. Senda, T. Senda and I. Abe, Nat. Commun., 2018, 9, 104 CrossRef PubMed
- P. Schrepfer, A. Buettner, C. Goerner, M. Hertel, J. Van Rijn, F. Wallrapp, W. Eisenreich, V. Sieber, R. Kourist and T. Brück, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E958–E967 CrossRef CAS PubMed
- H. Sato, K. Narita, A. Minami, M. Yamazaki, C. Wang, H. Suemune, S. Nagano, T. Tomita, H. Oikawa and M. Uchiyama, Sci. Rep., 2018, 8, 1–9 Search PubMed
- Y. Matsuda, T. Wakimoto, T. Mori, T. Awakawa and I. Abe, J. Am. Chem. Soc., 2014, 136, 15326–15336 CrossRef CAS
- Y. Nakashima, T. Mitsuhashi, Y. Matsuda, M. Senda, H. Sato, M. Yamazaki, M. Uchiyama, T. Senda and I. Abe, J. Am. Chem. Soc., 2018, 140, 9743–9750 CrossRef CAS PubMed
- H. J. Liao, J. Li, J. L. Huang, M. Davidson, I. Kurnikov, T. S. Lin, J. L. Lee, M. Kurnikova, Y. Guo, N. L. Chan and W. C. Chang, Angew. Chem., Int. Ed., 2018, 57, 1831–1835 CrossRef CAS PubMed
- W. Yan, H. Song, F. Song, Y. Guo, C. H. Wu, A. Sae Her, Y. Pu, S. Wang, N. Naowarojna, A. Weitz, M. P. Hendrich, C. E. Costello, L. Zhang, P. Liu and Y. Jessie Zhang, Nature, 2015, 527, 539–543 CrossRef CAS
- N. P. Dunham, J. M. Del Río Pantoja, B. Zhang, L. J. Rajakovich, B. D. Allen, C. Krebs, A. K. Boal and J. M. Bollinger, J. Am. Chem. Soc., 2019, 141, 9964–9979 CrossRef CAS
- L. Wu, Z. Wang, Y. Cen, B. Wang and J. Zhou, Angew. Chem., Int. Ed., 2022, 134, e202112063 Search PubMed
- W. C. Chang, Y. Guo, C. Wang, S. E. Butch, A. C. Rosenzweig, A. K. Boal, C. Krebs and J. M. Bollinger, Science, 2014, 343, 1140–1144 CrossRef CAS PubMed
- V. Siitonen, B. Selvaraj, L. Niiranen, Y. Lindqvist, G. Schneider and M. Metsä-Ketelä, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5251–5256 CrossRef CAS
- Y. Matsuda, T. Bai, C. B. W. Phippen, C. S. Nødvig, I. Kjærbølling, T. C. Vesth, M. R. Andersen, U. H. Mortensen, C. H. Gotfredsen, I. Abe and T. O. Larsen, Nat. Commun., 2018, 9, 2587 CrossRef
- T. Mori, R. Zhai, R. Ushimaru, Y. Matsuda and I. Abe, Nat. Commun., 2021, 12, 4417 CrossRef CAS PubMed
- V. J. Chen, A. M. Orville, M. R. Harpel, C. A. Frolik, K. K. Surerus, E. Munck and J. D. Lipscomb, J. Biol. Chem., 1989, 264, 21677–21681 CrossRef CAS
- J. E. Dotzlaf and W. K. Yeh, J. Biol. Chem., 1989, 264, 10219–10227 CrossRef CAS
- W. A. Van der Donk, A. L. Tsai and R. J. Kulmacz, Biochemistry, 2002, 41, 15451–15458 CrossRef CAS
- J. Pan, E. S. Wenger, M. L. Matthews, C. J. Pollock, M. Bhardwaj, A. J. Kim, B. D. Allen, R. B. Grossman, C. Krebs and J. Martin Bollinger, J. Am. Chem. Soc., 2019, 141, 15153–15165 CrossRef CAS PubMed
- X. Cao, Y. Shi, X. Wu, K. Wang, S. Huang, H. Sun, J. S. Dickschat and B. Wu, Org. Lett., 2019, 21, 6539–6542 CrossRef CAS
- X. Cao, Y. Shi, S. Wu, X. Wu, K. Wang, H. Sun, S. He, J. S. Dickschat and B. Wu, Tetrahedron, 2020, 76, 131349 CrossRef CAS
- X. Li, T. Awakawa, T. Mori, M. Ling, D. Hu, B. Wu and I. Abe, J. Am. Chem. Soc., 2021, 143, 21425–21432 CrossRef CAS PubMed
- C. C. Liaw, Y. L. Yang, C. K. Lin, J. C. Lee, W. Y. Liao, C. N. Shen, J. H. Sheuand and S. H. Wu, Org. Lett., 2015, 17, 2330–2333 CrossRef CAS
- W. D. Nes, Chem. Rev., 2011, 111, 6423–6451 CrossRef CAS PubMed
- J. M. Lv, D. Hu, H. Gao, T. Kushiro, T. Awakawa, G. D. Chen, C. X. Wang, I. Abe and X. S. Yao, Nat. Commun., 2017, 8, 1644 CrossRef PubMed
- C. F. Lee, L. X. Chen, C. Y. Chiang, C. Y. Lai and H. C. Lin, Angew. Chem., Int. Ed., 2019, 58, 18414–18418 CrossRef CAS
- O. W. Choroba, D. H. Williams and J. B. Spencer, J. Am. Chem. Soc., 2000, 122, 5389–5390 CrossRef CAS
- B. K. Hubbard, M. G. Thomas and C. T. Walsh, Chem. Biol., 2000, 7, 931–942 CrossRef CAS
- E. Kalliri, P. K. Grzyska and R. P. Hausinger, Biochem. Biophys. Res. Commun., 2005, 338, 191–197 CrossRef CAS PubMed
- T. Bai, Y. Matsuda, H. Tao, T. Mori, T. Mori, Y. Zhang and I. Abe, Org. Lett., 2020, 22, 4311–4315 CrossRef CAS
- H. Tao, T. Mori, H. Chen, S. Lyu, A. Nonoyama, S. Lee and I. Abe, Nat. Commun., 2022, 13, 95 CrossRef CAS PubMed
- D. Yan and Y. Matsuda, Org. Lett., 2021, 23, 3211–3215 CrossRef CAS PubMed
- J. Davison, A. Al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642–7647 CrossRef CAS
- N. Ishikawa, H. Tanaka, F. Koyama, H. Noguchi, C. C. C. Wang, K. Hotta and K. Watanabe, Angew. Chem., Int. Ed., 2014, 53, 12880–12884 CrossRef CAS PubMed
- W. C. Chang, J. Li, J. L. Lee, A. A. Cronican and Y. Guo, J. Am. Chem. Soc., 2016, 138, 10390–10393 CrossRef CAS
- X. Song, J. Lu and W. Lai, Phys. Chem. Chem. Phys., 2017, 19, 20188–20197 RSC
- S. L. Mader, A. Bräuer, M. Groll and V. R. I. Kaila, Nat. Commun., 2018, 9, 1–8 CrossRef CAS
- J. Pan, E. S. Wenger, M. L. Matthews, C. J. Pollock, M. Bhardwaj, A. J. Kim, B. D. Allen, R. B. Grossman, C. Krebs and J. Martin Bollinger, J. Am. Chem. Soc., 2019, 141, 15153–15165 CrossRef CAS
- J. Li, H. J. Liao, Y. Tang, J. L. Huang, L. Cha, T. S. Lin, J. L. Lee, I. V. Kurnikov, M. G. Kurnikova, W. C. Chang, N. L. Chan and Y. Guo, J. Am. Chem. Soc., 2020, 142, 6268–6284 CrossRef CAS PubMed
- M. E. Neugebauer, E. N. Kissman, J. A. Marchand, J. G. Pelton, N. A. Sambold, D. C. Millar and M. C. Y. Chang, Nat. Chem. Biol., 2022, 18, 171–179 CrossRef CAS
- T. Mori, Y. Nakashima, H. Chen, S. Hoshino, T. Mitsuhashi and I. Abe, Chem. Commun., 2022, 58, 5510–5513 RSC
- T. Mori, Z. Yu, H. Tao and I. Abe, Org. Lett., 2022, 24, 1737–1741 CrossRef CAS
- K. Tsukada, S. Shinki, A. Kaneko, K. Murakami, K. Irie, M. Murai, H. Miyoshi, S. Dan, K. Kawaji, H. Hayashi, E. N. Kodama, A. Hori, E. Salim, T. Kuraishi, N. Hirata, Y. Kanda and T. Asai, Nat. Commun., 2020, 11, 1830 CrossRef CAS PubMed
- Y. Morishita, K. Tsukada, K. Murakami, K. Irie and T. Asai, J. Nat. Prod., 2022, 85, 384–390 CrossRef CAS PubMed
- Z.-H. Xiao, J.-Y. Dong, A. Li, J.-M. Dai, Y.-P. Li, Q.-F. Hu, L.-D. Shao, Y. Matsuda and W.-G. Wang, Org. Chem. Front., 2022, 9, 1837–1843 RSC
| This journal is © The Royal Society of Chemistry 2023 |