
A DEHP plasticizer alters synaptic proteins via peroxidation
Shaohui
Wang†
,
Pengyan
Zhang†
,
Ruifang
Liu
,
Yuan
Li
,
Chao
Liu
and
Xiaomei
Liao
*
School of Life Sciences, Central China Normal University, Hubei Key Lab of Genetic Regulation and Integrative Biology, Wuhan, P.R. China. E-mail: liaodebox@mail.ccnu.edu.cn; Fax: +86 027 67861936; Tel: +86 027 67867229
First published on 2nd November 2016
Abstract
Di-(2-ethylhexyl) phthalate (DEHP) is a widely used commercial plasticizer. DEHP exposure has a negative impact on brain development and cognition, but the mechanisms responsible for DEHP-induced neurotoxicity are not well understood. Here we showed that DEHP exposure increased maleic dialdehyde and reactive oxygen species contents and decreased endogenous superoxide dismutase activity in a mouse neuroblastoma cell line (N2a cell line). DEHP exposure not only induced reduction of neurite outgrowth, but also led to microtubule-associated protein tau hyperphosphorylation and dissociation from microtubules. Furthermore, DEHP exposure decreased the levels of synapsin-1 and postsynaptic density protein 95 (PSD95), which play critical roles in synaptic function. Antioxidant vitamin E pretreatment prevented DEHP-induced abnormalities in the cells. These results indicate that DEHP exposure could induce abnormal action of proteins including tau, synapsin-1 and PSD95, which play critical roles in the synaptic structure and function, and that these alterations might be mediated by peroxidative damage.
Introduction
Di-2-ethylhexyl phthalate (DEHP) is widely used as a plasticizer in common products made of polyvinyl chloride (PVC), such as toys, food packaging, cosmetic products, and medical devices.1 Because of the loose covalent bond between DEHP and PVC, DEHP can easily seep from products2 into the environment, thus making it a widely spread environmental contaminant. DEHP is also thought to enter the body through ingestion, inhalation and dermal contact with plastic products,3,4 which raises the concerns about the health risks of DEHP.Mounting evidence indicates negative effects of DEHP on neurodevelopment and behavior.5–7 Epidemiological investigations have demonstrated that developmental exposure to DEHP decreases the intelligence of school-age children.8 Perinatal DEHP exposure can impair spatial cognition in 21-month-old rats.9 Furthermore, early developmental exposure to DEHP can reduce hippocampal dendritic spine density, and electrophysiological recordings of Drosophila projection neurons show that DEHP exposure can decrease the mini excitatory postsynaptic currents through inhibition of calcium channels.10 These results suggest that DEHP exposure can impair the synaptic plasticity and cognition. Neuronal cells have long processes and form synaptic connections that may be vulnerable to toxic proteins and insults. Indeed, synaptic dysfunction is a common pathological event in neurotoxic diseases. However, the action and mechanisms involved in DEHP-induced synaptic impairment are still not clear.
Tau is a major microtubule-associated protein, and is abundantly present in the neuronal axons of the central nervous system. By binding to microtubules, tau can promote their assembly and stability. Tau plays a key role in the outgrowth of neuronal processes, the development of neuronal polarity and the maintenance of synaptic plasticity.11 Changes in phosphorylation can modify the biological function of tau; when tau protein is abnormally hyperphosphorylated, it dissociates from microtubules, which disturbs microtubule assembly and axon transportation, and finally destroys the synaptic structure and function.12 Hyperphosphorylation of tau is mainly induced by the imbalance of protein kinases and phosphatases. Glycogen synthase kinase 3 (GSK-3) is the first identified tau kinase13 that can strongly phosphorylate tau at multiple microtubule binding sites.14,15 GSK-3 can be activated by reactive oxygen species (ROS),16 which suggests the possibility of aberrant tau phosphorylation induced by oxidative damage.
In addition, neurotransmitter release and the alterations of the postsynaptic density (PSD) area play important roles in synaptic plasticity. Synapsin-1 is a presynaptic protein that is critical for synaptic transmitter release. Postsynaptic density protein 95 (PSD95) is one of the most abundant proteins in the PSD area, which plays important roles in maintaining the plasticity of the synaptic structure, neurotransmission and signal transduction.17 The reduction of these pre- and post-synaptic proteins can directly impair synaptic transmission, thus leading to memory deficit.18,19
Taken together, microtubule-associated protein tau, synapsin-1 and PSD95 are critical proteins involved in the synaptic structure and function. It is reported that DEHP exposure may interfere with normal synaptogenesis and connectivity in the hippocampus.20 DEHP can induce neurotoxic effects through oxidative damage,21,22 and the brain is extremely sensitive to oxidative stress because of its elevated levels of peroxidable fatty acids, high requirement for oxygen, and relative paucity of antioxidant systems.23
In this study, we measured the effects of DEHP exposure on tau, synapsin-1 and PSD95 in N2a cells to explore the possible mechanism involved in DEHP-induced synaptic abnormalities. Our results indicate that DEHP treatment decreases the length of neurites in N2a cells. DEHP exposure activated GSK-3β, which hyperphosphorylated tau protein at the Thr231 and Ser262 sites and caused tau-microtubule binding ability impairment. Furthermore, DEHP exposure reduced the levels of synapsin-1 and PSD95. Lastly, the results showed that DEHP exposure caused dramatic oxidative damage in N2a cells. Exogenous antioxidant vitamin E pretreatment prevented DEHP-induced abnormalities, suggesting that peroxidation is a mechanism involved in DEHP-induced synaptic impairment.
Materials and methods
Reagent and antibodies
DEHP, vitamin E and dichloro-dihydro-fluorescein diacetate (DCFH-DA; used to detect the ROS content) were purchased from Sigma (St Louis, MO, USA). The Maleic Dialdehyde (MDA) assay kit and the Superoxide Dismutase (SOD) assay kit were purchased from NanJing JianCheng Bioengineering Institute (Nanjing, Jiang Su, China). Rabbit polyclonal (pAb) antibodies pT231 and pS262 against phosphorylation of tau at Thr231 and Ser262, respectively, were purchased from Bioworld Technology (Louis Park, MN, USA). Mouse monoclonal (mAb) Tau-5 was purchased from Abcam (New Territories, HK) and used to probe the total tau protein. Mouse mAb total GSK-3β and rabbit pAb synapsin-1 were purchased from Cell Signaling (Beverly, MA, USA). Rabbit pAb phospho-GSK-3β at Ser-9 (GSK-3β-Ser9), PSD95 and β-actin were obtained from Bioworld Technology.Cell culture and drug treatments
Mouse neuroblastoma Neuro2a (N2a) cells were maintained in 45% Dulbecco's modified Eagle's medium and 45% Opti-MEM, supplemented with 10% (v/v) fetal calf serum, in a 5% CO2 humidified incubator at 37 °C. DEHP and vitamin E were dissolved in dimethyl sulfoxide (DMSO). The cells were plated in 6 well plates at densities of 1 × 105 and incubated for 48 h. Then they were treated with 500 μM DEHP for 24 h alone, or pretreated with 25 μM or 50 μM vitamin E for 1 h, and then treated with 500 μM DEHP for 24 h. The vehicle control was treated with 0.1% DMSO.Cell viability assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was applied to detect the cell viability. Briefly, N2a cells were seeded onto 96-well culture plates at an initial density of 5000 cells per well, and allowed to grow for 48 h in an incubator maintained at 37 °C. The cells were treated with the appropriate drugs and were subsequently incubated with 20 μl MTT (0.5 mg ml−1) for 4 h and then lysed with 0.1% DMSO. After 30 min incubation at room temperature, the plates were scanned with a microplate reader (Bio-Rad, Hercules, CA, USA) at 560 nm for measuring the absorbance. Cells treated with 0.1% DMSO were defined as the control group.Peroxidation damage assay
N2a cells were treated with different concentrations of DEHP (125, 250, 500 and 1000 μM) for 24 h. The MDA and ROS content and the SOD activity were tested as indicators of peroxidative damage. The MDA content and SOD activity were measured in whole-cell extracts using the assay kit, according to the manufacturer's instructions. The ROS content was measured by the DCF method, as described previously.24Cell morphology observation
N2a cells were seeded in a 24-well plate and treated with 500 μM DEHP for 24 h. When the cells grew to 80% confluence, the morphology of the N2a cells was observed by using a phase contrast microscope. In each experiment, at least 5 randomly selected fields per zone in each well, with 3 wells per experiment and 3–5 experiments per group, were taken. Images were then processed in image processing software, ImageJ, where neurites were manually traced with a Wacom tablet to measure the length and branch number. Average lengths of the neurites were then obtained by dividing the total length of neurites by the number of neurons measured. If the cells failed to elaborate neurites in our selected fields, they were discarded.25Western blotting
Following DEHP treatment, cells were harvested on ice and lysed in a cell lysis buffer [50 mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.1% (w/v) sodium dodecyl sulfate (SDS), 1% (v/v) NP-40, 0.5% sodium deoxycholate, 0.02% NaN3, supplemented with protease inhibitors including 100 μg ml−1 phenylmethysulfonyl fluoride (PMSF) and 2 μg ml−1 aprotinin]. The protein concentration was tested by the method of BCA (Pierce, Thermo Fisher Scientific Inc., Rockford, IL, USA). Equal amounts of sample proteins were separated according to their molecular weights by 10% SDS-polyacrylamide gel electrophoresis and then electrically transferred onto the nitrocellulose membrane (Millipore, Billerica, MA, USA). The membranes were blocked with 5% defatted milk dissolved in 0.1% tris-buffered saline with Tween 20 solution [10 mM Tris-HCl, 150 mM NaCl, 0.1% (v/v) Tween-20, pH 7.6] for 1 h at room temperature and probed for β-actin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000), Tau-5 (1:500), pS262 (1:500), pT231 (1:500), GSK-3β (1:500), GSK-3β-Ser9 (1:300), synapsin-1 (1:800) and PSD95 (1:800), overnight at 4 °C. After incubating with IRDye 800CW conjugated goat pAb anti-rabbit IgG or anti-mouse IgG (LI-COR Biosciences, NE, USA) (1:10000) for 1 h at room temperature, the membranes were visualized using the Odyssey Infrared Imaging System (LI-COR Biosciences).
:1000), Tau-5 (1:500), pS262 (1:500), pT231 (1:500), GSK-3β (1:500), GSK-3β-Ser9 (1:300), synapsin-1 (1:800) and PSD95 (1:800), overnight at 4 °C. After incubating with IRDye 800CW conjugated goat pAb anti-rabbit IgG or anti-mouse IgG (LI-COR Biosciences, NE, USA) (1:10000) for 1 h at room temperature, the membranes were visualized using the Odyssey Infrared Imaging System (LI-COR Biosciences).
Tau microtubule-binding activity assay
The capacity of tau protein binding to microtubules was measured by the microtubule-binding assay, as previously described.26 Briefly, N2a cells were harvested in a high-salt reassembly buffer (100 mM Tris, 0.5 mM MgSO4, 1 mM EGTA, 2 mM dithiothreitol, and 750 mM NaCl, pH 6.8) supplemented with 0.1% Triton X-100, 20 μM taxol, 2 mM GTP, and a mixture of protease inhibitors (2 mM PMSF and 1 μg ml−1 aprotinin) at 37 °C and homogenized with 15 strokes in a warm Dounce homogenizer and then centrifuged at 50000g for 20 min at 25 °C. The supernatant was removed, and the remaining pellet was resuspended in the sample buffer. After measurement of the protein concentration, the samples were subjected to western blot analysis. The supernatant contains unbound tau, and the pellet contains microtubule-bound tau. The levels of unbound tau and microtubule-bound tau were assessed by the immunoreactivity of tau protein in the supernatant and pellet fractions, respectively.
Statistical analysis
All data were analyzed with the statistical software SPSS 13.0 and expressed as means ± SD. The images were analyzed with Quantity One software. Multiple group comparisons were done using one-way analysis of variance. Differences were considered statistically significant at p < 0.05.Results
Cell viability and metabolic activity upon treatment with DEHP
To test whether DEHP has the toxicity to affect cell growth, we detected cell viability using the MTT assay. DEHP did not induce significant cell toxicity at any dose tested (125, 250, 500 μM), but there was a significant toxicity in cells with the highest dose (p < 0.05, 1000 μM) (Fig. 1).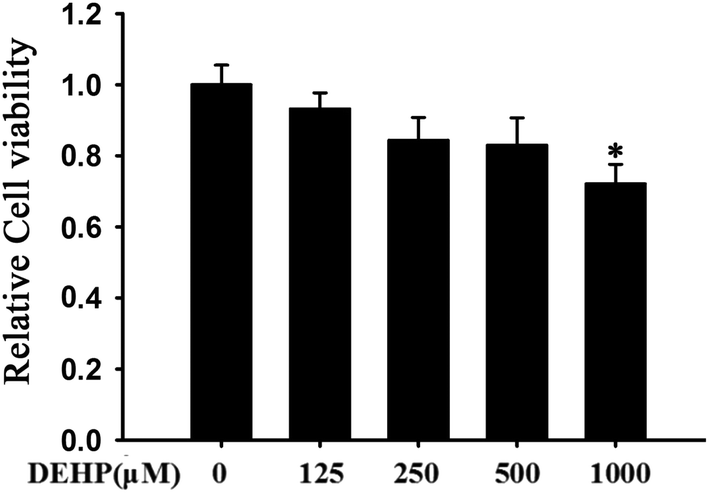 | ||
| Fig. 1 MTT-assay of N2a cells treated with different doses of DEHP (125, 250, 500 and 1000 μM). Cell viability was determined by MTT assay. Results are represented as mean ± SD, n = 3 independent observations. | ||
DEHP-induced peroxidation in N2a cells
N2a cells were treated with different doses of DEHP (125, 250, 500 and 1000 μM) for 24 h. The content of MDA and ROS, and the activity of SOD were measured to monitor the peroxidative damage in N2a cells. After DEHP exposure, the relative content of MDA (Fig. 2A) and ROS (Fig. 2B) increased with a trend of DEHP dose-dependency; the relative SOD activity decreased in a DEHP dose-dependent manner (Fig. 2C). Compared with the control group, 500 μM DEHP treatment dramatically increased the content of MDA and ROS (p < 0.05) and significantly decreased the activity of SOD (p < 0.01). These results indicate that DEHP exposure induces oxidative damage in N2a cells.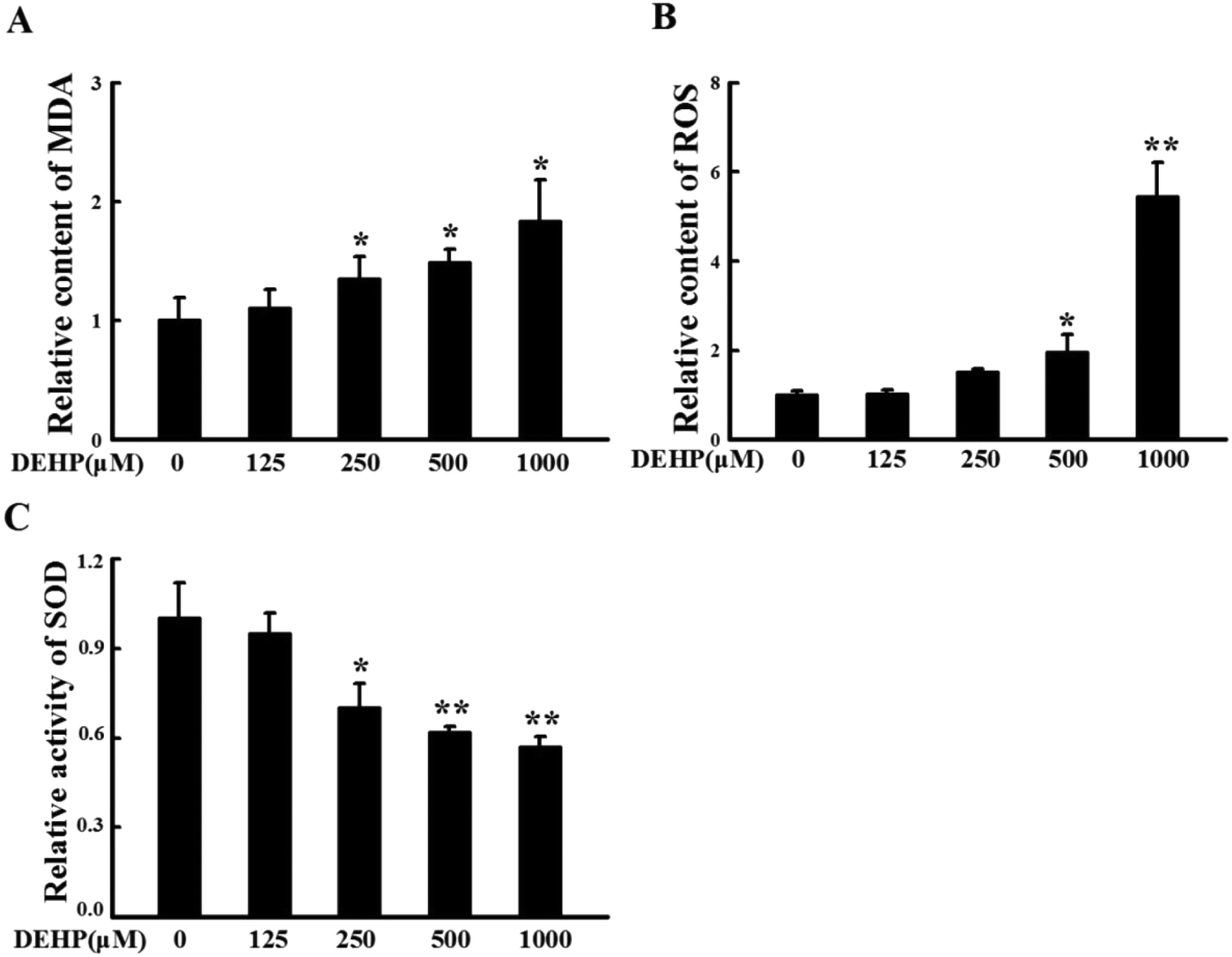 | ||
| Fig. 2 N2a cells were treated with DEHP (125, 250, 500 and 1000 μM) for 24 h. The relative content of MDA (A) and ROS (B), and the relative activity of SOD (C) were expressed as fold changes over DEHP-untreated cells and are represented as mean ± SD (n = 5). *p < 0.05, **p < 0.01 vs. control group. | ||
Vitamin E pretreatment prevents N2a cell from DEHP-induced peroxidative damage
Vitamin E is a fat-soluble antioxidant with a strong ability to scavenge lipophilic free radicals produced by oxidative stress.27 We examined the effect of vitamin E (25 and 50 μM) pretreatment on DEHP-induced peroxidative damage in N2a cells. Compared with the control group, 500 μM DEHP treatment alone increased the MDA (Fig. 3A) and ROS content (Fig. 3B, p < 0.05), and decreased SOD activity (Fig. 3C, p < 0.05). However, 25 or 50 μM vitamin E pretreatment for 1 h effectively prevented DEHP-induced MDA, ROS and SOD abnormalities.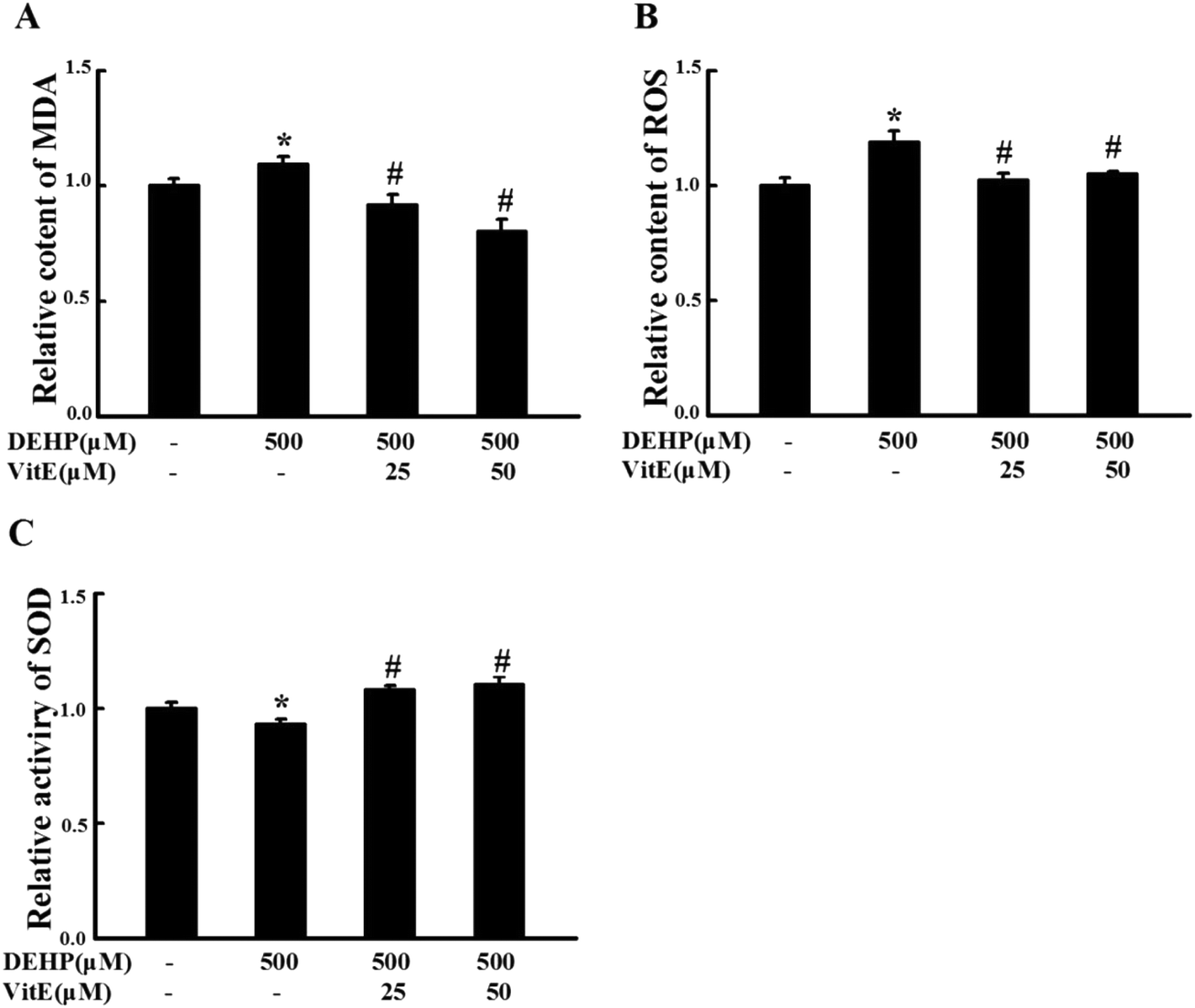 | ||
| Fig. 3 Vitamin E pretreatment prevented DEHP-induced oxidative damage in N2a cells. 25 and 50 μM vitamin E pretreatment for 1 h protected N2a cells against DEHP-induced MDA (A) and ROS (B) deficits and SOD activity deficits (C). All data are expressed as fold changes over control cells and are represented as mean ± SD (n = 5). *p < 0.05 vs. control group; #p < 0.05, ##p < 0.01 vs. DEHP treatment group. | ||
DEHP treatment induces neurite retraction
Neurite growth is the structural basis of synaptic formation,28 thus determining that the effect of DEHP exposure on the morphology of neurites is critical for unraveling the neurotoxic effects of DEHP on the synaptic structure and function. Previous studies have shown that oxidative stress could lead to the reduction of neurite outgrowth.29 We observed the morphology of N2a cells after DEHP treatment by using a phase contrast microscope. Compared with control cells, 500 μM DEHP treatment induced obvious reduction of neurite outgrowth and caused a significant decrease in the length of neurites (Fig. 4). However, upon vitamin E pretreatment, the length of neurites in DEHP-treated cells showed no observable difference from that of the control cells (Fig. 4Ac, d).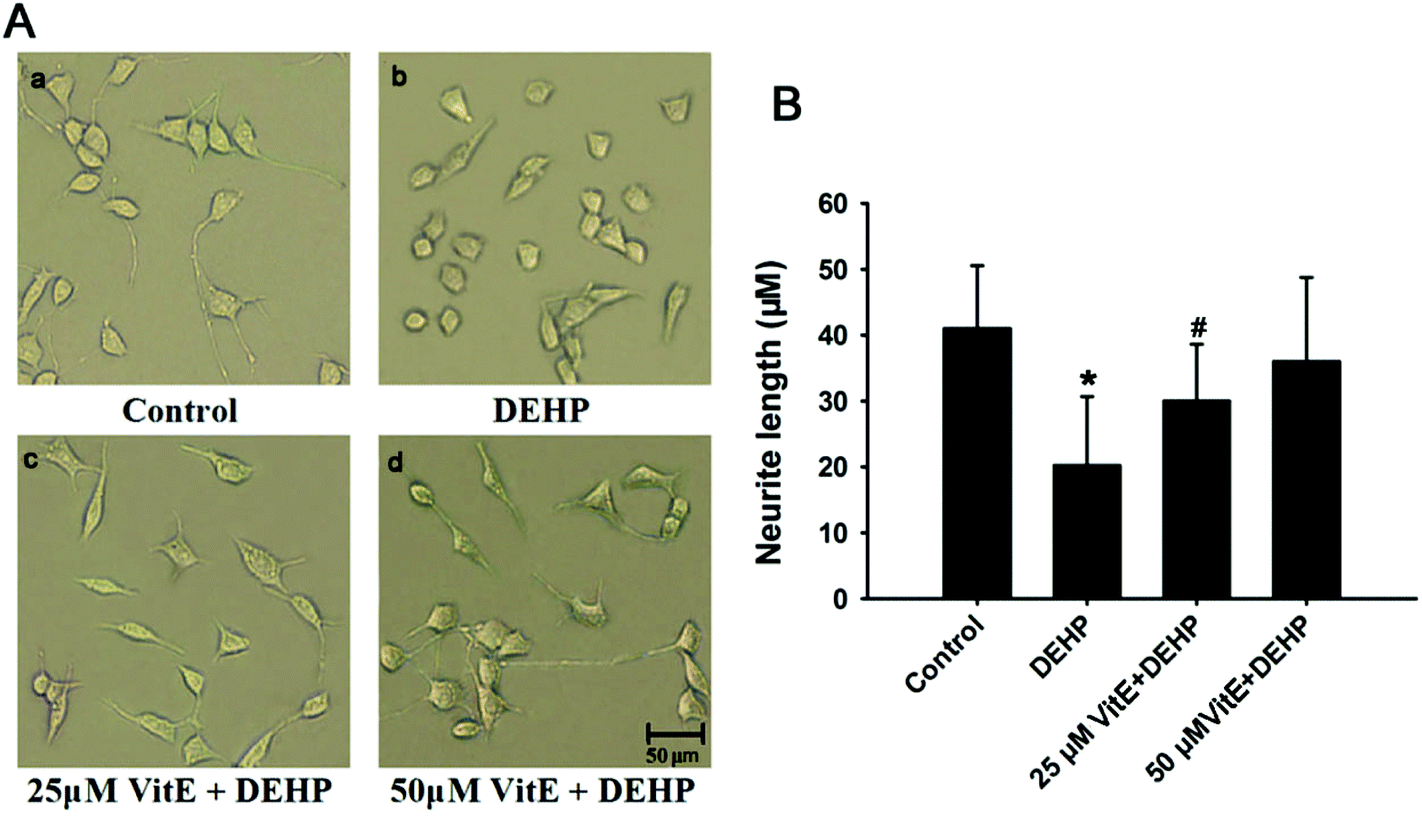 | ||
| Fig. 4 N2a cell neurite changes following DEHP, or vitamin E plus DEHP (VitE + DEHP) treatment. A, The morphology of N2a cells in the control group (a), DEHP exposure group (b), 25 μM vitamin E (c) and 50 μM vitamin E (d) pretreatment plus DEHP exposure group. B, The quantification of neurite length. Scale bar = 50 μm. n = 30 cells. *p < 0.05 vs. control group; #p < 0.05 vs. DEHP treatment group. | ||
DEHP exposure decreased tau-microtubule binding ability
Tau protein binds with microtubules and plays important roles in the cytoskeleton structure and neurite outgrowth. After our observation that DEHP exposure could induce the reduction of neurite outgrowth in N2a cells, we investigated the effect of DEHP treatment on the microtubule binding capacity of tau protein. We treated N2a cells with 500 μM DEHP for 24 h alone, or pretreated cells with vitamin E (25 and 50 μM) for 1 h and then 500 μM DEHP for 24 h. The levels of unbound tau and the microtubule-bound tau were measured by western blot analysis. DEHP treatment increased the immunostaining of unbound tau, and decreased the immunostaining of microtubule-bound tau. However, vitamin E pretreatment prevented DEHP-induced tau-microtubule binding function impairment of N2a cells in a dose-dependent manner (Fig. 5A and B).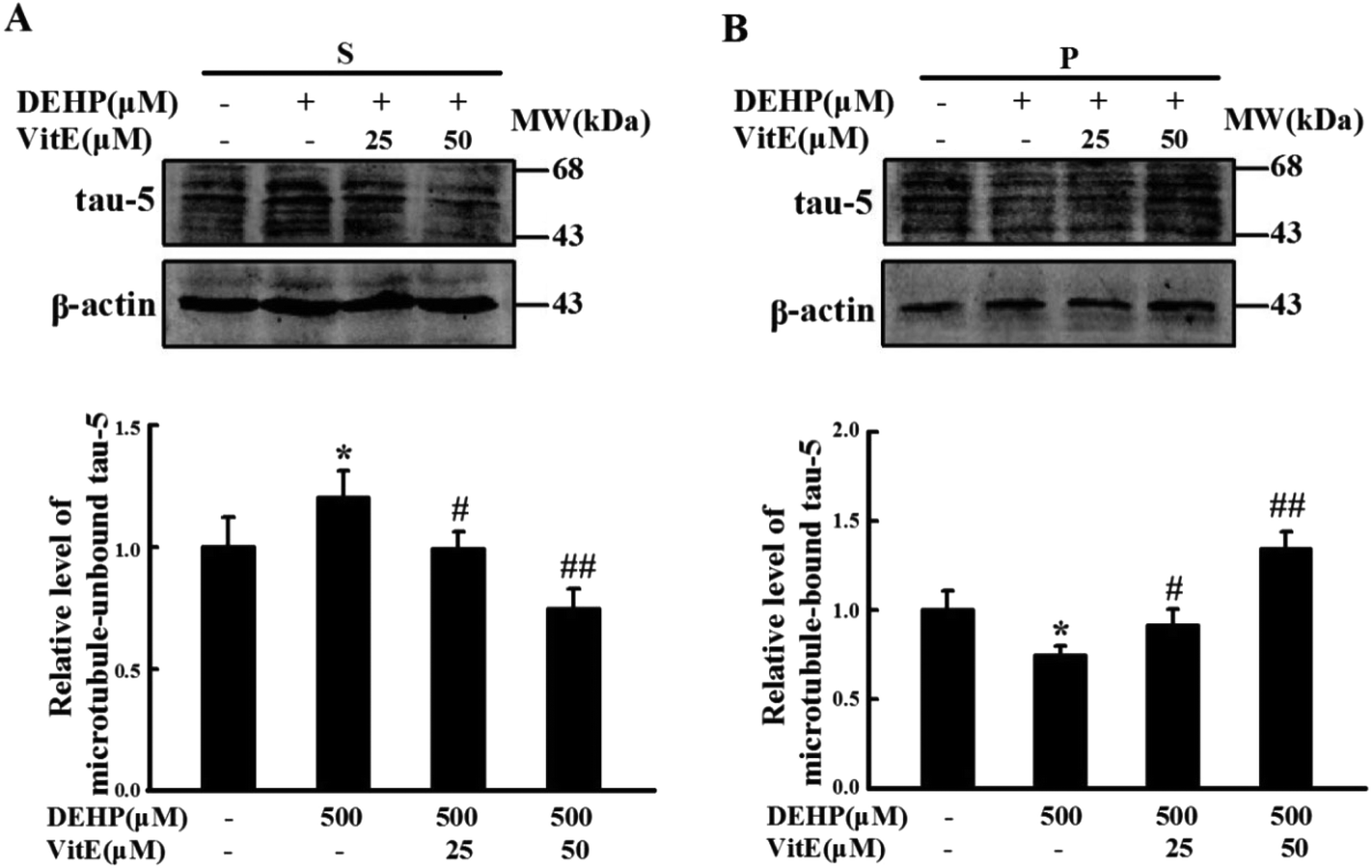 | ||
| Fig. 5 Vitamin E pretreatment mitigates DEHP-induced tau-microtubule binding function impairment in N2a cells. Immunostaining of unbound tau in (A) the supernatant fraction (S) and (B) microtubule-bound tau in the pellet fraction (P) were detected by western blot analysis. Values were normalized to those of the control group. The protein levels were expressed as fold changes over untreated cells and are represented as mean ± SD, *p < 0.05 vs. control group; #p < 0.05, ##p < 0.01 vs. DEHP treatment group. | ||
DEHP treatment induces abnormal phosphorylation of tau protein
Abnormal phosphorylation is the major cause of impairment in the tau-microtubule binding ability. We detected the phosphorylation state of tau protein at Thr231 and Ser262 epitopes, which play important roles in tau-microtubule binding. Compared with the control group, DEHP-treated cells were hyperphosphorylated at tau-Ser262 and Thr231 epitopes. However, 25 and 50 μM vitamin E pretreatment effectively prevented DEHP-induced hyperphosphorylation of tau at these two sites. The total level of tau protein (detected by the Tau-5 antibody) had no observable change (Fig. 6A and B).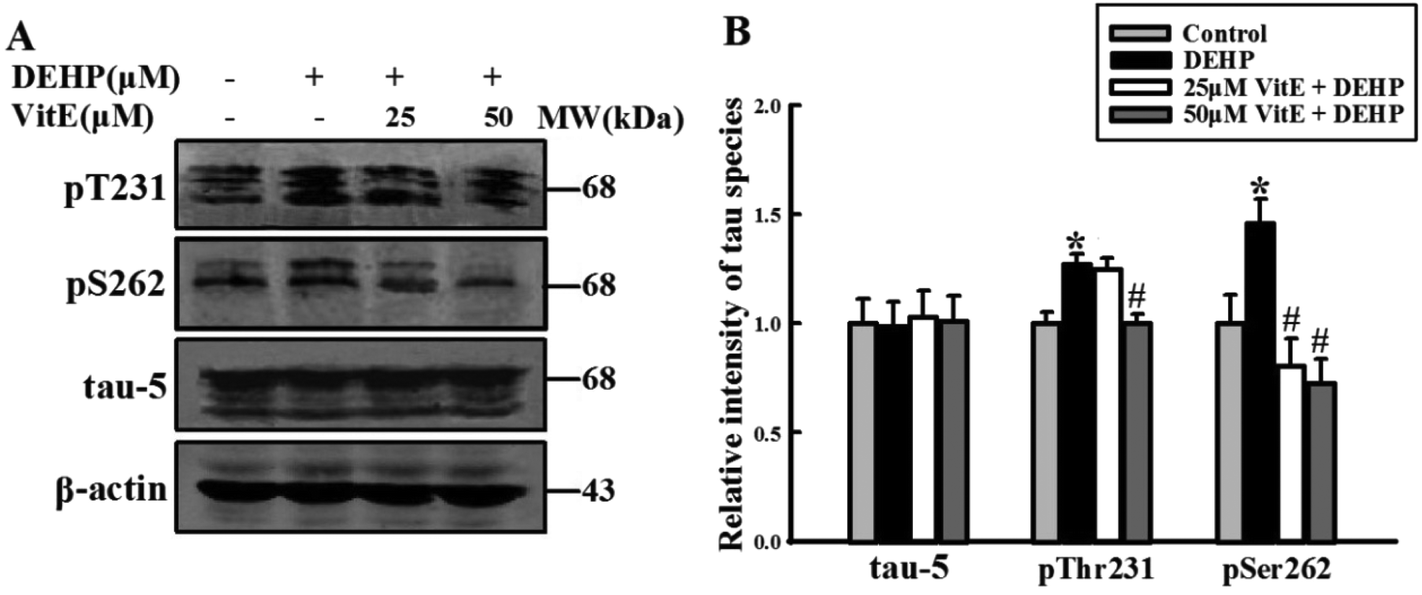 | ||
| Fig. 6 Tau protein was hyperphosphorylated at pT231 and pS262 sites after DEHP treatment. (A) 500 μM DEHP exposure for 24 h hyperphosphorylated tau at pT231 and pS262 sites in N2a cells; 25 and 50 μM vitamin E (VitE) pretreatment prevented DEHP-induced tau hyperphosphorylation in N2a cells in a dose-dependent manner. The total level of tau had no observable change. (B) Quantification of the total tau and phosphorylated tau levels. The protein levels were expressed as fold changes over untreated cells and are represented as mean ± SD, *p < 0.05, **p < 0.01 vs. control cells; #p < 0.05, ##p < 0.01 vs. DEHP treatment cells. | ||
DEHP treatment increases the activity of GSK-3β
GSK-3β is one of the critical Ser/Thr kinases responsible for abnormal tau phosphorylation, and GSK-3β-Ser9 phosphorylation is negatively correlated with its activity. Therefore, we examined the activity change of GSK3β following DEHP treatment (Fig. 7). We found that DEHP treatment significantly (p < 0.01) decreased the immunostaining of phosphorylated GSK-3β at the Ser9 site (inactivated form). Vitamin E pretreatment could mitigate DEHP-induced GSK-3β activation in a dose-dependent manner. The total level of GSK-3β had no observable change in any of the four groups.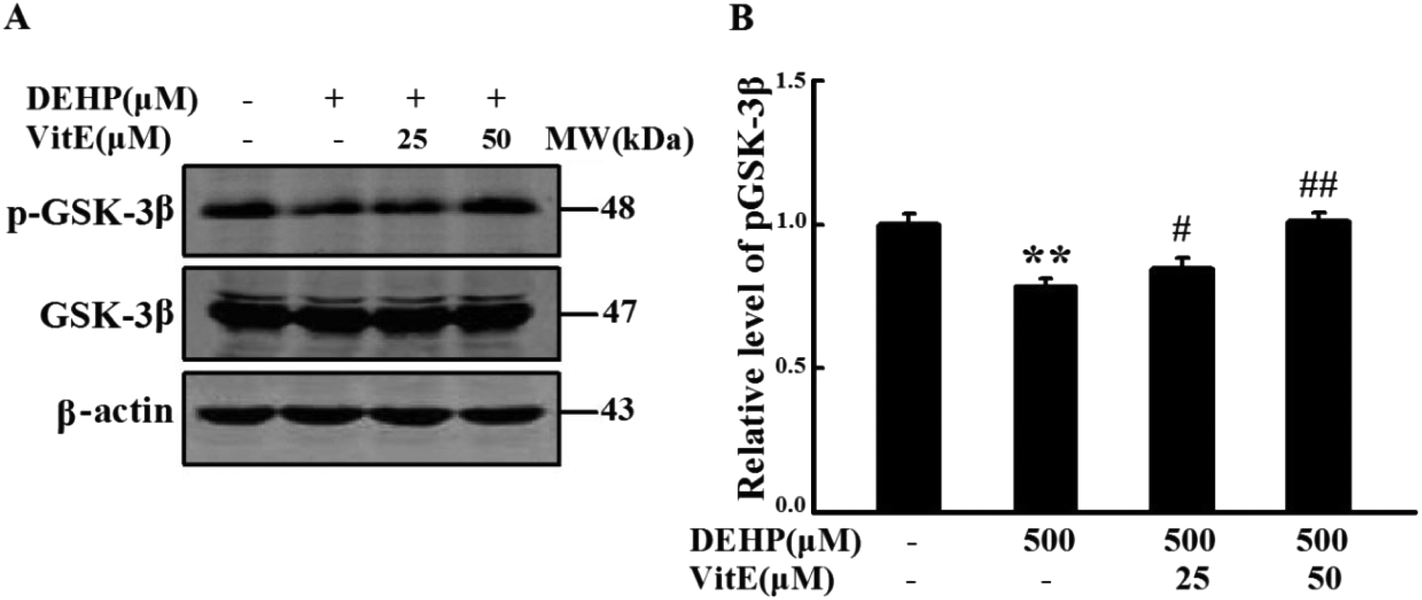 | ||
| Fig. 7 Vitamin E pretreatment mitigated DEHP induced GSK-3β activation. (A) The level of phosphorylated GSK-3β at the Ser9 site and the total level of GSK-3β were detected by western blot analysis. (B) Quantification of the relative intensity of phosphorylated GSK-3β at the Ser9 site. The values were expressed as fold changes over control and are represented as mean ± SD. Significance is denoted as **p < 0.01 vs. control group, and #p < 0.05, ##p < 0.01 vs. DEHP treatment group. | ||
DEHP treatment decreased the levels of synapsin-1 and PSD95
Both neurotransmitter release and the structure of PSD play important roles in synaptic plasticity30,31 and the transduction pathways of neurons.32 We analyzed the level of synapsin-1, a presynaptic vesicle protein that plays an important role in neurotransmitter release from vesicles33,34 and PSD95, a marker of synapse activation. Levels of synapsin-1 and PSD95 were lower in the DEHP treated group than in the control group (Fig. 8). However, vitamin E pretreatment reversed DEHP-induced synapsin-1 and PSD95 deficits.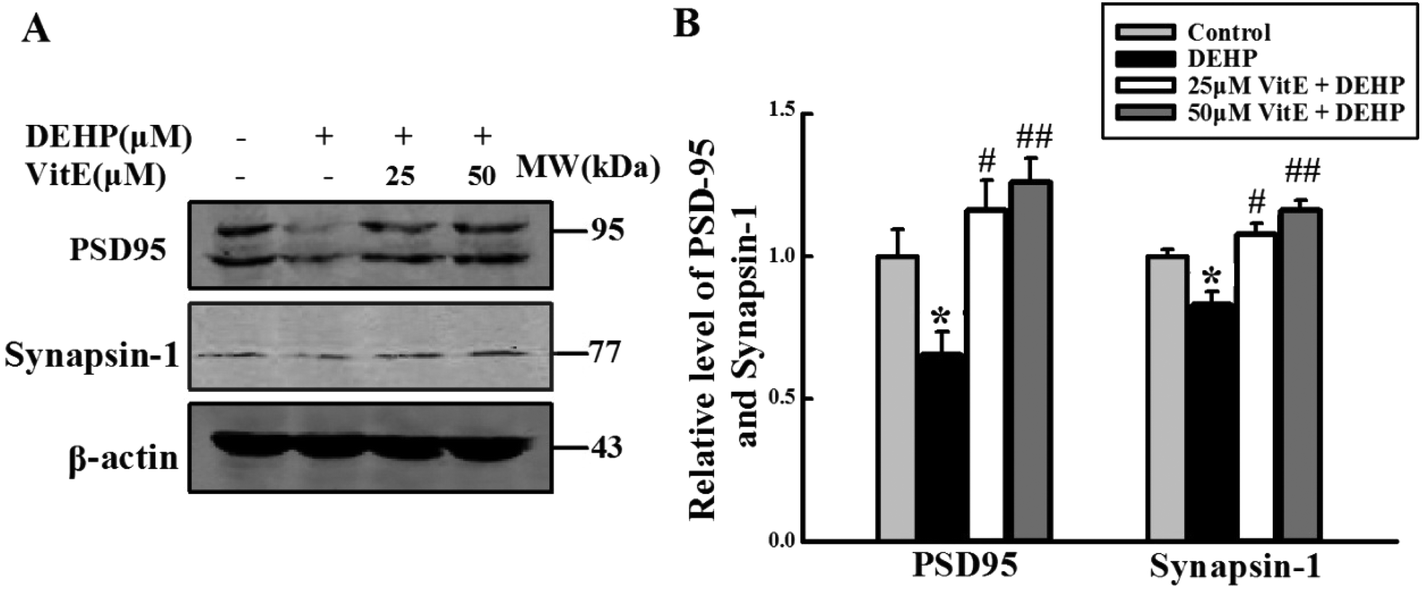 | ||
| Fig. 8 DEHP treatment decreased the levels of synapsin-1 and PSD95. (A) The expressions of synapsin-1 and PSD95 were detected by western blot analysis. (B) Quantification of the relative intensity of synapsin-1 and PSD95; the values were expressed as fold changes over control and are represented as mean ± SD. Significance is denoted as **p < 0.01 vs. control group, and ##p < 0.01 vs. DEHP treatment group. | ||
Discussion
DEHP is widely used as a plasticizer in a variety of consumer products. It has been found that DEHP can have toxic effects on the human body, and accumulating evidence has shown that DEHP exposure has negative effects on neurodevelopment, and synaptic formation and maintenance.5,20 Although the connection between DEHP exposure and synaptic impairment has been identified, the molecular link is not clear.DEHP exposure can increase the contents of ROS and MDA in mice,35 and decrease the levels of antioxidants.36 In this study, we demonstrated that in N2a cells, DEHP exposure for 24 h increased the intracellular content of MDA and ROS and decreased the activity of SOD in a dose-dependent manner. Vitamin E pretreatment (25 and 50 μM), which acts as an exogenous antioxidant, could effectively protect the cells from DEHP-induced ROS, MDA and SOD abnormalities. These findings are consistent with previous research and suggest that DHEP can cause peroxidative damage in N2a cells.
In the central nervous system, peroxidation can cause axonal degeneration. Additionally, abnormal phosphorylation of cytoskeletal proteins as a result of oxidative stress-induced cell injury can destroy the cytoskeletal structure and function.37 Our results showed that after DEHP treatment, the neurite length decreased. Furthermore, vitamin E pretreatment could prevent DEHP-induced morphology changes. The results indicate that DEHP can impair the structural basis of synapse formation via decreasing the length of neurites, and that peroxidative damage plays a role in neurite changes induced by DEHP.
A possible mechanism for the DEHP-induced changes in the neurite length is abnormal phosphorylation of tau, which plays an important role in neurite outgrowth. Our study demonstrated that after DEHP treatment, tau was hyperphosphorylated at the Ser262 and Thr231 epitopes. Hyperphosphorylation at the Thr231 epitope causes a local conformational change that allows access by kinases to further phosphorylate tau,38 and diminishes the ability of tau protein to bind microtubules in situ.39 The Ser262 site of tau protein is a critical site that mediates tau-microtubule binding; hyperphosphorylation of tau at Ser262 can promote the dissociation of tau from the microtubule.40 In this study, the tau-microtubule binding assay showed that DEHP treatment attenuated the binding between tau protein and microtubule, and vitamin E pretreatment efficiently prevented the dissociation of tau protein from microtubules. These results indicate that DEHP-induced oxidation damage could hyperphosphorylate tau and decrease tau-microtubule binding.
The phosphorylation of tau is regulated by the kinase and phosphate systems, and GSK-3β is one of the critical kinases that can regulate tau phosphorylation at the Thr231 and Ser262 sites.39,41 Oxidative stress could maintain GSK-3β in a highly activated state.42 In this study, the results showed that upon DEHP treatment, the phosphorylation of GSK-3β at the Ser9 site (inactivated form) decreased, which suggests increased activity of GSK-3β. Furthermore, vitamin E pretreatment could prevent the augmented activity of GSK-3β induced by DEHP. These data suggest that DEHP treatment increases the activation of GSK-3β, which could then lead to tau hyperphosphorylation.
Oxidative stress may reduce the expression of synaptic proteins;43 in the hippocampus, destruction of the antioxidant system resulting from trauma reduces the levels of synapsin-1 and PSD95.44 In this study, we found that the levels of synapsin-1 and PSD95 decreased significantly following DEHP treatment. However, with vitamin E pretreatment, the levels of synapsin-1 and PSD95 were higher than the DEHP treatment group. The results of the previous study and our study together indicate that DEHP-induced peroxidation could reduce the expression of synaptic proteins, synapsin-1 and PSD95.
Conclusions
In conclusion, the present results revealed that DEHP exposure caused peroxidative damage in N2a cells, which could alter neurite growth of N2a cells. The changes in neurites strongly suggest that DEHP exposure can induce synaptic structural impairment. DEHP exposure also caused microtubule associated protein tau hyperphosphorylation, tau and microtubule dissociation, and synapsin-1 and PSD95 deficits. These findings provide initial evidence of the possible molecular mechanisms involved in DEHP-induced synaptic impairment. The finding that vitamin E pretreatment can protect cells from DEHP-induced abnormalities highlights the importance of peroxidation in DEHP-induced neurotoxicity, and suggests a targeted prevention strategy for DEHP exposure.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Professor Wang Jianzhi at the Tongji Medical College of Huazhong University of Sciences and Technology, Wuhan, China, for providing the N2a cell line used in this study. This work was financially supported by grants obtained from self-determined research funds of Central China Normal University (CCNU) from the colleges’ basic research and operation of MOE (CCNU15A02024 and CCNU15A02029) and the National Natural Science Foundation of China (no. 81361120245).Notes and references
- D. Anderson, T. W. Yu and F. Hincal, Effect of some phthalate esters in human cells in the comet assay, Teratog., Carcinog., Mutagen., 1999, 19, 275–280 Search PubMed.
- R. J. Rubin and C. A. Schiffer, Fate in humans of the plasticizer, di-2-ethylhexyl phthalate, arising from transfusion of platelets stored in vinyl plastic bags, Transfusion, 1976, 16, 330–335 Search PubMed.
- K. Hashizume, J. Nanya, C. Toda, T. Yasui, H. Nagano and N. Kojima, Phthalate esters detected in various water samples and biodegradation of the phthalates by microbes isolated from river water, Biol. Pharm. Bull., 2002, 25, 209–214 Search PubMed.
- R. A. Rudel, D. E. Camann, J. D. Spengler, L. R. Korn and J. G. Brody, Phthalates, alkylphenols, pesticides, polybrominated diphenyl ethers, and other endocrine-disrupting compounds in indoor air and dust, Environ. Sci. Technol., 2003, 37, 4543–4553 Search PubMed.
- T. Chen, W. Yang, Y. Li, X. Chen and S. Xu, Mono-(2-ethylhexyl) phthalate impairs neurodevelopment: inhibition of proliferation and promotion of differentiation in PC12 cells, Toxicol. Lett., 2011, 201, 34–41 Search PubMed.
- R. Hokanson, W. Hanneman, M. Hennessey, K. C. Donnelly, T. McDonald, R. Chowdhary and D. L. Busbee, DEHP, bis(2)-ethylhexyl phthalate, alters gene expression in human cells: possible correlation with initiation of fetal developmental abnormalities, Hum. Exp. Toxicol., 2006, 25, 687–695 Search PubMed.
- H. Lin, K. Yuan, L. Li, S. Liu, S. Li, G. Hu, Q. Q. Lian and R. S. Ge, In Utero Exposure to Diethylhexyl Phthalate Affects Rat Brain Development: A Behavioral and Genomic Approach, Int. J. Environ. Res. Public Health, 2015, 12, 13696–13710 Search PubMed.
- S. C. Cho, S. Y. Bhang, Y. C. Hong, M. S. Shin, B. N. Kim, J. W. Kim, H. J. Yoo, I. H. Cho and H. W. Kim, Relationship between environmental phthalate exposure and the intelligence of school-age children, Environ. Health Perspect., 2010, 118, 1027–1032 Search PubMed.
- W. Sun, J. B. Ban, N. Zhang, Y. K. Zu and W. X. Sun, Perinatal exposure to Di-(2-ethylhexyl)-Phthalate leads to cognitive dysfunction and phospho-tau level increase in aged rats, Environ. Toxicol., 2014, 29, 596–603 Search PubMed.
- D. Ran, S. Cai, H. Wu and H. Gu, Di (2-ethylhexyl) phthalate modulates cholinergic mini-presynaptic transmission of projection neurons in Drosophila antennal lobe, Food Chem. Toxicol., 2012, 50, 3291–3297 Search PubMed.
- J. Gotz, D. Xia, G. Leinenga, Y. L. Chew and H. Nicholas, What Renders TAU Toxic, Front. Neurol., 2013, 4, 72 Search PubMed.
- M. A. Meraz-Rios, K. I. Lira-De Leon, V. Campos-Pena, M. A. De Anda-Hernandez and R. Mena-Lopez, Tau oligomers and aggregation in Alzheimer's disease, J. Neurochem., 2010, 112, 1353–1367 Search PubMed.
- A. Takashima, GSK-3 is essential in the pathogenesis of Alzheimer's disease, J. Alzheimers Dis., 2006, 9, 309–317 Search PubMed.
- S. J. Liu, A. H. Zhang, H. L. Li, Q. Wang, H. M. Deng, W. J. Netzer, H. Xu and J. Z. Wang, Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory, J. Neurochem., 2003, 87, 1333–1344 Search PubMed.
- E. Rockenstein, M. Torrance, A. Adame, M. Mante, P. Bar-on, J. B. Rose, L. Crews and E. Masliah, Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation, J. Neurosci., 2007, 27, 1981–1991 Search PubMed.
- S. Y. Liu, C. L. Chen, T. T. Yang, W. C. Huang, C. Y. Hsieh, W. J. Shen, T. T. Tsai, C. C. Shieh and C. F. Lin, Albumin prevents reactive oxygen species-induced mitochondrial damage, autophagy, and apoptosis during serum starvation, Apoptosis, 2012, 17, 1156–1169 Search PubMed.
- S. E. Craven, A. E. El-Husseini and D. S. Bredt, Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs, Neuron, 1999, 22, 497–509 Search PubMed.
- S. Chaudhury, S. Jain and S. Wadhwa, Expression of synaptic proteins in the hippocampus and spatial learning in chicks following prenatal auditory stimulation, Dev. Neurosci., 2010, 32, 114–124 Search PubMed.
- M. B. Kennedy, Signal-processing machines at the postsynaptic density, Science, 2000, 290, 750–754 Search PubMed.
- C. A. Smith and M. R. Holahan, Reduced hippocampal dendritic spine density and BDNF expression following acute postnatal exposure to di(2-ethylhexyl) phthalate in male Long Evans rats, PLoS One, 2014, 9, e109522 Search PubMed.
- Y. Wu, K. Li, H. Zuo, Y. Yuan, Y. Sun and X. Yang, Primary neuronal-astrocytic co-culture platform for neurotoxicity assessment of di-(2-ethylhexyl) phthalate, J. Environ. Sci., 2014, 26, 1145–1153 Search PubMed.
- H. You, S. Chen, L. Mao, B. Li, Y. Yuan, R. Li and X. Yang, The adjuvant effect induced by di-(2-ethylhexyl) phthalate (DEHP) is mediated through oxidative stress in a mouse model of asthma, Food Chem. Toxicol., 2014, 71, 272–281 Search PubMed.
- D. A. Butterfield, Oxidative stress in neurodegenerative disorders, Antioxid. Redox Signaling, 2006, 8, 1971–1973 Search PubMed.
- J. Tang, Y. Yuan, C. Wei, X. Liao, J. Yuan, E. Nanberg, Y. Zhang, C.-G. Bornehag and X. Yang, Neurobehavioral changes induced by di (2-ethylhexyl) phthalate and the protective effects of vitamin E in Kunming mice, Toxicol. Res., 2015, 4, 1006–1015 Search PubMed.
- C. Y. Lin, W. J. Huang, K. Li, R. Swanson, B. Cheung, V. W. Lin and Y. S. Lee, Differential intensity-dependent effects of magnetic stimulation on the longest neurites and shorter dendrites in neuroscreen-1 cells, J. Neural Eng., 2015, 12, 026013 Search PubMed.
- Q. G. Ren, X. M. Liao, Z. F. Wang, Z. S. Qu and J. Z. Wang, The involvement of glycogen synthase kinase-3 and protein phosphatase-2A in lactacystin-induced tau accumulation, FEBS Lett., 2006, 580, 2503–2511 CrossRef CAS PubMed.
- G. W. Burton and M. G. Traber, Vitamin E: antioxidant activity, biokinetics, and bioavailability, Annu. Rev. Nutr., 1990, 10, 357–382 Search PubMed.
- J. L. McGaugh, Memory–a century of consolidation, Science, 2000, 287, 248–251 Search PubMed.
- S. M. de la Monte, T. R. Neely, J. Cannon and J. R. Wands, Oxidative stress and hypoxia-like injury cause Alzheimer-type molecular abnormalities in central nervous system neurons, Cell. Mol. Life Sci., 2000, 57, 1471–1481 Search PubMed.
- L. Foa and R. Gasperini, Developmental roles for Homer: more than just a pretty scaffold, J. Neurochem., 2009, 108, 1–10 CrossRef CAS PubMed.
- C. Y. Zheng, G. K. Seabold, M. Horak and R. S. Petralia, MAGUKs, synaptic development, and synaptic plasticity, Neuroscientist, 2011, 17, 493–512 Search PubMed.
- G. Romero, M. von Zastrow and P. A. Friedman, Role of PDZ proteins in regulating trafficking, signaling, and function of GPCRs: means, motif, and opportunity, Adv. Pharmacol., 2011, 62, 279–314 Search PubMed.
- E. F. Fornasiero, D. Bonanomi, F. Benfenati and F. Valtorta, The role of synapsins in neuronal development, Cell. Mol. Life Sci., 2010, 67, 1383–1396 Search PubMed.
- O. Shupliakov, V. Haucke and A. Pechstein, How synapsin I may cluster synaptic vesicles, Semin. Cell Dev. Biol., 2011, 22, 393–399 Search PubMed.
- E. Kasahara, E. F. Sato, M. Miyoshi, R. Konaka, K. Hiramoto, J. Sasaki, M. Tokuda, Y. Nakano and M. Inoue, Role of oxidative stress in germ cell apoptosis induced by di(2-ethylhexyl)phthalate, Biochem. J., 2002, 365, 849–856 Search PubMed.
- V. Manojkumar, K. G. Padmakumaran Nair, A. Santhosh, K. V. Deepadevi, P. Arun, L. R. Lakshmi and P. A. Kurup, Decrease in the concentration of vitamin E in blood and tissues caused by di(2-ethylhexyl) phthalate, a commonly used plasticizer in blood storage bags and medical tubing, Vox Sang., 1998, 75, 139–144 Search PubMed.
- G. Bellomo and F. Mirabelli, Oxidative stress and cytoskeletal alterations, Ann. N. Y. Acad. Sci., 1992, 663, 97–109 Search PubMed.
- M. Kolarova, F. Garcia-Sierra, A. Bartos, J. Ricny and D. Ripova, Structure and pathology of tau protein in Alzheimer disease, Int. J. Alzheimers Dis., 2012, 2012, 731526 Search PubMed.
- J. H. Cho and G. V. Johnson, Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau's ability to bind and stabilize microtubules, J. Neurochem., 2004, 88, 349–358 Search PubMed.
- J. Biernat, N. Gustke, G. Drewes, E. M. Mandelkow and E. Mandelkow, Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding, Neuron, 1993, 11, 153–163 Search PubMed.
- C. X. Gong, F. Liu, I. Grundke-Iqbal and K. Iqbal, Post-translational modifications of tau protein in Alzheimer's disease, J. Neural Transm., 2005, 112, 813–838 Search PubMed.
- G. Chen, K. A. Bower, C. Ma, S. Fang, C. J. Thiele and J. Luo, Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death, FASEB J., 2004, 18, 1162–1164 Search PubMed.
- D. A. Forero, G. Casadesus, G. Perry and H. Arboleda, Synaptic dysfunction and oxidative stress in Alzheimer's disease: emerging mechanisms, J. Cell. Mol. Med., 2006, 10, 796–805 Search PubMed.
- M. A. Ansari, K. N. Roberts and S. W. Scheff, A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI, J. Neurotrauma, 2008, 25, 513–526 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |