
Improved hepatic physiology in hepatic cytochrome P450 reductase null (HRN™) mice dosed orally with fenclozic acid
James A.
Akingbasote
a,
Alison J.
Foster
*b,
Huw B.
Jones
b,
Rhiannon
David
b,
Nigel J.
Gooderham
c,
Ian D.
Wilson
c and
J. Gerry
Kenna
d
aMRC Centre for Drug Safety Science, University of Liverpool, Liverpool, L69 3GE, UK. E-mail: j.akingbasote@liverpool.ac.uk
bDrug Safety and Metabolism, Unit 310 – Darwin Building, Cambridge Science Park, Milton Road, Cambridge, CB4 0WG, UK. E-mail: alison.foster2@astrazeneca.com; huwbjoneshb@gmail.com; Rhiannon.David@astrazeneca.com
cSection of Computational and Systems Medicine, Department of Surgery and Cancer Faculty of Medicine, Imperial College London, South Kensington Campus, London, SW7 2AZ UK. E-mail: n.gooderham@imperial.ac.uk; iwilson@imperial.ac.uk
dDrug Safety Consultant, Macclesfield, UK. E-mail: j.gerrykenna@gmail.com; Tel: +44 (0)1625432113
First published on 9th November 2016
Abstract
Hepatic NADPH-cytochrome P450 oxidoreductase null (HRN™) mice exhibit no functional expression of hepatic cytochrome P450 (P450) when compared to wild type (WT) mice, but have normal hepatic and extrahepatic expression of other biotransformation enzymes. We have assessed the utility of HRN™ mice for investigation of the role of metabolic bioactivation in liver toxicity caused by the nonsteroidal anti-inflammatory drug (NSAID) fenclozic acid. In vitro studies revealed significant NADPH-dependent (i.e. P450-mediated) covalent binding of [14C]-fenclozic acid to liver microsomes from WT mice and HRN™ mice, whereas no in vitro covalent binding was observed in the presence of the UDP-glucuronyltransferase cofactor UDPGA. Oral fenclozic acid administration did not alter the liver histopathology or elevate the plasma liver enzyme activities of WT mice, or affect their hepatic miRNA contents. Livers from HRN™ mice exhibited abnormal liver histopathology (enhanced lipid accumulation, bile duct proliferation, hepatocellular degeneration, necrosis, inflammatory cell infiltration) and plasma clinical chemistry (elevated alanine aminotransferase, glutamate dehydrogenase and alkaline phosphatase activities). Modest apparent improvements in these abnormalities were observed when HRN™ mice were dosed orally with fenclozic acid for 7 days at 100 mg kg−1 day−1. Previously we observed more marked effects on liver histopathology and integrity in HRN™ mice dosed orally with the NSAID diclofenac for 7 days at 30 mg kg−1 day−1. We conclude that HRN™ mice are valuable for assessing P450-related hepatic drug biotransformation, but not for drug toxicity studies due to underlying liver dysfunction. Nonetheless, HRN™ mice may provide novel insights into the role of inflammation in liver injury, thereby aiding its treatment.
Introduction
HRN™ mice lack the gene encoding hepatic NADPH-cytochrome P450 oxidoreductase (CYP P450 reductase; POR; EC 1.6.2.4),1 which catalyses the transfer of electrons from cytochrome P450 (P450) and other endoplasmic reticulum enzymes which use NADPH as co-factor.2,3 Hence HRN™ mice are deficient in hepatic P450 activities, whereas the activities of other hepatic drug metabolizing enzymes and extra-hepatic P450 enzymes are unaffected.1,4In vivo biotransformation studies have shown that HRN™ mice can provide valuable insights into the role played by hepatic P450s in xenobiotic biotransformation. For example, formation of the toxic quinoneimine metabolite was not observed when HRN™ mice were dosed with acetaminophen (paracetamol).1 This model may also prove especially useful in circumstances where both oxidative P450-mediated metabolism and conjugation reactions result in the production of reactive metabolites that might cause toxicity. For example, the nonsteroidal anti-inflammatory drug (NSAID) diclofenac causes hepatotoxicity in patients and is metabolised in vivo via both P450-mediated oxidative and UDP-glucuronyltransferase (UGT)-mediated conjugative routes in wild type mice,5 humans and other animal species.6 Currently it is unclear whether oxidation or conjugation of diclofenac, or a combination of both, is responsible for the hepatotoxicity caused by the drug in patients.When administered to HRN™ mice, diclofenac was metabolized overwhelmingly to conjugates (mainly to taurine and glucuronic acid), with little evidence of oxidative metabolism.7 In a previous study8 we undertook toxicological studies in wild type (WT) and HRN™ mice dosed orally with diclofenac to assess whether use of this model might provide novel insights into the possible role played by conjugation of diclofenac to chemically reactive acyl glucuronides in the liver toxicity observed in human patients. HRN™ mice treated with dose vehicle alone exhibited enhanced hepatic lipid accumulation and degenerative liver histopathologies that primarily affected bile ducts and periportal hepatocytes, plus markedly elevated activities in plasma of various liver-derived enzymes, which were not present in livers from dose vehicle treated WT mice. Unexpectedly, the severities of the abnormal liver histopathology and plasma liver enzyme findings in HRN™ mice were reduced markedly when HRN™ mice were dosed orally with diclofenac for 7 days at 30 mg kg−1 day−1.8
A possible explanation for the positive effect of diclofenac administration to HRN™ mice on liver pathophysiology is via a reduction/inhibition of hepatic inflammation.9,10 To gain further insight into whether this is a plausible hypothesis, and determine whether the effects were unique to diclofenac or were seen with other drugs in this class, in the present studies we have explored tissue histopathology and plasma clinical chemistry in HRN™ and WT mice dosed orally for seven days with dose vehicle alone or fenclozic acid (Myalex®). Fenclozic acid is a carboxylic acid NSAID with potent analgesic and antipyretic activities11 whose clinical development was terminated due to liver injury observed in clinical trials12 and which also is metabolised to chemically reactive metabolites.13,14 Recently it has recently been reported that miRNA could be sensitive and informative biomarkers of hepatotoxicity; for example, paracetamol induced toxicity in mice rapidly resulted in a decline in hepatic miRNAs such as miR-122 and miR-192.15 This observation prompted us also to investigate whether altered amounts of hepatic miRNAs were evident when HRN™ and WT mice were treated with fenclozic acid.
Materials and methods
Chemicals
Fluothane™, fenclozic acid and [14C]-fenclozic acid (purity >99%, specific activity 60.5 mCi mmol−1) were obtained from AstraZeneca. Pierce™ BCA Protein Assay Kit was from Thermo Scientific (Rockford, IL). The EnzyChrom™ Alanine Transaminase Assay Kit was from BioAssay Systems (Hayward, CA). The bile acid reagent was from Diazyme Laboratories (Dresden, Germany) and other clinical chemistry reagents were from Roche Diagnostics (Burgess Hill, UK). All other reagents and solvents were from Sigma-Aldrich Company Ltd (Poole, UK) or Fisher Scientific (Loughborough, UK).In vivo studies
The experimentation, transportation and care of the mice were performed in accordance with approved project license, the relevant national guidelines (UK Animals and Scientific Procedures Act, 1986) and in compliance with AstraZeneca regulations on the ethical treatment of animals. All experiments were approved by AstraZeneca's ethical committee. Statistical analysis was undertaken to determine the minimum number of animals to give 80% power to detect fold changes of approximately 2–3 fold in ALT/ALP via pairwise comparison of groups with 5% significance level, using a t-test approach (2-sided) via ANOVA. The animals were individually housed in standard conditions with freely available access to drinking water and food. All animals were thoroughly examined before dosing commenced and at weekly intervals from the start of dosing through to termination. They were also visually inspected at least once daily after dosing, paying particular attention up to 3 hours after dosing.The dose levels and route of administration of fenclozic acid were based on information available in the literature.12,16 Male C57/Bl6 WT mice (8–10 weeks old (21–25 g), Charles River, Margate, UK) were treated daily for 7 days, by oral gavage, with dose vehicle alone (0.1 M sodium phosphate buffer, pH 7.7; Group 1), or fenclozic acid at doses of 50 or 100 mg kg−1 day−1 (in the same vehicle), 10 mL kg−1 (Groups 2 and 3 respectively; n = 5 per group). Male HRN™ mice (4–11 weeks old (20–27 g); Taconic Farms Inc. Germantown, NY, USA) were treated by daily oral gavage for 7 days with fenclozic acid at a dose of 100 mg kg−1 day−1 (Group 5; n = 5), or with equivalent volumes of dose vehicle alone (Group 4; n = 4). The HRN™ mice had been developed using C57BL/6J background strain mice.1 Blood samples (50 μL) were obtained from the animals by tail bleeding on days -1, 2 and 4 of the study and plasma ALT activities were determined. At 24 h after the final administration of fenclozic acid or dose vehicle, animals were euthanized under fluothane™ anaesthesia. Terminal blood samples were collected for immediate analysis of plasma alanine aminotransferase (ALT), alkaline phosphatase (ALP) and glutamate dehydrogenase (GLDH), which was undertaken using a Roche Modular P800 Chemistry Analyser (Roche Diagnostics Ltd, Burgess Hill, UK). Additionally, ALT activities of plasma samples were determined using an EnzyChrom™ Alanine Transaminase Assay Kit (Bioassays Systems, Hayward, CA).
The liver, intestines, oesophagus and stomach/duodenum were examined for any abnormalities and weighed and tissue samples were processed to paraffin blocks using standard histologic procedures. Sections were cut and were stained with haematoxylin and eosin (H&E) for histopathologic examination using light microscopy. The severities of tissue histopathology findings were scored qualitatively, using the following scheme: Grade 1 = Minimal/very few/very small; Grade 2 = Slight/few/small; Grade 3 = Moderate/moderate number/moderate size; Grade 4 = Marked/many/large; Grade 5 = Massive/extensive number/extensive size.
Statistical analysis of data was performed by paired or unpaired t-testing, using GraphPad Prism version 4.03 (GraphPad Software, San Diego, CA).
To quantify hepatic miRNAs, total RNA was extracted from liver tissue using the MiRVana PARIS kit (Life Technologies, Paisley, UK) and was quantified using a NanoPhotometer (Implen GmbH, Munchen, Germany). The ratios A260/A280 and A260/A230 were used to assess RNA quality. Total RNA was reverse transcribed using the TaqMan MicroRNA Reverse Transcription Kit (Life Technologies, Paisley, UK). Mature miRNA TaqMan assays (Life Technologies) were used to generate cDNA, which was amplified using the Taqman 2× Universal PCR master mix, No AmpErase UNG (Life Technologies), with each reaction performed in triplicate. The qPCR data were analysed using the ABI 7500 Sequence Detection System (Life Technologies) and the comparative Ct Method (ΔΔCT Method).17 Calibration was based on the expression of snoRNA, as per manufacturer's instructions.
Preparation of liver microsomes and determination of microsomal cytochrome c reductase activity
The preparation of liver microsomes from both WT and HRN™ mouse livers was undertaken as described by Akingbasote et al.8 Protein concentrations were determined using the Pierce BCA Protein Assay Kit®.In vitro covalent binding (CVB) of [14C]-fenclozic acid to liver microsomes
Determination of CVB for [14C]-fenclozic acid was performed as described by Day et al.18 Liver microsomes (1 mg mL−1 final protein concentration) were incubated at 37 °C with 10 μM [14C]-fenclozic acid in 0.1 M phosphate buffer supplemented with 10 mM MgCl2, pH 7.0, in the presence or absence of NADPH (2 mM) or UDPGA (4 mM). In some experiments, 1-aminobenzotriazole (ABT; 1 mM) was also added to the incubation mixture. The final assay volume was 500 μL. After incubation for 0 and 60 min, aliquots (200 μL) were mixed with 300 μL of chilled acetone and then a further 500 μL acetone was added. Precipitated proteins were collected onto filter disks using a Brandel 96-sample cell harvester (Gaithersburg MD, USA), washed with 80% methanol, then protein-bound [14C] was quantified by liquid scintillation counting as described by Foster et al.19 Non-specific binding (NSB) was determined by incubating microsomes in the absence of fenclozic acid for 1 hour at 37 °C, then ‘back-adding’ 10 μM [14C]-fenclozic acid immediately prior to processing the samples. Levels of CVB were expressed as pmol equivalents per mg protein.Results
Abnormal liver histopathology and plasma clinical chemistry of vehicle treated HRN™ mice
WT mice which were dosed orally for 7 days with dose vehicle alone exhibited no abnormalities in liver histology, other than minimal extents of non-periportal hepatocyte necrosis with inflammatory cell infiltration and minimal mixed inflammatory cell aggregates (Fig. 1A and Table 1). The frequencies and severities of these findings were in line with those observed historically in WT control mice. As we have reported previously,8 a variety of histopathological findings were observed in livers from dose vehicle treated HRN™ mice that were not evident in livers from WT mice. Elevated numbers and sizes of hepatocellular fat vacuoles, increased extents of bile duct proliferation and periportal hepatocyte necrosis with accompanying inflammatory cell infiltration were observed in each of four livers from vehicle-treated HRN™ mice, while a few necrotic hepatocyte foci were also observed in one liver (Fig. 1A, C and Table 1). Furthermore, plasma from mice exhibited markedly elevated ALT and GLDH activities and less markedly elevated ALP activity, when compared with plasma from vehicle treated WT mice (Table 2).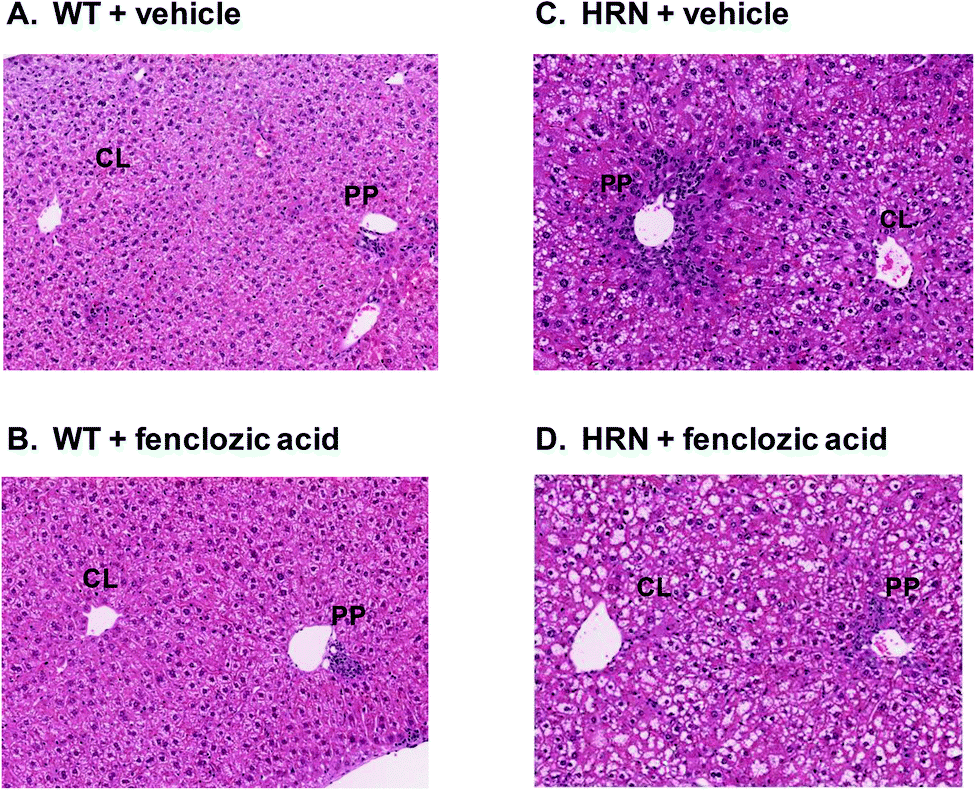 | ||
| Fig. 1 Liver histopathology in wild type (A, B) and HRN™ (B, D) mice treated for 7 days with dose vehicle (A, B) or 100 mg kg−1 day−1 fenclozic acid (C, D). Representative photomicrographs from formalin fixed and H&E stained liver sections at ×20 magnification are shown. The positions of centrilobular (CL) and periportal (PP) hepatocytes are indicated. | ||
| Histopathology finding | Severity | Number of WT mice affected | Number of HRN™ mice affected | |||
|---|---|---|---|---|---|---|
| Vehicle control (n = 5) | 50 mg kg−1 day−1 fenclozic acid (n = 5) | 100 mg kg−1 day−1 fenclozic acid (n = 5) | Vehicle control (n = 4) | 100 mg kg−1 day−1 fenclozic acid (n = 5) | ||
| Hepatocellular fat vacuolation | Grade 2 | 0 | 0 | 0 | 0 | 0 |
| Grade 3 | 0 | 0 | 0 | 4 | 4 | |
| Grade 4 | 0 | 0 | 0 | 0 | 1 | |
| Multifocal hepatocellular necrosis | Grade 2 | 0 | 0 | 0 | 1 | 1 |
| Bile duct proliferation | Grade 1 | 0 | 0 | 0 | 0 | 2 |
| Grade 2 | 0 | 0 | 0 | 1 | 1 | |
| Grade 3 | 0 | 0 | 0 | 3 | 2 | |
| Periportal hepatocyte necrosis/inflammatory cell infiltration | Grade 1 | 0 | 0 | 0 | 0 | 3 |
| Grade 2 | 0 | 0 | 0 | 4 | 1 | |
| Hepatocyte necrosis with inflammatory cell infiltration | Grade 1 | 1 | 0 | 0 | 2 | 4 |
| Grade 2 | 0 | 0 | 0 | 0 | 1 | |
| Mixed inflammatory cell aggregates | Grade 1 | 2 | 3 | 3 | 0 | 0 |
| Parameter | WT mice treated with: | HRN™ mice treated with: | ||
|---|---|---|---|---|
| Dose vehicle (n = 5) | Fenclozic acid, 100 mg kg−1 (n = 5) | Dose vehicle (n = 4) | Fenclozic acid, 100 mg kg−1 (n = 5) | |
| *p < 0.05, compared with vehicle control value. | ||||
| ALT (IU L−1) | 37.6 ± 12.6 | 66.4 ± 25.8 | 1,764.0 ± 859.7 | 1,131.6 ± 1056.3 |
| AST (IU L−1) | 56.0 ± 21.5 | 71.2 ± 22.3 | 1,153.0 ± 656.2 | 965.6 ± 1,022.7 |
| ALP (IU L−1) | 162.4 ± 35.1 | 128.0 ± 24.7 | 444.0 ± 72.7 | 325.6 ± 31.1* |
| GLDH (IU L−1) | 15.2 ± 9.8 | 16.8 ± 5.2 | 521.5 ± 332.08 | 174.4 ± 152.8 |
| miR-122 (relative expression) | 1.0 | 0.96 | 1.0 | 0.86 |
Hepatic effects of oral administration of fenclozic acid to HRN™ and WT mice
Oral daily administration of fenclozic acid at doses of 50 or 100 mg kg−1 day−1 to WT mice did not result in any compound-related alterations in liver histopathology (Fig. 1B and Table 1), in hepatic expression of miR-122 or in plasma ALT or GLDH activities (Table 2). However, a dose dependent reduction in plasma ALP activity was observed in fenclozic acid treated WT mice (Table 2).When compared with HRN™ mice treated with dose vehicle alone, HRN™ mice dosed orally for 7 days with fenclozic acid at 100 mg kg−1 day−1 exhibited less severe bile duct proliferation and periportal hepatocyte necrosis with accompanying inflammatory cell infiltration (Fig. 1, compare panels D and C; Table 1). The modest apparent reduction in severity of liver histopathology in fenclozic acid dosed HRN™ mice was consistent with plasma clinical chemistry data, which demonstrated a statistically significant (p < 0.05) reduction in plasma ALP activity and by reductions in plasma ALT and GLDH activities that did not achieve statistical significance (Table 2). However, the relative expression of hepatic miR-122 in these animals was unaffected by fenclozic acid treatment (Table 2).
Upper GI histopathology in fenclozic acid treated HRN™ and WT mice
Neither HRN™ nor WT mice dosed orally for 7 days with dose vehicle alone exhibited abnormalities in upper gastrointestinal (GI) histopathology, apart from minimal or slight oesophageal muscle cell alterations and minimal glandular epithelial cysts at the stomach limiting ridge in several animals (Table 3). The frequencies and severities of these upper GI findings in these animals were in line with those observed historically in WT control mice.| Organ | Histopathology finding | Severity | Number of WT mice affected | Number of HRN™ mice affected | |||
|---|---|---|---|---|---|---|---|
| Vehicle control (n = 5) | 50 mg kg−1 day−1 fenclozic acid (n = 5) | 100 mg kg−1 day−1 fenclozic acid (n = 5) | Vehicle control (n = 4) | 100 mg kg−1 day−1 fenclozic acid (n = 5) | |||
| a Tissues from 4 animals were evaluated. | |||||||
| Oesophagus | No abnormalities | 0 | 2 | 0 | 4 | 3 | 3 |
| Adjacent muscle degeneration/necrosis/inflammation | Grade 1 | 2 | 0 | 0 | 1 | 0 | |
| Grade 2 | 1 | 0 | 1 | 0 | 1 | ||
| Stomach | No abnormalities | 4 | 4 | 1a | 4 | 2 | |
| Pyloric ulceration | Grade 2 | 0 | 0 | 0 | 0 | 1 | |
| Pyloric erosion | Grade 1 | 0 | 0 | 2 | 0 | 0 | |
| Grade 2 | 0 | 0 | 1 | 0 | 2 | ||
| Pyloric gastritis | Grade 1 | 0 | 0 | 1 | 0 | 0 | |
| Grade 2 | 0 | 0 | 0 | 0 | 2 | ||
| Glandular epithelial cyst(s) at limiting ridge | Grade 2 | 1 | 1 | 0 | 0 | 0 | |
No alterations in upper GI histopathology were observed in WT mice dosed with fenclozic acid at 50 mg kg−1 day−1 for 7 days. However, three of four WT mice dosed with fenclozic acid at 100 mg kg−1 day−1 exhibited evidence of stomach pyloric erosion and pyloric gastritis was observed in one of these animals (Table 3). Similar stomach lesions were observed in three of five HRN™ mice dosed with fenclozic acid at 100 mg kg−1 day−1 (Table 3).
In vitro CVB of [14C]-fenclozic acid to liver microsomes from WT and HRN™ mice
CVB of radiolabel to proteins (>170 pmol mg−1 protein) was observed when [14C]-fenclozic acid was incubated for 60 min with liver microsomes from WT mice that were supplemented with NADPH (Table 4). In contrast, CVB to proteins was not observed when equivalent incubations were undertaken either in the absence of NADPH, or in the presence of UDPGA in place of NADPH. Similarly, no CVB to proteins was seen when the incubation with NADPH was undertaken for 0 min or when [14C]-fenclozic acid was “back-added” to microsomes that had been incubated with NADPH for 60 min. A marked and statistically significant reduction in the amount of NADPH-dependent CVB to proteins (to <50 pmol mg−1 protein) was observed when [14C]-fenclozic acid was incubated for 60 min with WT liver microsomes in the presence of the CYP inhibitor ABT. Detectable CVB was also observed when [14C]-fenclozic acid was incubated for 60 min with liver microsomes from HRN™ mice in the presence of NADPH. However, the amount of CVB to HRN™ mouse liver microsomes was approximately 6-fold lower than the amount of CVB to WT liver microsomes. Furthermore, the CVB to HRN™ mouse liver microsomes was not altered in the presence of ABT. A detectable amount of CVB was not observed when [14C]-fenclozic acid was incubated for 60 min with liver microsomes from HRN™ mice in the presence of UDPGA (Table 4).| pmol equivalents per mg protein | ||||
|---|---|---|---|---|
| WT microsomes | HRN™ microsomes | |||
| Mean | SD | Mean | SD | |
| *Significantly elevated vs. NADPH background value (+NADPH, T = 0); p < 0.001. #Significantly reduced vs. CVB in absence of ABT (+NADPH, T = 60); p < 0.001. | ||||
| Mic − NADPH, T = 0 | 7.7 | 2.3 | 6.0 | 0.8 |
| Mic − NADPH, T = 60 | 9.7 | 1.9 | 9.8 | 1.7 |
| Mic + NADPH, T = 0 | 19.4 | 4.6 | 9.0 | 2.9 |
| Mic + NADPH, T = 60 | 187.7* | 30.4 | 32.2* | 7.0 |
| Mic + ABT, T = 0 | 9.7 | 1.7 | 10.4 | 0.0 |
| Mic + ABT, T = 60 | 39.5# | 10.0 | 29.2 | 4.2 |
| Mic + UDPGA, T = 0 | 7.1 | 1.9 | 8.2 | 1.9 |
| Mic + UDPGA, T = 60 | 10.4 | 2.3 | 9.2 | 2.8 |
| Back added | 33.0 | 8.5 | 16.3 | 4.6 |
Discussion
The clinical development of fenclozic acid was terminated due to liver injury in clinical trials,12 which was not observed in any of the animal species used in its preclinical safety testing. In order to gain insight into the mechanisms responsible for the human-specific hepatotoxicity of this drug, previously we investigated its potential for bioactivation in vitro and in vivo. The in vitro studies were undertaken using isolated human hepatocytes13 and also rat, dog and human liver microsomes.14 These revealed relatively high levels of NADPH-dependent CVB to liver proteins, implying that oxidative bioactivation had occurred. In addition, in vivo metabolite profiling studies identified a variety of oxidized and glutathione-derived metabolites in urine and bile from fenclozic acid treated rats20 and WT mice (Pickup et al., in preparation). These findings provided evidence that metabolic bioactivation of fenclozic acid also occurs in vivo and support our suggestion13,14 that this process could be responsible for the hepatotoxicity caused by the drug in humans in vivo. Such oxidative metabolites were not seen in urine or faeces obtained from HRN™ mice that had been dosed with fenclozic acid, where its metabolism was via the carboxylic acid moiety and resulted in the formation of an acyl glucuronide as well as glycine and taurine conjugates.7 These various metabolic pathways are summarised in Fig. 2.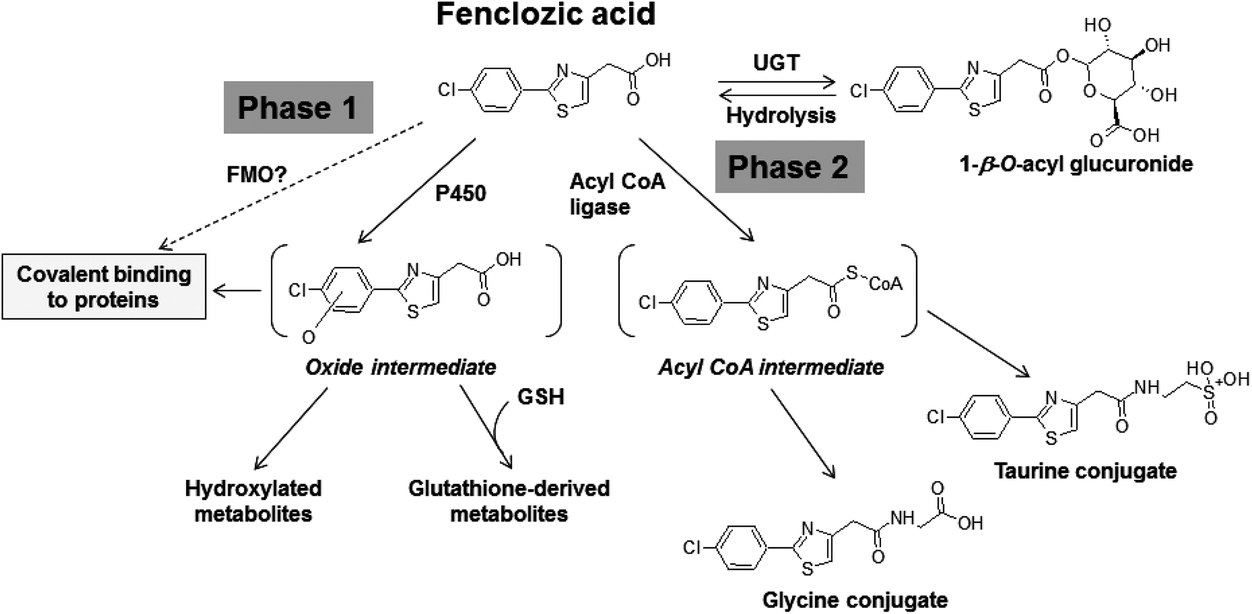 | ||
| Fig. 2 Metabolism of fenclozic acid. FMO, flavin monooxygenase; CYP, cytochrome P450; UGT, UDP-glucuronyltransferase; GSH, reduced glutathione. | ||
The NADPH-dependent CVB of radiolabelled fenclozic acid to WT liver microsomal proteins that we observed in our present investigations are consistent with these earlier studies. However, the small but statistically significant amount of CVB to proteins in liver microsomes from HRN™ mice, which was unaffected by addition of the P450 inhibitor ABT, was a novel and unexpected finding. A plausible explanation is that fenclozic acid is bioactivated in livers from HRN™ mice by a novel mechanism, which is distinct from the P450 mediated process evident in livers of rats and WT mice and shown in Fig. 2. Additional studies will be required to characterise the responsible enzyme, although a possible candidate is flavin monoxygenase (FMO).21 Since we observed no evidence of compound related liver toxicity following in vivo administration of fenclozic acid to either WT or HRN™ mice, our current investigations were unable to provide insight into the possible relevance of metabolic bioactivation to its human hepatotoxicity.
Similar patterns and severities of adverse stomach pathology were observed when both WT and HRN™ mice were dosed orally with fenclozic acid for 7 days at 100 mg kg−1 day−1, which was not observed in the vehicle treated animals. The likely explanation for this toxicity is impaired wound healing which arises as a consequence of cyclooxygenase inhibition, since such toxicity is a common finding in animals and humans dosed with numerous other NSAIDs.22 However, whereas vehicle-treated HRN™ mice exhibited no abnormalities in stomach and gastrointestinal pathology when compared with WT mice, as we have noted previously8 these animals have a variety of underlying abnormalities in liver pathology and function (resulting in markedly abnormal plasma clinical chemistry). Therefore whilst HRN™ mice are useful for dissecting out the role of hepatic P450 dependent processes in drug metabolism, they appear to be less well suited to investigations of mechanisms of hepatic drug toxicity.
Interestingly, we found no changes in the relative expression of hepatic miR-122 in either mouse strain, irrespective of treatment. Paracetamol-induced hepatotoxicity has been reported to reduce the expression of the abundant hepatic enriched miR-122 with a concomitant release of miR-122 into the plasma.15 This finding prompted the proposal that changes in miRNA expression could be sensitive and informative biomarkers of drug-induced liver injury. Although the data reported herein fail to support this hypothesis, there are significant experimental differences between our study and paracetamol induced toxicity. Specifically, the acute response to paracetamol toxicity and the slower onset of hepatotoxicity induced by fenclozic acid in humans.12 In addition, the hepatic miR-122 levels were assessed after 7 days of fenclozic acid administration to mice. It is also pertinent to note that the hepatic miR-122 levels were unaffected by fenclozic acid treatment of the HRN™ mice, despite the apparent drug-induced improvement of hepatic histology; again hepatic levels of miR-122 were assessed after 7 days of treatment. This lack of effect on hepatic miR-122 expression in both mouse strains may be a reflection of the regulatory function of the miRs and their role in maintaining homeostasis. In this context, it would be interesting to measure circulating and hepatic miR-122 expression during the first 24 h of treatment of mice with fenclozic acid.
The abnormal and degenerative liver histopathology and high plasma activities of liver-derived enzymes present in vehicle treated HRN™ mice are likely to be a consequence of impaired lipid metabolism and bile acid homeostasis, which arise due to the absence of hepatic P450 activities in these animals.1–3 Previously we reported a marked reduction in both degenerative liver histopathology, and plasma liver-derived enzyme activities (ALT, GLDH and ALP), when HRN™ mice were dosed orally for 7 days with diclofenac.8 Our present results demonstrate more modest apparent improvements in plasma liver-derived enzyme activities and liver histopathology when HRN™ mice were dosed orally for 7 days with a second NSAID, fenclozic acid, at a dose of 100 mg kg−1 day−1. Additional experiments are required to clarify whether the apparent effects observed in HRN™ mice dosed with fenclozic acid can indeed be attributed to compound administration. Nonetheless, both diclofenac and fenclozic acid have, by design, anti-inflammatory properties and are potent inhibitors of the enzyme cyclooxygenase, which converts arachidonic acid to prostanoid inflammatory mediators.23 Inflammatory responses in the liver have been found to mediate hepatic injury caused by endotoxins and various other xenobiotics.24,25 Hepatic inflammation involves activation of Kupffer cells, which release biologically active compounds (including cytokines, growth factors, arachidonic-acid-derived inflammatory mediators and lysosomal enzymes) that cause hepatocyte necrosis and liver inflammation. Inhibition of cyclooxygenase in Kupffer cells has been found to impair their activation and it has been proposed that NSAIDs may provide a promising strategy for mitigation of the liver inflammation, and its resulting liver pathologies, that occurs in human non-alcoholic fatty liver disease (NAFLD)26,27 and other liver types of liver disease.28
Therefore our current findings are consistent with our previous proposal8 that inflammatory processes may play important roles in the liver injury evident in HRN™ mice, and that the positive effects of diclofenac on the liver histopathology and function of these animals could be due to the anti-inflammatory activity of the drug. It is also conceivable that the liver abnormalities evident in HRN™ mice might provide a useful model system for exploration of mechanisms of NAFLD and other clinically important human liver diseases, which could aid the assessment of the potential therapeutic value of NSAIDs or other novel strategies for treatment of liver inflammation.
Conflicts of interest
The authors declare no conflicts of interest.Abbreviations
| HRN™ | Hepatic cytochrome P450 reductase null mice |
| WT | Wild type C57BL6J mice |
| P450 | Cytochrome P450 |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| UDPGA | Uridine 5′-diphospho-glucuronic acid |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| GLDH | Glutamate dehydrogenase |
| ALP | Alkaline phosphatase |
| NAFLD | Non-alcoholic fatty liver disease |
| miR-122 | Liver specific miRNA 122 |
| snoRNA | Small nucleolar RNAs. |
Acknowledgements
We thank Jonathan Greenall, Elizabeth Johnson and Peter Cotton for their excellent technical assistance, James Noakes for his directing of the in vivo study and Marie South for her statistical advice.References
- C. J. Henderson, D. M. Otto, D. Carrie, M. A. Magnuson, A. W. Mclaren, I. Rosewell and C. R. Wolf, Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase, J. Biol. Chem., 2003, 278, 13480–13486 CrossRef CAS PubMed.
- X. J. Wang, M. Chamberlain, O. Vassieva, C. J. Henderson and C. R. Wolf, Relationship between hepatic phenotype and changes in gene expression in cytochrome P450 reductase (POR) null mice, Biochem. J., 2005, 388, 857–867 CrossRef CAS PubMed.
- D. M. Mutch, B. Klocke, P. Morrison, C. A. Murray, C. J. Henderson, M. Seifert and G. Williamson, The disruption of hepatic cytochrome p450 reductase alters mouse lipid metabolism, J. Proteome Res., 2007, 6, 3976–3984 CrossRef CAS PubMed.
- M. Stiborová, V. M. Arlt, C. J. Henderson, C. R. Wolf, V. Kotrbová, M. Moserová, J. Hudecek, D. H. Phillips and E. Frei, Role of hepatic cytochromes P450 in bioactivation of the anticancer drug ellipticine: studies with the hepatic NADPH:cytochrome P450 reductase null mouse, Toxicol. Appl. Pharmacol., 2008, 226(3), 318–327 CrossRef PubMed.
- S. Sarda, C. Page, K. Pickup, T. Schulz-Utermoehl and I. Wilson, Diclofenac metabolism in the mouse: Novel in vivo metabolites identified by high performance liquid chromatography coupled to linear ion trap mass spectrometry, Xenobiotica, 2012, 42, 179–194 CrossRef CAS PubMed.
- U. A. Boelsterli, Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity, Toxicol. Appl. Pharmacol., 2003, 192, 307–322 CrossRef CAS PubMed.
- K. Pickup, J. Wills, A. Rodrigues, H. B. Jones, C. Page, S. Martin, S. Sarda and I. Wilson, The metabolic fate of [14C]-fenclozic acid in the hepatic reductase null (HRN) mouse, Xenobiotica, 2014, 44, 164–173 CrossRef CAS PubMed.
- J. A. Akingbasote, A. J. Foster, I. Wilson, S. Sarda, H. B. Jones and J. G. Kenna, Hepatic effects of repeated oral administration of diclofenac to hepatic cytochrome P450 reductase null (HRN™) and wild–type mice, Arch. Toxicol., 2016, 90, 853–862 CrossRef CAS PubMed.
- N. Alkhouri, L. J. Dixon and A. E. Feldstein, Lipotoxicity in non-alcoholic fatty liver disease: not all lipids are created equal, Expert Rev. Gastroenterol. Hepatol., 2009, 3(4), 445–451 CrossRef CAS PubMed.
- V. Vaish and S. N. Sanyal, Chemopreventive effects of NSAIDs on cytokines and transcription factors during the early stages of colorectal cancer, Pharmacol. Rep., 2011, 63(5), 1210–1221 CrossRef CAS PubMed.
- T. M. Chalmers, J. H. Kellgren and D. S. Platt, Evaluation in man of fenclozic acid (I.C.I. 54,450: Myalex), a new anti-inflammatory agent. II. Clinical trial in patients with rheumatoid arthritis, Ann. Rheum. Dis., 1969, 28(6), 595–601 CrossRef CAS PubMed.
- S. Alcock, An anti-inflammatory compound: non-toxic to animals but with an adverse action in man, Proc. Eur. Soc. Study Drug Toxic, 1971, 12, 7 Search PubMed.
- R. A. Thompson, E. M. Isin, Y. Li, L. Weidolf, K. Page, I. Wilson, S. Swallow, B. Middleton, S. Stahl, A. J. Foster, H. Dolgos, R. Weaver and J. G. Kenna, In Vitro Approach to Assess the Potential for Risk of Idiosyncratic Adverse Reactions Caused by Candidate Drugs, Chem. Res. Toxicol., 2012, 25(8), 1616–1632 CrossRef CAS PubMed.
- A. V. Rodrigues, H. E. Rollison, S. Martin, S. Sarda, T. Schulz-Utermoehl, S. Stahl, F. Gustafsson, J. Eakins, J. G. Kenna and I. D. Wilson, In vitro exploration of potential mechanisms of toxicity of the human hepatotoxic drug fenclozic acid, Arch. Toxicol., 2013, 87, 1569–1579 CrossRef CAS PubMed.
- K. Wang, S. Zhang, B. Marzolf, P. Troisch, A. Brightman, Z. Hu, L. E. Hood and D. J. Galas, Circulating microRNAs, potential biomarkers for drug-induced liver injury, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(11), 4402–4407 CrossRef CAS PubMed.
- B. B. Newbould, The pharmacology of fenclozic acid (2-(4-chlorophenyl)-thiazol-4-ylacetic acid; I.C.I. 54,450; ‘Myalex’); a new compound with anti-inflammatory, analgesic and antipyretic activity, Br. J. Pharmacol., 1969, 35(3), 487–497 CrossRef CAS PubMed.
- K. J. Livak and T. D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods, 2001, 25(4), 402–408 CrossRef CAS PubMed.
- S. H. Day, A. Mao, R. White, T. Schulz-Utermoehl, R. Miller and M. G. Beconi, A semi-automated method for measuring the potential for protein covalent binding in drug discovery, J. Pharmacol. Toxicol. Methods, 2005, 52, 278–285 CrossRef CAS PubMed.
- A. J. Foster, L. H. Prime, F. Gustafsson, D. G. Temesi, E. M. Isin, J. Midlöv, N. Castagnoli Jr. and J. G. Kenna, Bioactivation of the cannabinoid receptor antagonist rimonabant to a cytotoxic iminium ion metabolite, Chem. Res. Toxicol., 2013, 26(1), 124–135 CrossRef CAS PubMed.
- S. Martin, E. M. Lenz, W. Keene and M. R. Clench, Identification of the reactive metabolites of fenclozic acid in bile duct cannulated rats, Anal. Chem., 2014, 86(22), 11281–11289 CrossRef CAS PubMed.
- R. S. Foti and D. K. Dalvie, Cytochrome P450 and Non-Cytochrome P450 Oxidative Metabolism: Contributions to the Pharmacokinetics, Safety, and Efficacy of Xenobiotics, Drug Metab. Dispos., 2016, 44(8), 1229–1245 CrossRef CAS PubMed.
- H. Mizuno, C. Sakamoto, K. Matsuda, K. Wada, T. Uchida, H. Noguchi, T. Akamatsu and M. Kasuga, Induction of cyclooxygenase 2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice, Gastroenterology, 1997, 112, 387–397 CrossRef CAS.
- T. J. Gan, Diclofenac: an update on its mechanism of action and safety profile, Curr. Med. Res. Opin., 2010, 26, 1715–1731 CrossRef CAS PubMed.
- J. Claria and E. Titos, Kupffer cell, Gastroenterol. Hepatol., 2004, 27, 264–273 CrossRef CAS PubMed.
- M. D. Wheeler, Endotoxin and Kupffer cell activation in alcoholic liver disease, Alcohol Res. Health, 2003, 27, 300–306 Search PubMed.
- A. Planaguma, J. Claria, R. Miquel, M. Lopez-Parra, E. Titos, J. L. Masferrer, V. Arroyo and J. Rodes, The selective cyclooxygenase-2 inhibitor SC-236 reduces liver fibrosis by mechanisms involving non-parenchymal cell apoptosis and PPARgamma activation, FASEB J., 2005, 19(9), 1120–1122 CAS.
- B. J. Park, Y. J. Lee and H. R. Lee, Chronic liver inflammation: clinical implications beyond alcoholic liver disease, World J. Gastroenterol., 2014, 20(9), 2168–2175 CrossRef CAS PubMed.
- N. Ferre and J. Claria, New insights into the regulation of liver inflammation and oxidative stress, Mini–Rev. Med. Chem., 2006, 6, 1321–1330 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |