DOI:
10.1039/C6RA01802E
(Paper)
RSC Adv., 2016,
6, 34110-34119
Novel solid forms of oxaprozin: cocrystals and an extended release drug–drug salt of salbutamol†
Received
21st January 2016
, Accepted 28th March 2016
First published on 30th March 2016
Abstract
Oxaprozin (OXP) is a non-steroidal anti-inflammatory drug mainly used to relieve the inflammation, swelling, and joint pain associated with osteoarthritis and rheumatoid arthritis. OXP shows poor aqueous solubility (0.0325 mg mL−1) that limits its bioavailability. The current study was aimed at identifying novel solid forms that could impact the physicochemical properties of OXP. Our solid form screening resulted in cocrystals with 4,4′-bipyridine and 1,2-bis(4-pyridyl)ethane and molecular salts with piperazine, 2-amino-3-picoline, and an antiasthmatic drug, salbutamol (SAL). Differential scanning calorimetry and X-ray diffraction techniques were employed to characterize the salts and cocrystals, and stability of the pharmaceutically acceptable salts was evaluated by storing at accelerated test conditions and slurry and dynamic vapour sorption techniques. Evaluation of the solubility and dissolution rate of piperazine and SAL salts revealed a modest effect of salt formation on the physicochemical properties of the OXP. The OXP−–SAL+–H salt provides an alternative solution to the short half-life of existing SAL formulations, which need to be dosed frequently to maintain therapeutic plasma levels. The drug–drug salt dissolves much slower compared to SAL and could potentially be developed as controlled release formulations of SAL. In addition, the presence of OXP as an integral part of the molecular salt enables exploitation of the molecular salt as a novel composition for development of combination drug therapy for treating inflammation associated with asthma.
Introduction
Oxaprozin (4,5-diphenyl-2-2 oxazole propionic acid, OXP, Fig. 1) is one of the widely used non-steroidal anti-inflammatory drugs (NSAIDs).1 OXP has been therapeutically used in inflammatory and painful diseases of rheumatic and nonrheumatic origin.1 It has recently been recognized that OXP inhibits the enzymes protein tyrosine kinase Syk, ZAP-70 and phosphodiesterase IV in pharmacologically relevant doses and thus effectively reduces the symptoms and inflammation associated with contact dermatitis in humans.2 OXP is a class II drug of the Biopharmaceutics Classification System (BCS), showing low aqueous solubility (0.0325 mg mL−1) and high permeability (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 4.19).3 In addition, OXP was found to comply with the BCS low solubility criteria over the entire pH range from 1.2 to 7.4.3 The use of substituted cyclodextrins for improving OXP dissolution properties has been reported recently.4 It has also been demonstrated that further improvement in dissolution rates of OXP could be obtained by combined use of substituted cyclodextrins, chitosan and bile acids.5 Formation of salts with alkali metals is an alternative route to enhance the solubility of OXP.6 For example, pharmaceutical formulations that contain sodium and potassium salts of OXP have been shown to possess improved solubility compared to the parent OXP.6 However, these salts have been found to interact with excipients, such as magnesium stearate, and form insoluble gel which prevents the tablets from disintegrating and dissolving.6 OXP is currently being marketed under the trade names DAYPRO® and OXAPROZIN, and both contain the OXP in its parent form.7
P = 4.19).3 In addition, OXP was found to comply with the BCS low solubility criteria over the entire pH range from 1.2 to 7.4.3 The use of substituted cyclodextrins for improving OXP dissolution properties has been reported recently.4 It has also been demonstrated that further improvement in dissolution rates of OXP could be obtained by combined use of substituted cyclodextrins, chitosan and bile acids.5 Formation of salts with alkali metals is an alternative route to enhance the solubility of OXP.6 For example, pharmaceutical formulations that contain sodium and potassium salts of OXP have been shown to possess improved solubility compared to the parent OXP.6 However, these salts have been found to interact with excipients, such as magnesium stearate, and form insoluble gel which prevents the tablets from disintegrating and dissolving.6 OXP is currently being marketed under the trade names DAYPRO® and OXAPROZIN, and both contain the OXP in its parent form.7
 |
| | Fig. 1 Molecular diagrams of OXP and cocrystal/salt formers. | |
Our study was aimed at exploring novel cocrystals and molecular salts of OXP with organic compounds. In this paper we report molecular salts of OXP with piperazine (PIP), an antiasthmatic drug, salbutamol (SAL), and 2-amino-3-picoline (2A3P), and cocrystals with 4,4′-bipyridine (BP) and 1,2-bis(4-pyridyl)ethane (BPE). As PIP and SAL are relevant in terms of drug development, more emphasis was given to evaluation of stability, solubility, and dissolution rate of these solids. The potential advantage of the drug–drug salt (OXP−–SAL+–H) in development of extended release formulations of SAL was highlighted.
Results
Nature of a multi-component crystal involving an acid and a base, whether it is a molecular salt or cocrystal, can be predicted based on pKa difference between the base and acid.8 It is generally accepted that when ΔpKa is less than 0 then a cocrystal is expected, on the other hand, when the ΔpKa is greater than 3 a salt is expected to result. However, in the intermediate ΔpKa range of 0 and 3, the nature of a multi-component crystal is difficult to predict.8,9 In these cases, experimental techniques such as single crystal X-ray diffraction and Fourier-transform infrared (FT-IR) spectroscopy provide accurate confirmation of salt/cocrystal formation. Table 1 lists the pKa and ΔpKa values for the combinations of OXP and coformers used in our study. ΔpKa values in Table 1 suggest that the PIP and SAL should result in salts and BP result in a cocrystal. The nature of molecular complexes involving BPE and 2A3P is difficult to predict based on the ΔpKa. In these cases, single crystal X-ray diffraction provided an additional means of confirming these molecular complexes as cocrystal in the case of BPE and a salt in the case of 2A3P.
Table 1 ΔpKa value of combinations of OXP (pKa = 4.3) and coformers
| Compound |
pKa |
ΔpKa |
Nature of molecular complex |
| PIP |
9.73 |
5.43 |
Salt |
| SAL |
10.3 |
6.0 |
Salt |
| 2A3P |
7.21 |
2.91 |
Salt |
| BP |
3.27 |
−1.03 |
Cocrystal |
| BPE |
6.13 |
1.83 |
Cocrystal |
Crystal structure analysis
A search for crystal structure of OXP in the Cambridge Structural Database (CSD) has not resulted in any reported crystal structures. However, the crystal structure has been recently determined by Sagdinc and Esme and the experimental crystal structure has been compared with geometry optimized structure.10 It was noted that the OXP molecules form a dimer mediated by acid–acid dimer synthon in the crystal structure.
OXP. The crystal structure of OXP was redetermined to compare with the other crystal structures reported in this work. Needle shaped crystals grown from 1,4-dioxane were found to belong to orthorhombic, Pbca space group (Table 2) with one molecule of OXP in the asymmetric unit. Interestingly, the crystal structure is significantly different from the structure reported in the previous report.10 The carboxylic acid group now forms an O–H⋯N hydrogen bond (Table 3) with the N of the oxazol ring, resulting in hydrogen bonded chains along the crystallographic a-axis (Fig. 2). Crystal structure also features several C–H⋯O and C–H⋯π interactions.
Table 2 Crystallographic parameters for the crystal structures reported in this paper
| Compound reference |
OXP |
OXP−–PIP+–H |
OXP−–SAL+–H |
OXP−–2A3P+–H |
OXP–BP |
OXP–BPE |
| Chemical formula |
C18H15NO3 |
C20H20N2O3 |
C31H36N2O6 |
C24H23N3O3 |
C23H19N2O3 |
C24H21N2O3 |
| Formula mass |
293.32 |
336.38 |
532.62 |
401.45 |
371.40 |
385.43 |
| Crystal system |
Orthorhombic |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
Triclinic |
| a/Å |
10.3129(5) |
5.8155(12) |
10.453(2) |
6.0156(12) |
13.422(3) |
9.7069(19) |
| b/Å |
7.6460(3) |
7.3355(15) |
10.627(2) |
8.8644(18) |
15.305(3) |
10.878(2) |
| c/Å |
36.7642(15) |
19.744(4) |
13.379(3) |
19.296(4) |
18.618(4) |
10.907(2) |
| α/° |
90 |
90.14(3) |
76.02(3) |
81.31(3) |
90.00 |
76.50(3) |
| β/° |
90 |
91.13(3) |
69.82(3) |
85.29(3) |
103.53(3) |
68.03(3) |
| γ/° |
90 |
99.08(3) |
88.05(3) |
80.23(3) |
90.00 |
66.04(3) |
| Unit cell volume/Å3 |
2899.0(2) |
831.5(3) |
1351.8(5) |
1000.7(3) |
3718.3(13) |
971.4(3) |
| Temperature/K |
100 |
110(2) |
110(2) |
110(2) |
110(2) |
110(2) |
| Space group |
Pbca |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
C2/c |
P |
| Radiation type |
Mo-Kα |
Mo-Kα |
Mo-Kα |
Mo-Kα |
Mo-Kα |
Mo-Kα |
| Absorption coefficient, μ/mm−1 |
0.092 |
0.091 |
0.091 |
0.089 |
0.089 |
0.088 |
| No. of reflections measured |
28658 |
12253 |
19467 |
13861 |
13232 |
13713 |
| No. of independent reflections |
4065 |
4093 |
6630 |
4810 |
4540 |
4686 |
| Rint |
0.0383 |
0.0302 |
0.0301 |
0.0211 |
0.0210 |
0.0193 |
| Final R1 values (I > 2σ(I)) |
0.0641 |
0.0637 |
0.0792 |
0.0730 |
0.0570 |
0.0505 |
| Final wR(F2) values (I > 2σ(I)) |
0.1690 |
0.1409 |
0.1910 |
0.2056 |
0.1430 |
0.1342 |
| Final R1 values (all data) |
0.0717 |
0.0698 |
0.0871 |
0.0744 |
0.0605 |
0.0578 |
| Final wR(F2) values (all data) |
0.1741 |
0.1445 |
0.1973 |
0.2074 |
0.1462 |
0.1406 |
| Goodness of fit on F2 |
1.069 |
1.181 |
1.179 |
1.155 |
1.141 |
1.058 |
Table 3 Neutron normalized intermolecular interactions in the crystal structures of OXP salts and cocrystals
| Crystal forms |
D–H⋯Aa |
H⋯A/Å |
D⋯A/Å |
D–H⋯A/° |
Symmetry code |
| D = donor, A = acceptor. |
| OXP |
O3–H3⋯N1 |
1.85 |
2.779(1) |
156 |
1/2 + x, 3/2 − y, 1 − z |
| C4–H4A⋯O2 |
2.37 |
3.349(2) |
150 |
3/2 − x, 1/2 + y, z |
| C4–H4B⋯O2 |
2.39 |
3.428(2) |
160 |
−1/2 + x, 3/2 − y, 1 − z |
| C14–H14⋯O3 |
2.46 |
3.291(2) |
133 |
−1/2 + x, 3/2 − y, 1 − z |
| OXP−–PIP+–H |
N2–H1⋯O3 |
1.63 |
2.636(2) |
177 |
1 + x, −1 + y, z |
| N2–H2⋯O2 |
1.80 |
2.768(2) |
160 |
x, −1 + y, z |
| N2–H2⋯O3 |
2.65 |
3.023(2) |
102 |
−x, 1 − y, 1 − z |
| C4–H4B⋯O3 |
2.51 |
3.577(2) |
170 |
1 + x, y, z |
| C16–H16⋯O1 |
2.59 |
3.634(3) |
161 |
1 + x, −1 + y, z |
| C20–H20A⋯O2 |
2.61 |
3.359(3) |
126 |
1 − x, 2 − y, 1 − z |
| C20–H20B⋯O3 |
2.48 |
3.202(3) |
123 |
−x, 1 − y, 1 − z |
| C20–H20B⋯N1 |
2.42 |
3.185(2) |
127 |
1 − x, 1 − y, 1 − z |
| OXP−–SAL+–H |
N2–H1⋯O2 |
1.79 |
2.797(2) |
176 |
1 + x, y, z |
| N2–H2⋯O3 |
1.81 |
2.795(3) |
166 |
−x, 1 − y, 1 − z |
| O6–H4⋯O5 |
2.00 |
2.827(2) |
140 |
|
| O5–H5⋯O2 |
1.63 |
2.609(2) |
179 |
1 + x, −1 + y, z |
| O4–H7⋯O3 |
1.80 |
2.704(2) |
152 |
1 + x, y, z |
| C4–H4B⋯O5 |
2.48 |
3.366(3) |
138 |
−1 + x, 1 + y, z |
| C8–H8⋯O6 |
2.66 |
3.709(3) |
164 |
−1 + x, 1 + y, z |
| C16–H16⋯O6 |
2.65 |
3.333(4) |
121 |
−1 + x, y, z |
| C29–H29B⋯O3 |
2.54 |
3.343(4) |
130 |
−x, 1 − y, 1 − z |
| C30–H30A⋯O6 |
2.64 |
3.713(3) |
169 |
2 − x, −y, 1 − z |
| C31–H31A⋯O5 |
2.62 |
3.702(3) |
178 |
2 − x, −y, 1 − z |
| OXP–BP |
O3–H3⋯N2 |
1.69 |
2.675(2) |
177 |
−x, −y, 1 − z |
| C4–H4B⋯O1 |
2.68 |
3.493(2) |
131 |
−x, y, 3/2 − z |
| C10–H10⋯O2 |
2.27 |
3.225(2) |
146 |
1/2 + x, 1/2 − y, 1/2 + z |
| C11–H11⋯O3 |
2.66 |
3.550(2) |
139 |
1/2 − x, 1/2 + y, 3/2 − z |
| C15–H15⋯O1 |
2.61 |
3.454(2) |
134 |
1/2 − x, −1/2 + y, 3/2 − z |
| C23–H23⋯O2 |
2.52 |
3.259(2) |
125 |
−x, −y, 1 − z |
| OXP–BPE |
O3–H1⋯N2 |
1.66 |
2.637(2) |
174 |
−1 + x, y, z |
| C4–H4B⋯O2 |
2.42 |
3.445(2) |
158 |
−x, 1 − y, 1 − z |
| C5–H5B⋯O1 |
2.51 |
2.952(2) |
103 |
|
| C15–H15⋯O2 |
2.58 |
3.525(2) |
146 |
1 − x, 1 − y, 1 − z |
| C16–H16⋯O1 |
2.54 |
3.443(2) |
140 |
1 + x, y, z |
| OXP−–2A3P+–H |
N3–H2⋯O2 |
1.97 |
2.868(2) |
146 |
−1 + x, −1 + y, z |
| N2–H5⋯O2 |
2.76 |
3.541(2) |
134 |
−x, 1 − y, 1 − z |
| N2–H5⋯O3 |
1.64 |
2.644(2) |
172 |
−x, 1 − y, 1 − z |
| N3–H6⋯O2 |
1.79 |
2.797(2) |
175 |
−x, 1 − y, 1 − z |
| C4–H4B⋯N2 |
2.63 |
3.346(2) |
123 |
1 + x, y, z |
| C22–H22⋯N1 |
2.38 |
3.462(2) |
176 |
|
| C23–H23⋯O3 |
2.63 |
3.633(3) |
154 |
|
| C24–H24C⋯O3 |
2.53 |
3.473(3) |
145 |
x, −1 + y, z |
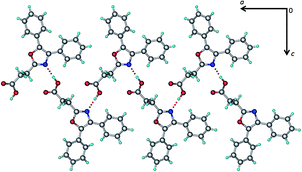 |
| | Fig. 2 Crystal structure of OXP. | |
OXP−–PIP+–H (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0.5). PIP is a pharmaceutically acceptable salt former and PIP-citrate and PIP salts of many active pharmaceutical ingredients are currently on the market.11 OXP and PIP were crystallized in a 1:0.5 molar ratio from methanol and obtained the crystals that belong to the salt. The salt can also be prepared in bulk by solid-state grinding technique (see ESI, Fig. S2†). The crystal structure of the salt belongs to triclinic, P space group with one molecule of OXP− and half a molecule of PIP+–H in the asymmetric unit. Proton transfer from carboxyl of the OXP to the N of the PIP is clearly evident. As shown in Fig. 3, the crystal structure is made up of hydrogen bonded ladder network along the crystallographic a-axis. The ladders are built up by OXP− and PIP+–H which are connected to each other via N+–H⋯O− hydrogen bonds. Symmetry related ladder networks are interconnected to each other via C–H⋯O and C–H⋯π interactions.
:0.5). PIP is a pharmaceutically acceptable salt former and PIP-citrate and PIP salts of many active pharmaceutical ingredients are currently on the market.11 OXP and PIP were crystallized in a 1:0.5 molar ratio from methanol and obtained the crystals that belong to the salt. The salt can also be prepared in bulk by solid-state grinding technique (see ESI, Fig. S2†). The crystal structure of the salt belongs to triclinic, P space group with one molecule of OXP− and half a molecule of PIP+–H in the asymmetric unit. Proton transfer from carboxyl of the OXP to the N of the PIP is clearly evident. As shown in Fig. 3, the crystal structure is made up of hydrogen bonded ladder network along the crystallographic a-axis. The ladders are built up by OXP− and PIP+–H which are connected to each other via N+–H⋯O− hydrogen bonds. Symmetry related ladder networks are interconnected to each other via C–H⋯O and C–H⋯π interactions.
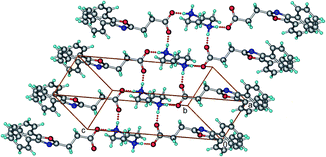 |
| | Fig. 3 Crystal structure of OXP−–PIP+–H salt. Notice the hydrogen bonded chain. | |
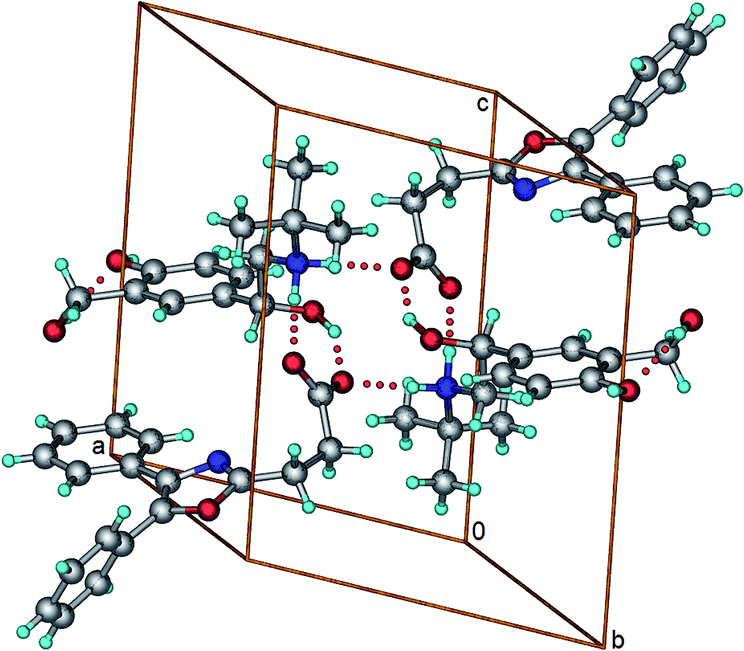
OXP−–SAL+–H (1:1). SAL is a bronchodilator and it has been used commonly in the treatment of respiratory diseases.12 Crystallization of OXP and SAL (1:1 molar ratio) from methanol resulted in a salt. The salt can also be obtained in quantitative yield by solid state grinding of OXP and SAL in 1:1 molar ratio (see ESI, Fig. S3†). The crystal structure of the salt has a triclinic unit cell with P space group. Each one molecule of OXP− and SAL–H+ are present in the crystallographic asymmetric unit. Presence of multiple hydrogen bond donors and acceptors in SAL–H+ resulted in a complex crystal packing. In the crystal structure, a four component supramolecular unit consisting of two molecules each of OXP− and SAL+–H mediated by O–H⋯O− and N+–H⋯O− hydrogen bonds was identified as the recurring supramolecular unit (Fig. 4). Such translation related supramolecular units generate hydrogen bonded ladders along the crystallographic b-axis. These ladders are interconnected to each other via C–H⋯O and π⋯π interactions.
 |
| | Fig. 4 A four-component supramolecular unit in the crystal structure of OXP−–SAL+–H salt. | |
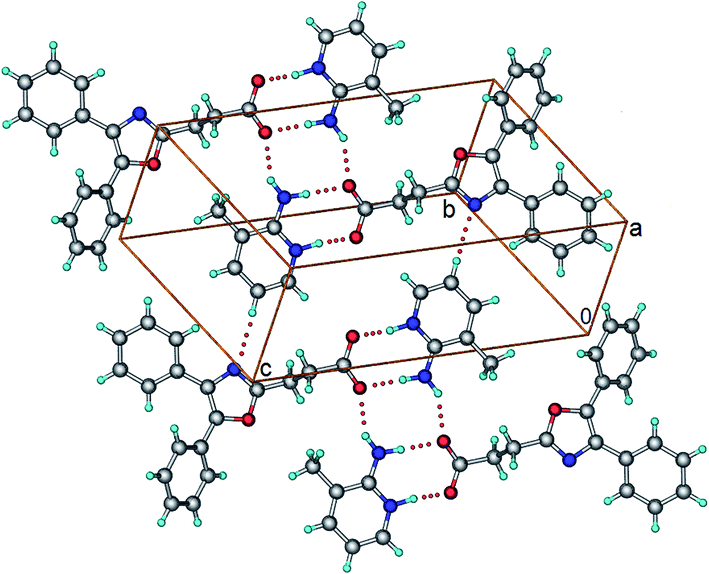
OXP−–2A3P+–H (1:1). The salt was obtained when OXP and 2A3P were crystallized in 1:1 molar ratio from acetonitrile. Crystal structure belongs to triclinic, P space group. One molecule each of OXP− and 2A3P+–H constitute the asymmetric unit. In the crystal structure, the carboxylate ion forms N+–H⋯O− and N–H⋯O− hydrogen bonds with the pyridinium and amine groups, respectively. The resulting supramolecular unit dimerizes involving the second N–H of the amine via N–H⋯O− hydrogen bond to result in a four-component supramolecular unit (Fig. 5). Several C–H⋯O, C–H⋯N, and C–H⋯π interactions stabilize the supramolecular units in the crystal structure.
 |
| | Fig. 5 Crystal structure of OXP−–2A3P+–H salt showing the four-component supramolecular units connected via C–H⋯N interactions. | |
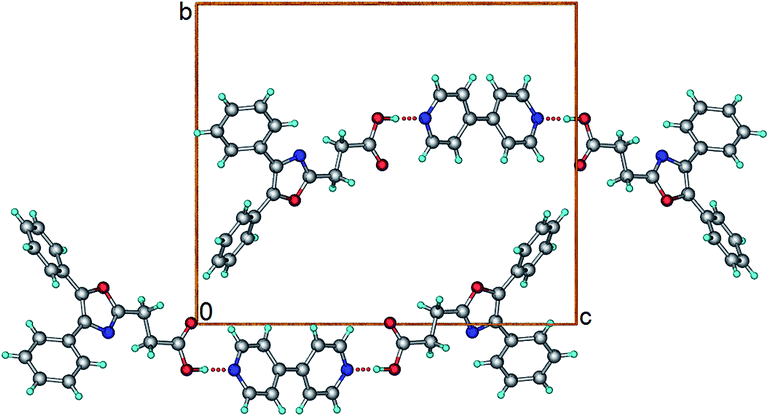
OXP–BP (1:0.5). Crystallization of OXP and BP from methanol in 1:0.5 molar ratio gave crystals that were confirmed as a 1:0.5 cocrystal. Crystal structure was solved and refined in monoclinic, C2/c space group. One molecule of OXP and half a molecule of BP present in the asymmetric unit. The structure features a 3-component supramolecular unit consisting of two molecules of OXP and one molecule of BP connected by O–H⋯N hydrogen bond (Fig. 6). The crystallographic inversion centre resides at the centre of the BP molecule. Crystal structure is built up by self-assembly of the 3-component supramolecular units by several C–H⋯O and C–H⋯π interactions.
 |
| | Fig. 6 Crystal structure of OXP–BP cocrystal. | |
OXP–BPE (1:0.5). Crystallization of OXP and BPE from methanol in 1:0.5 molar ratio gave a cocrystal. Crystal structure was solved in triclinic, P space group with one molecule of OXP and half a molecule of BPE in the asymmetric unit. The crystal structure is similar to the crystal structure of OXP–BP cocrystal (Fig. 7). The 3-component supramolecular units self-assemble in the crystal structure via several π⋯π, C–H⋯O, and C–H⋯π interactions.
 |
| | Fig. 7 Crystal structure of OXP–BPE cocrystal. | |
Conformational analysis
OXP (Fig. 1) is a flexible molecule and can adopt different conformations in the solid-state. Overlaying the conformers of OXP from crystal structures revealed a significant conformational flexibility (Fig. 8). Conformational differences are mainly due to free rotation of the propyl chain. Selected torsion angles and calculated conformer energies are tabulated in Table 4. Interestingly, the conformer in the parent crystal structure was not found in any of its salts and cocrystals. Comparison of the conformer energies revealed that the conformer in the parent crystal structure of OXP has the lowest energy (Table 4), suggesting that the higher energy of the OXP conformers in salts/cocrystals is compensated by the strong intermolecular interactions.
 |
| | Fig. 8 Overlay of OXP conformers retrieved from the crystal structures reported in this paper. Color codes: OXP-brown, OXP−–PIP+–H-red, OXP−–SAL+–H-blue, OXP–BP-green, OXP–BPE-cyan, and OXP−–2A3P+–H-magenta. | |
Table 4 Selected torsion angles and conformer energies of OXP molecule in the crystal structures of its salts and cocrystals
| Conformer (colour code) |
τ1 (O1–C1–C4–C5) |
τ2 (O1–C2–C7–C8) |
τ3 (N1–C3–C13–C14) |
Conformer energya (kcal mol−1) |
| Computed in Materials Studio 7.0 using Compass force field. Lowest energy of the conformer was arbitrarily set to 0. |
| OXP (brown) |
−69.38 |
−28.49 |
−29.30 |
0 |
| OXP−–PIP+–H (red) |
78.4 |
−28.3 |
−30.4 |
30.25 |
| OXP−–SAL+–H (blue) |
166.8 |
51.5 |
13.3 |
39.98 |
| OXP–BP (green) |
−156.36 |
32.3 |
30.4 |
1.31 |
| OXP–BPE (cyan) |
−51.66 |
37.26 |
23.92 |
7.84 |
| OXP−–2A3P+–H (magenta) |
92.2 |
39.4 |
28.9 |
34.61 |
Thermal analysis
Melting points of the salts and cocrystals were determined by differential scanning calorimetry (DSC) and compared with the melting points of the components of salts and cocrystals (Table 5). As shown in Fig. 9, a single melting endotherm for all the salts/cocrystals confirmed the purity and homogeneity of the bulk samples. All the samples showed a distinct melting point compared to the melting points of OXP and salt/cocrystal former, suggesting that the DSC can be used to identify cocrystal or salt formation. Salts with PIP and SAL showed higher melting point than the melting point of OXP which could be due to stronger ionic hydrogen bonds in their respective crystal structures. The lower melting point of the OXP−–2A3P+–H salt compared to the melting point of OXP is due to the lower melting point of 2A3P (Table 5). Melting point of both the cocrystals (OXP–BP and OXP–BPE) is intermediate compared to the melting point of starting materials. A recent survey on melting points of cocrystals revealed that a majority of the cocrystals had melting points between those of starting materials.13
Table 5 Melting point (MP) of OXP, crystalline forms, and salt/cocrystal formers used in this study
| Drug/coformer |
MP of drug/coformer (°C) |
Salt/cocrystal |
MP of salt/cocrystal (Tonset, °C) |
| Taken from http://www.ChemSpider.com. |
| OXP |
164.3 |
— |
|
| SAL |
161.5 |
OXP−–SAL+–H |
172.7 |
| PIP |
108–113a |
OXP−–PIP+–H |
204.8 |
| 2A3P |
29–32a |
OXP−–2A3P+–H |
111.7 |
| BP |
111–11a |
OXP–BP |
148.5 |
| BPE |
110–112a |
OXP–BPE |
134.5 |
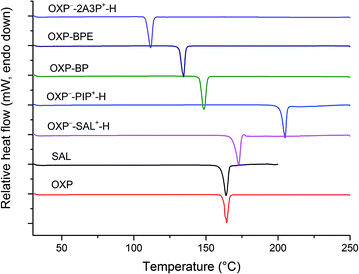 |
| | Fig. 9 DSC plots of OXP, SAL and the molecular salts and cocrystals reported in this paper. | |
Stability of OXP, SAL, OXP−–PIP+–H, and OXP−–SAL+–H
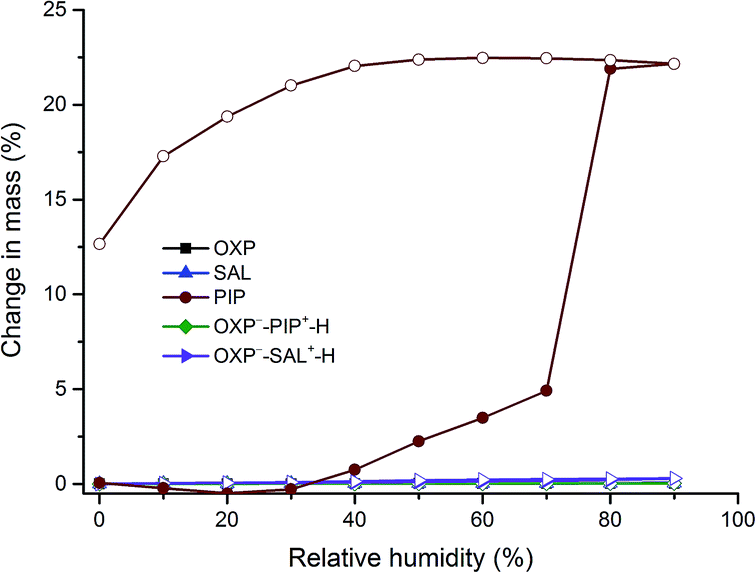
Among the various salt/cocrystal formers used in this study, PIP and SAL are pharmaceutically relevant11,12 and hence stability of these salts was tested using dynamic vapour sorption (DVS), slurry method, and at accelerated stability condition (45 °C, 75% relative humidity (RH)). The DVS profiles in Fig. 10 revealed that both the salts, OXP, and SAL are nonhygroscopic which was evidenced by a negligible moisture uptake (<1%). In contrast, PIP which is known to form a hexahydrate upon exposure to moisture,14 showed ∼22% moisture uptake corresponding to conversion of part of the PIP to PIP hexahydrate (PIP hexahydrate contains 55.6% of water). All other solid forms reported in this paper were also analyzed by DVS and found that they are nonhygroscopic, except that BP showed moisture uptake corresponding to monohydrate formation (see ESI, Fig. S4†). In the slurry experiments, excess solids of the salts and their individual components were slurried for 24 h at ambient condition. PXRD was used to identify the powder samples which suggested that all the samples are stable under slurry condition (see ESI, Fig. S5–S7†). In the stability experiments at accelerated condition, OXP, SAL, OXP−–PIP+–H, and OXP−–SAL+–H were stored at 45 °C and 75% RH for 13 weeks and stability of the solids was monitored by PXRD. The results suggested that there was no phase transformation or degradation of the samples at this test condition (see ESI, Fig. S8–S10†). Therefore, it could be concluded that the stability of the salts is comparable to the stability of the individual components.
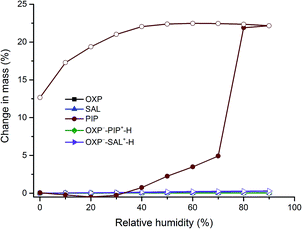 |
| | Fig. 10 DVS profiles of OXP, SAL, OXP−–PIP+–H, and OXP−–SAL+–H. Solid and open legends correspond to the data of sorption and desorption, respectively. DVS profiles of other solids and salt/cocrystal formers are provided in ESI (Fig. S4†). | |
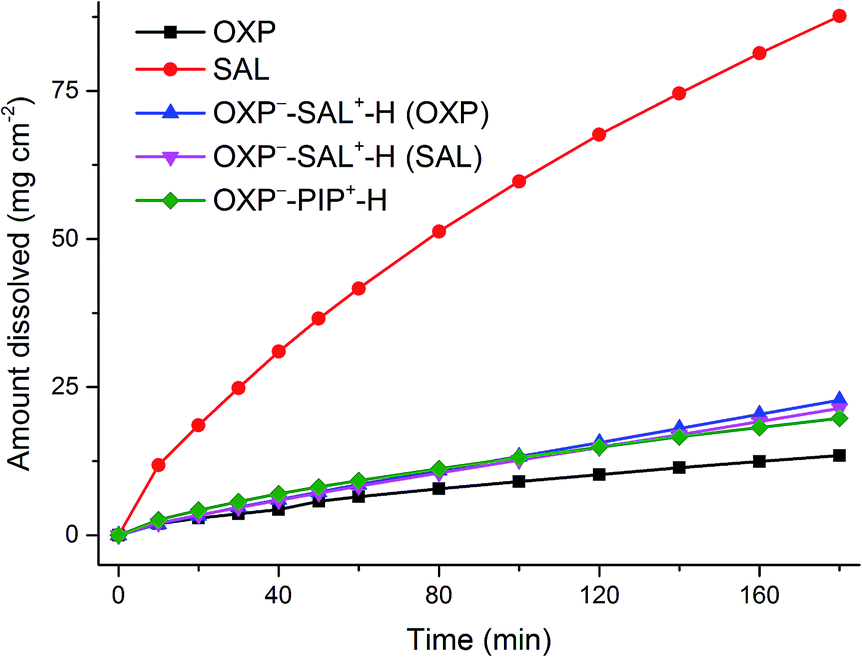
Dissolution rate and solubility
Intrinsic dissolution rate (IDR) and solubility of OXP−–PIP+–H and OXP−–SAL+–H were measured and compared with the solubility and dissolution rate of OXP and SAL. Solubility and dissolution experiments were conducted in pH 7.4 phosphate buffer (with the addition of 0.5% Tween 80). The results are tabulated in Table 6. It is clear that salt formation with organic salt formers has only a modest effect on the solubility and dissolution rate of OXP. The observed solubility follows the trend: OXP > OXP−–PIP+–H > OXP−–SAL+–H. In the IDR experiments, OXP−–PIP–H+ salt showed the highest IDR and the observed dissolution rate follows the trend: OXP−–PIP+–H > OXP > OXP−–SAL+–H (Fig. 11). With respect to solubility and dissolution rate of SAL, the drug–drug salt OXP−–SAL+–H, significantly lowers the solubility and dissolution rate of SAL. The solids recovered after the solubility and dissolution experiments confirm that the solids remain stable and, therefore, the observed solubility and dissolution rate correspond to the true solubility and dissolution rate of the solids reported (see ESI, Fig. S11–S13†).
Table 6 Solubility and dissolution rate of OXP, SAL and pharmaceutically acceptable salts in pH 7.4 phosphate buffer (with 0.5% of Tween 80) at 37 °C
| Solid form |
Equilibrium solubility (mg mL−1) |
IDR (×10−3) (mg cm−2 min−1) |
| Based on OXP concentration. Based on SAL concentration. |
| OXP |
2.30 ± 0.01 |
5.98 |
| SAL |
21.20 ± 0.08 |
41.22 |
| OXP−–PIP+–H |
2.11 ± 0.01 |
7.09 |
| OXP−–SAL+–Ha |
2.02 ± 0.01 |
4.25 |
| OXP−–SAL+–Hb |
1.39 ± 0.01 |
2.85 |
 |
| | Fig. 11 IDR profiles of SAL, OXP and its molecular salts. | |
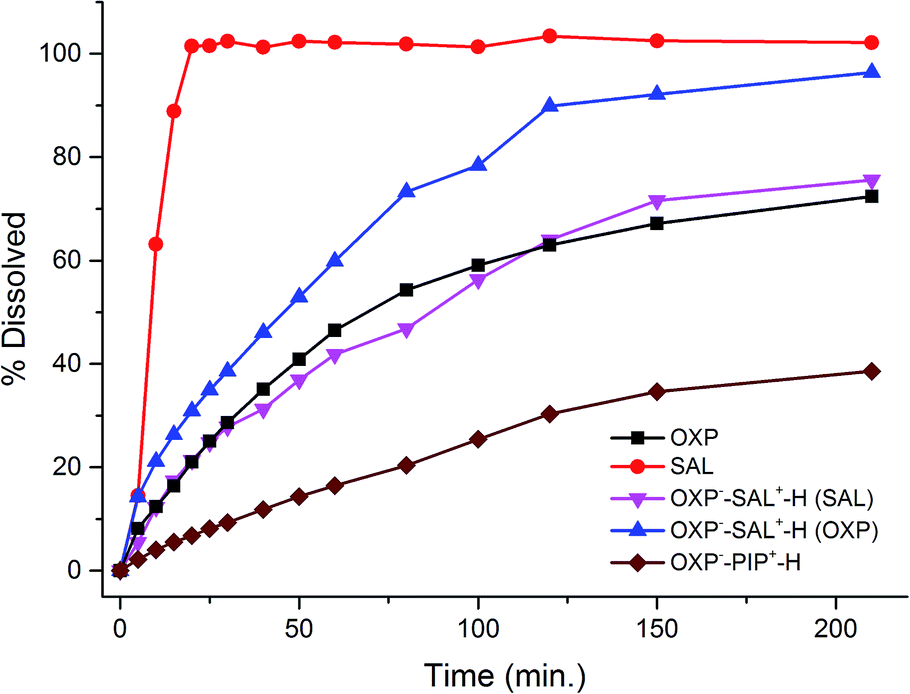
The observed slower IDR of OXP−–SAL+–H prompted us to conduct dissolution experiments on compressed tablets. The drug–drug salt, OXP−–SAL+–H, dissolves much slower compared to the SAL base (15% vs. 89% in 15 min). In addition, SAL base was completely dissolved in 25 min, on the other hand, 37% of the molecular salt remained undissolved even after 200 min. OXP−–PIP–H+ salt is the slowest dissolving solid which showed dissolution of only 38% after 200 min (Fig. 12).
 |
| | Fig. 12 Dissolution profiles of compressed tablets of SAL, OXP, and the molecular salts. | |
Discussion
OXP is a BCS class II drug and has documented evidence of solubility and dissolution issues.3 Previous attempts to make more soluble OXP solids were based on complexation with cyclodextrins and salt formation with alkali metals.4–6 In general, salt formation is expected to yield significant improvement in physicochemical properties such as, dissolution rate and solubility due to their greater affinity towards water.15 Therefore, we have reasoned that the salts reported herein with organic salt formers could offer additional means of modifying the solubility and dissolution rate of OXP. Both the pharmaceutically acceptable salts showed only a modest effect on the solubility/dissolution rate. While both the salts showed lower solubility than OXP, IDR of OXP−–PIP+–H is higher than OXP, but the IDR of OXP−–SAL+–H is lower than OXP (Table 6). We attempt here to rationalize these observations.
Solubility of a solid depends on a combination of entropic and enthalpic factors. Attempts have been made to correlate solubility of salts with their melting point, crystal structure, solubility of counterion, etc.16 However, there are no reliable general rules to correlate these properties with solubility of the salts. In general, higher melting salts show lower solubility due to greater stability of the high melting salt.16c In the case of OXP salts, no correlation was found between melting point of the salt and solubility, as higher melting OXP−–PIP+–H salt (205 °C) showed lower solubility than the low melting OXP−–SAL+–H (173 °C). This suggests that alternative factors such as solubility of counterion could impact the solubility of the salts. PIP is freely soluble in water and has a significantly higher solubility than the SAL. Therefore, the marginally higher solubility of OXP−–PIP+–H compared to the OXP−–SAL+–H could be attributed to the higher solubility of PIP. A detailed analysis of the intermolecular interactions in the crystal structures of salts and OXP provided a greater insight into structure–property correlation. Crystal structure of OXP features a 1-D chain of OXP molecules connected by O–H⋯N hydrogen bond. The carbonyl–O is currently involved in a weaker C–H⋯O interaction which can be replaced by a stronger hydrogen bond by interacting with water. However, in both the salts, all the potential hydrogen bonding sites are involved in strong ionic and neutral hydrogen bonds and there is no free hydrogen bond donor or acceptor site with which water may interact (Table 3). Comparison of packing coefficients revealed a better close packing in salts than the individual components. The packing coefficients of OXP and SAL (0.70, 0.64) are lower than the packing coefficients of the salts (0.71, 0.71). Therefore, perhaps the lower solubility of the salts is an account of their more extensive hydrogen-bond arrangement and efficient packing of molecular fragments in the crystal lattice.
In general, IDR should be proportional to the solubility if the measurement conditions for dissolution rate are kept the same (constant surface area, stirring speed, pH and ionic strength of the dissolution).17 In our studies, all the IDR experiments were performed under the same stirring speed and in the same dissolution media. However, the observed dissolution rate and solubility do not strictly follow the theoretical dissolution rate vs. solubility trend. This could be due to differences in their particle size distribution which is not the same for the samples used for dissolution experiments and hence the surface areas exposed to the dissolution medium could be slightly different. The impact of various particle properties and experimental variables such as, preferred orientation of crystallites, size, pressure, etc. on IDR and tablet dissolution rate of several drugs has been studied.18 For example, it has been found that the samples of aspirin and tolbutamide with higher degree of preferred orientation showed lower IDR.18d
SAL is an antiasthmatic drug and SAL-sulfate is currently one of the most prescribed bronchodilators for the treatment of bronchial asthma.12 SAL-sulfate has a short plasma half-life which is reported to be in the range of 2–3 h.20 Due to this, frequent dosing of typically three or four times a day orally at a dose of 2–4 mg is necessary to maintain therapeutic plasma levels.19 In addition, inflammation associated with asthma necessitates the use of anti-inflammatory drug as adjunct therapy and combination use of a bronchodilator and anti-inflammatory drug has become an effective treatment for asthma management in the recent times.20–24 Controlled release/slow-release tablet formulations of the SAL-sulfate have been developed to address the issues associated with short half-life. These include mainly polymeric excipients, such as Eudragit RS®, Kollicoat SR®, hydroxypropyl methylcellulose, sodium alginate, polyvinyl alcohol, etc.25–28 While these attempts have been proved successful only for development of extended release tablet formulations, the need for anti-inflammatory drug as adjunct therapy for treating inflammation associated with asthma has not been addressed fully. The drug–drug salt, OXP−–SAL+–H, reported herein shows reduced solubility of SAL from 21.20 mg mL−1 to 1.39 mg mL−1 in pH 7.4 phosphate buffer (with 0.5% Tween 80), and IDR of SAL was reduced from 41.22 mg cm−2 min−1 to 2.85 mg cm−2 min−1. The fact that the solubility and dissolution rate of SAL could be reduced by salt formation with OXP could mean that the molecular salt may help to prolong duration of effective concentration of the drug. This ultimately minimizes the number of doses required to achieve the effective concentration. Therefore, OXP−–SAL+–H salt provides an alternative route to develop controlled release/slow-release tablet formulations of SAL that possibly address issues associated with short half-life of existing SAL formulations. In addition, being an anti-inflammatory drug and an integral part of the molecular salt, OXP may provide synergistic effect in the treatment of inflammation associated with asthma.
Conclusions
We have reported two cocrystals and three salts of OXP. The salt formation is in agreement with the ΔpKa rule of 3. All the cocrystals and salts were characterized by X-ray diffraction and DSC. In all the crystal structures, the OXP molecule forms heteromeric interactions with the salt/cocrystal former. Physicochemical properties such as stability, dissolution rate, and solubility of the pharmaceutically acceptable salts were evaluated and compared with parent OXP. While the stability of the novel salts is comparable with the stability of OXP, the salt formers did not show significant impact on the solubility and dissolution rate of OXP. Lower solubility of the salts was attributed to their more extensive hydrogen-bond arrangement and efficient packing of molecular fragments in the crystal lattice. Knowing the difficulties in development of formulations with alkali metal salts of OXP,6 the pharmaceutically acceptable salts reported herein may provide additional means of exploring novel routes for development of OXP and SAL formulations. The drug–drug salt, OXP−–SAL+–H, showed slower dissolution rate and low solubility, and thus suggests that the molecular salt could be a possible alternative for development of extended release tablet formulations of SAL to address the problems of short half-life and frequent dosing of existing SAL drug formulations. In addition, the presence of OXP as an integral part of the molecular salt enables possible exploitation of the molecular sat as a novel formulation for treating inflammations associated with asthma. Cocrystals containing two or more drugs have been recently suggested as relevant to development of combination drug products.29–37 As preparation of salts is the most common and effective way to modify physicochemical properties of ionizable drugs,16 drug–drug salts38–40 of the kind reported herein warrant potential applications in the development of combination drug products of ionizable APIs.
Experimental section
OXP and SAL were purchased from Junda Pharmaceuticals, China. All other salt/cocrystal formers were procured from Alfa-Aesar®, Singapore. Analytical grade solvents were used for the crystallization experiments.
Solid form screening
Cocrystals and salts were primarily screened using solvent-evaporative crystallization techniques at ambient experiments.
OXP. OXP (100 mg) was dissolved in 5 mL of 1,4-dioxane at 60 °C and slow evaporation of the solvent at room temperature resulted in needle-shaped single crystals of OXP in 2 days.
SAL. SAL (100 mg) was dissolved in 5 mL of methanol at 60 °C and slow evaporation of the solvent at room temperature resulted in block-shaped crystals in 2 days.
OXP−–PIP+–H (1:0.5) salt. OXP (100 mg, 0.34 mmol) and PIP (14.7 mg, 0.17 mmol) were dissolved in 10 mL of methanol at 60 °C and slow evaporation of the solvent at ambient condition resulted in single crystals of the OXP−–PIP–H+ (1:0.5) salt in 1:1 stoichiometry as elongated needles after 7 days.
OXP−–SAL+–H (1:0.5) salt. OXP (100 mg, 0.34 mmol) and SAL (81.6 mg, 0.34 mmol) were dissolved in 10 mL of methanol at 60 °C and slow evaporation of the solvent at ambient condition resulted in block-shaped crystals that belong to OXP−–SAL–H+ salt in approximately 3 days.
OXP−–2A3P+–H (1:0.5) salt. OXP (100 mg, 0.34 mmol) and 2A3P (36.8 mg, 0.34 mmol) were dissolved in 10 mL of methanol at 60 °C and slow evaporation of the solvent at ambient condition gave single crystals of the salt in 1:1 stoichiometry as colorless needles after 3 days.
OXP·BP. OXP (100 mg, 0.34 mmol) and BP (26.6 mg, 0.17 mmol) were dissolved in 10 mL of methanol at 60 °C and slow evaporation of the solvent at ambient condition gave thin yellow needles after 3 days.
OXP·BPE. OXP (100 mg, 0.34 mmol) and BPE (46.1 mg, 0.17 mmol) were dissolved in 10 mL of methanol at 60 °C and slow evaporation of the solvent at ambient condition gave thin yellow needles after 3 days.
Preparation of salts by grinding
Solid-state grinding experiments were performed using a Retsch Mixer Mill model MM301. In a typical experiment, 100 mg (0.34 mmol) of OXP and an appropriate amount of the salt former were taken in 10 mL stainless steel grinding jar with one 7 mm stainless steel grinding ball and ground at a rate of 20 Hz for 30 min.
Single crystal X-ray diffraction
Reflections for all the samples were collected on a Rigaku Saturn CCD area detector. Mo-Kα radiation (λ = 0.71073 Å) was used. CrystalClear (Rigaku) software was used for data collection and processing. Structures were solved and refined using SHELX-TL41 and non-hydrogen atoms were anisotropically refined. Hydrogen atoms on N and O atoms were experimentally located in all the structures. All other H atoms were positioned geometrically and refined using a riding model. All O–H, N–H and C–H distances are neutron normalized to 0.983, 1.009 and 1.083 Å, respectively. X-Seed42 was used to prepare images for graphical representation of the crystal structures. CCDC 1062956–1062961† contains the supplementary crystallographic data for this paper.
Powder X-ray diffraction (PXRD)
D8 Advance powder X-ray diffractometer (Bruker AXS GmbH, Germany) with Cu-Kα radiation (λ = 1.54056 Å) at 35 kV and 40 mA was used to identify the powder samples. Diffraction patterns were collected over a scan range of 2θ = 5° to 50° continuous scan, with a scan rate of 2 deg min−1.
Differential scanning calorimetry (DSC)
DSC was performed with a Mettler Toledo DSC 822e module. Bulk crystals harvested from crystallization experiments were ground to fine powder used for analysis. Samples were placed in crimped but vented aluminium sample pans. The sample size was typically 2–5 mg temperature range was of 30–250 °C @5 °C min−1. The DSC instrument was calibrated using indium as the reference material.
Dynamic vapour sorption (DVS) studies
Water vapour sorption isotherms were determined using Surface Measurement Systems (SMS) advantage dynamic vapour sorption (DVS) instrument at 25 °C. About 40 mg of the sample was subjected to relative humidity flux from 0% to 95% in 9 steps of around 10% each and back to 0% in a similar manner via desorption. The samples were allowed to equilibrate at one specific partial vapour pressure until the rate change of mass was less than 0.002% min−1. Samples were initially dried for 180 min at 45 °C and 0% partial vapor pressure.
Stability experiments
Stability of the OXP salts with PIP and SAL was tested over a 13 week period at accelerated storage condition (40 °C and 75% RH). Samples of 100 mg were stored at test condition and the samples were periodically analyzed by PXRD. In the slurry experiments, excess powder samples of the salts were slurried in water for 24 h at room temperature. PXRD was used to identify the filtered samples.
Dissolution and solubility
Concentrations of OXP and SAL were measured by HPLC (Agilent 1100 series). Methods were developed to make sure that the OXP and SAL components were observed at different retention time. The HPLC instrument was equipped with a ZORBAX Extended-C18 column (4.6 × 150 mm, 8 nm pore size, 5 μm). An injection volume of 10 μL and flow rate of 1 mL min−1 were used. Detection wavelength in the UV-visible range was set at 285 nm for OXP and 226 nm for SAL. Elution was achieved with a mobile phase containing a mixture of methanol and ultrapure water in a ratio (v/v) of 70:30. The observed retention times for SAL is 3.4 min, OXP is 11.8, and for PIP is 2.3 min. Shake-flask method was used to measure equilibrium solubility. In this method, an excess amount of the powdered material was added to 5 mL of pH 7.4 phosphate buffer (with 0.5% of Tween 80) and stirred at 37 °C for 24 h. The slurry was then filtered through 0.2 μm syringe filter and the concentration of the resultant solution was determined.
IDR experiments were carried out on a Varian VK7010 dissolution apparatus equipped with a VK750D heater/circulator. 500 mg of sample was taken in the intrinsic attachment and compressed to a 0.5 cm2 disk using a hydraulic press at a pressure of 5 ton for 5 min. The intrinsic attachment was placed in a jar of 900 mL of pH 7.4 phosphate buffer (with 0.5% of Tween 80) preheated at 37 °C and rotated at 150 rpm. Dissolution experiments on compressed tablets were conducted under sink conditions. The tablets were prepared by compressing the samples at a pressure of 0.4 ton for 2 min. Dissolution medium, temperature, and stirring rate were same as the IDR experiments. In all the dissolution experiments, two milliliter aliquots were collected at specific time intervals, and assayed for OXP and SAL concentrations from the respective calibration plots.
Acknowledgements
We thank Lucy K. Mapp for assistance in crystal structure analysis of OXP and we gratefully acknowledge the financial support from the Institute of Chemical and Engineering Sciences of A*STAR (Agency for Science, Technology and Research), Singapore.
Notes and references
- M. A. al-Faks and M. C. Pugh, Orthop. Rev., 1992, 21, 558–560 CAS.
- M. S. Weider, WO2006102900 A1, 5 October 2006.
- M. Yazdanian, K. Briggs, C. Jankovsky and A. Hawi, Pharm. Res., 2004, 21, 293–299 CrossRef CAS.
- F. Maestrelli, M. Cecchi, M. Cirri, G. Capasso, N. Mennini and P. Mura, J. Inclusion Phenom. Macrocyclic Chem., 2009, 63, 17–25 CrossRef CAS.
- F. Maestrelli, M. Cirri, N. Mennini, N. Zerrouk and P. Mura, Eur. J. Pharm. Biopharm., 2011, 78, 385–393 CrossRef CAS PubMed.
- M. E. Adams, S. Desai, A. Karim, K. Paul, D. J. Schaaf and D. J. Zold, US Pat., 6030643, 29 Feb. 2000.
- Accessed the data from http://www.accessdata.fda.gov/scripts/cder/ob/docs/tempai.cfm.
- B. Sarma, N. K. Nath, B. R. Bhogala and A. Nangia, Cryst. Growth Des., 2009, 9, 1546–1557 CAS.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS PubMed.
- S. G. Sagdinc and A. Esme, Spectrochim. Acta, Part A, 2010, 75, 1370–1376 CrossRef PubMed.
- M. Katz, Drugs, 1986, 32, 358–371 CrossRef CAS PubMed.
- S. N. Murthy and H. R. Shobharani, Int. J. Pharm., 2004, 287, 47–53 CrossRef CAS PubMed.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950–2967 CAS.
- D. Schwarzenbach, J. Chem. Phys., 1968, 48, 4134–4140 CrossRef CAS.
- Pharmaceutical Salts and Co-crystals, ed. J. Wouters and L. Quere, Royal Society of Chemistry, London, 2011 Search PubMed.
-
(a) R. Banerjee, P. M. Bhatt, N. V. Ravindra and G. R. Desiraju, Cryst. Growth Des., 2005, 5, 2299–2309 CrossRef CAS;
(b) J. Galcera and E. Molins, Cryst. Growth Des., 2009, 9, 327–334 CrossRef CAS;
(c) L. Kumar, C. L. Meena, Y. B. Pawar, B. Wahlang, K. Tikoo, R. Jain and A. K. Bansal, AAPS PharmSciTech, 2012, 14, 141–150 CrossRef PubMed.
- A. Dokoumetzidis and P. Macheras, Int. J. Pharm., 2006, 321, 1–11 CrossRef CAS PubMed.
-
(a) A. J. Jounela, P. J. Pentikäinen and A. Sothmann, Eur. J. Clin. Pharmacol., 1975, 8, 365–370 CrossRef CAS PubMed;
(b) M. Mosharraf and C. Nyström, Int. J. Pharm., 1995, 122, 35–47 CrossRef CAS;
(c) M. D. Tuladhar, J. E. Carless and M. P. Summers, J. Pharm. Pharmacol., 1983, 35, 269–274 CrossRef CAS PubMed;
(d) M. Tenho, J. Aaltonen, P. Heinänen, L. Peltonen and V.-P. Lehto, J. Appl. Crystallogr., 2007, 40, 857–864 CrossRef CAS.
- A. S. Vicente, R. M. Hernandez, A. R. Gascon, M. B. Calvo and J. L. Pedraz, Int. J. Pharm., 2000, 208, 13–21 CrossRef.
- C. C. G. Carling and J. W. Trofast, US Pat., 5972919, 26 Oct. 1999.
- C. McDonald, G. M. Pover and G. K. Crompton, Curr. Med. Res. Opin., 1988, 11, 116–122 CrossRef CAS PubMed.
- C. Schmelzer and F.-R. Weitzel, US Pat., 2014/0228397 A1, 14 Aug. 2014.
- N. El-Gendy, W. Pornputtapitak and C. Berkland, Eur. J. Pharm. Sci., 2011, 44, 522–533 CrossRef CAS PubMed.
- J. A. Short, C. A. Barr, C. D. Palmer, J. M. Goddard, C. G. Stack and R. A. Primhak, Anaesthesia, 2000, 55, 334–337 CrossRef CAS PubMed.
- M. Purushothaman, R. Dhanapal, S. Vijayakumar and J. V. Ratna, Int. J. Adv. Pharm. Sci., 2012, 2, 5–8 Search PubMed.
- K. Malodia, A. Kumar, S. Kumar and P. Rakha, Der Pharmacia Lettre, 2013, 5(1), 177–181 CAS.
- S. Sultana, I. Ahmad, M. R. Islam and M. H. Rahman, Dhaka Univ. J. Pharm. Sci., 2010, 9, 109–118 Search PubMed.
- L. Pachuau, S. Sarkar and B. Mazumder, Trop. J. Pharm. Res., 2008, 7, 995–1002 Search PubMed.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2009, 11, 1823–1827 RSC.
- A. O. Surov, A. P. Voronin, A. N. Manin, N. G. Manin, L. G. Kuzmina, A. V. Churakov and G. L. Perlovich, Mol. Pharmaceutics, 2014, 11, 3707–3715 CrossRef CAS PubMed.
- M. Žegarac, E. Lekšić, P. Šket, J. Plavec, M. D. Bogdanović, D.-K. Bučar, M. Dumiće and E. Meštrović, CrystEngComm, 2014, 16, 32–35 RSC.
- A. O. L. Évora, R. A. E. Castro, T. M. R. Maria, M. T. S. Rosado, M. R. Silva, A. M. Beja, J. Canotilho and M. E. S. Eusébio, Cryst. Growth Des., 2011, 11, 4780–4788 Search PubMed.
- S. Cherukuvada and A. Nangia, CrystEngComm, 2012, 14, 2579–2588 RSC.
- B. S. Sekhon, Daru, J. Pharm. Sci., 2012, 20, 45 CrossRef CAS PubMed.
- P. Grobelny, A. Mukherjee and G. R. Desiraju, CrystEngComm, 2011, 13, 4358–4364 RSC.
- H. G. Lee, G. G. Z. Zhang and D. R. Flanagan, J. Pharm. Sci., 2011, 100, 1736–1744 CrossRef CAS PubMed.
- M. L. Cheney, D. R. Weyna, N. Shan, M. Hanna, L. Wojtas and M. J. Zaworotko, J. Pharm. Sci., 2011, 100, 2172–2181 CrossRef CAS PubMed.
- P. P. Bag, S. Ghosh, H. Khan, R. Devarapalli and C. M. Reddy, CrystEngComm, 2014, 16, 7393–7396 RSC.
- S. H. Thorat, S. K. Sahu, M. V. Patwadkar, M. V. Badiger and R. G. Gonnade, J. Pharm. Sci., 2015, 104, 4207–4216 CrossRef CAS PubMed.
- D. Stepanos, M. Jure, M. Gosteva, J. Popelis, G. Kiselovs and A. Mishnev, CrystEngComm, 2016, 18, 1235–1241 RSC.
- G. M. Sheldrick, SHELXS-97 AND SHELXL-97, Programs for the Solution and Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, X-Seed, Graphical Interface to SHELX-97 and POV-RAY, a Program for Better Quality of Crystallographic Figures, University of Missouri-Columbia, Columbia, Mo, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP plots, DVS profiles, and PXRD patterns of the powders obtained from grinding and stability experiments. CCDC 1062956–1062961. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra01802e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.