
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual emission and multi-stimuli-response in iridium(III) complexes with aggregation-induced enhanced emission: applications for quantitative CO2 detection†
Clàudia
Climent‡
a,
Parvej
Alam‡
b,
Sheik Saleem
Pasha
b,
Gurpreet
Kaur
c,
Angshuman Roy
Choudhury
c,
Inamur Rahaman
Laskar
 *b,
Pere
Alemany
*a and
David
Casanova
*de
*b,
Pere
Alemany
*a and
David
Casanova
*de
aDepartament de Ciència de Materials i Química Física and Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, Martí i Franquès 1-11, Barcelona 08028, Spain
bDepartment of Chemistry, Birla Institute of Technology and Science, Pilani Campus, Pilani, Rajasthan, India
cDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER), Sector 81, S. A. S. Nagar, Manauli PO, Mohali, Punjab 140306, India
dKimika Facultatea, Euskal Herriko Unibertsitatea (UPV/EHU), Donostia International Physics Center (DIPC), Paseo Manuel de Lardizabal, 4, Donostia 20018, Spain. E-mail: david.casanova@ehu.es
eIKERBASQUE, Basque Foundation for Science, Bilbao 48013, Spain
First published on 10th July 2017
Abstract
Four new Ir(III) complexes with the general formula [IrHCl(C^N)(PPh3)2] containing different conjugated Schiff base ligands (C^N) have been synthesized and characterized by 1H, 13C, and 31P NMR, HRMS, and IR spectra and one of them by single crystal X-ray diffraction. Their photophysical properties in solution and in the solid state have been analyzed and three main practical results have been obtained: (i) a dual fluorescent and phosphorescent emissive complex in solution, (ii) successful acid/base sensing in the solid state and (iii) quantitative CO2 detection. Quantum chemical calculations have been employed to assign the character of the lowest excited states. A plausible explanation for the observed aggregation induced enhanced emission (AIEE) is given, based on the restriction of intramolecular motions due to the effect of intermolecular C–H⋯π and C–H⋯Cl type interactions upon aggregation.
1. Introduction
Ir(III) complexes with long luminescence lifetimes (from 100 ns to ms), large Stokes shifts, high quantum yields and versatile color tuning by modification of the coordinating ligands are renowned as phosphorescent materials for their potential applications in the fields of organic light-emitting diodes (OLEDs),1–3 photoredox catalysis,4 vapoluminescent materials5–7 or luminescent probes for biological systems.8–10In the last decade, several studies have demonstrated Ir(III) compounds to be excellent candidates for materials exhibiting ‘Aggregation-Induced Enhanced Emission’ (AIEE),11–28 which makes them appealing for applications such as solid state lighting, where the luminescent complexes are present in aggregated forms such as solids or thin films. A key step in developing new materials based on the AIEE effect is to understand its origin at a fundamental level. The mechanism behind AIEE is, however, still under debate29–32 since many types of different systems, ranging from purely organic molecules to organometallic complexes, sometimes containing highly flexible parts, but in other cases quite rigid, have been identified as AIEE active compounds. In order to understand the origin of this photophysical phenomenon and identify the key parameters controlling its efficiency to optimize the implementation of AIEE materials in technological applications, it is necessary to combine experimental investigations with theoretical and computational studies.
In the present work, we report the complete synthesis of four novel Ir(III) complexes (1–4) by aliphatic C–H activation of a Schiff base ligand, in line with recent reports on the synthesis of new Ir(III)11,19–21,33–37 and Pt(II)38 luminescent complexes. Using both experimental characterization techniques and ab initio quantum chemical calculations we explore the photophysical properties of the synthesized compounds. In particular, complex 4 has been found to exhibit dual fluorescence and phosphorescence emission with the particularity that the fluorescent emission band is solvatochromic. This property is indeed very suitable for the development of Ir(III) probes,39 in which switching of the luminescence by external stimuli is essential. In the literature, there are several examples of dual phosphorescent Ir(III) complexes,40–47 but dual fluorescent and phosphorescent Ir(III) complexes are still scarce.48–51 To the best of our knowledge, the Ir(III) complex that we report herein is the first example of facile emission tuning between a singlet and a triplet state by just varying the solvent's polarity. This certainly opens a whole new range of possibilities for the application of Ir(III) complexes as luminescent probes.
The capability of the synthesized complexes as acid–base sensors in the solid state and the potential use of complex 4 in the quantitative detection of CO2 has also been explored. Due to the well-known harmful effects that high concentrations of CO2 have on human beings, the development of molecular sensors for its quantitative detection is nowadays an intense research field.52–58 Current detection techniques59,60 based on electrochemical (EC), infrared (IR), or gas chromatography–mass spectroscopy (GC–MS) methods, or field-effect transistors are quite costly, and efficient molecular sensors based on AIEE materials could represent a real breakthrough in this area.
2. Materials, methods and characterization
2.1 Materials
Ir(III) chloride hydrate, triphenylphosphine, pyridine-2-amine, pyrimidine-2-amine, 5-methylpyridine-2-amine, benzaldehyde, triphenylamine, phosphorous oxychloride, dimethyl formamide and 2-ethoxyethanol were purchased from the Sigma Aldrich Chemical Company Ltd. All spectroscopic grade solvents: dichloromethane (DCM), methanol (MeOH), benzene, 1,4-dioxane, ethyl acetate, chloroform (CHCl3), tetrahydrofuran (THF), acetonitrile (ACN), dimethylformamide (DMF) and dimethylsulphoxide (DMSO) were procured from the Merck Company.2.2 Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). X-ray quality single crystals could only be obtained for complex 2. Single crystal X-ray diffraction (SCXRD) revealed that the one-pot reaction of Ir(III) chloride and the triphenylphosphine mixture with the Schiff base resulted in a structure in which the imine C–H position was activated by Ir(III).
:1). X-ray quality single crystals could only be obtained for complex 2. Single crystal X-ray diffraction (SCXRD) revealed that the one-pot reaction of Ir(III) chloride and the triphenylphosphine mixture with the Schiff base resulted in a structure in which the imine C–H position was activated by Ir(III).
2.3 Characterization
1H, 13C and 31P NMR spectra were recorded in a 400 MHz Bruker spectrometer using CDCl3 as a solvent. Tetramethylsilane (TMS, δ = 0 for 1H and 13C NMR) and phosphoric acid (H3PO4, δ = 0 for 31P NMR) were used as internal standards. UV-VIS absorption spectra were recorded in a Simadzu Spectrophotometer (model UV-1800). The photoluminescence (PL) spectra were recorded on a Fluoromax-4 (0406C-0809) spectrofluorophotometer. The steady state photoluminescence spectra were recorded on a Spectrofluorometer FLS920-s Edinburgh. The quantum efficiency (QE) in solution for 1–4 has been measured with respect to coumarin 153 (in degassed ethanol, QE = 0.38, excitation 400–420 nm). High-resolution MS (HRMS) experiments were carried out with a (TOF MS ES+1.38 eV) VG Analytical (70-S) spectrometer and a Q-Tof micro mass spectrometer. Infrared spectra were recorded with an FTIR Shimadzu (IR prestige-21) spectrometer. All the reactions were performed under a nitrogen atmosphere and the progress of the reaction was monitored using thin-layer chromatography (TLC) plates (pre-coated with 0.20 mm silica gel).Lifetime data have been acquired with time correlated single photon counting (TCSPC) instruments, suitable for a ns–ps time range and nano LEDs with a 375 nm (average pulse width of 60.4 ps) and 405 nm (average pulse width of 56.9 ps) excitation source at room temperature. The solid-state quantum yield of the powder samples was measured using a calibrated integrating sphere in a Gemini spectrophotometer (Gemini 180). The emission and absorption spectra of the complexes were recorded after purging the sample with N2 gas.
The highly shielded 1HNMR signal in the range of −14.39 to −14.53 ppm indicates the presence of hydride (Ir–H) in the complexes. This finding is further supported by the stretching frequency at 2128–2176 cm−1 recorded by FTIR and the value computed using density functional theory (DFT) based methods at ∼2210 cm−1 for all the complexes. The IR stretching frequency of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the Schiff base ligands appears within the 1579–1610 cm−1 range and the computed DFT frequencies have the same order of magnitude (Table S8, ESI†). A 31P NMR signal is observed in the range of 4.34–6.39 ppm. From the HRMS data, the major fragmented peak appears as [M + H]+ at m/z in the range of 936.2476–1102.2661.
N in the Schiff base ligands appears within the 1579–1610 cm−1 range and the computed DFT frequencies have the same order of magnitude (Table S8, ESI†). A 31P NMR signal is observed in the range of 4.34–6.39 ppm. From the HRMS data, the major fragmented peak appears as [M + H]+ at m/z in the range of 936.2476–1102.2661.
N); ESI-HRMS, calculated: ([M + H]+), m/z 936.4581, found ([M + H]+): m/z 936.2476. (Fig. S1, ESI†)
N); ESI-HRMS, calculated: ([M + H]+), m/z 935.2063, found ([M + H]+): m/z 935.3567 (Fig. S2, ESI†).
N) ESI-HRMS, calculated: ([M + H]+), m/z 949.2219, found ([M + H]+): m/z 949.2698 (Fig. S3, ESI†).
N); ESI-HRMS, calculated: ([M + H]+), m/z 1102.2798, found ([M + H]+): m/z 1102.2661 (Fig. S4, ESI†).
2.4 Single crystal X-ray diffraction study
Single crystal X-ray diffraction data for 2 were recorded on a Rigaku XtaLAB mini single crystal X-ray diffractometer equipped with a Mercury 375/M CCD detector and graphite monochromated Mo-Kα radiation. The data set was collected at 100.0(1) K by using an Oxford cryosystem. The data were processed using Rigaku CrystalClear suite 2.0.61 The crystal structure was solved by using SHELXS201362 and refined using SHELXL2013, available within Olex2.63 All the hydrogen atoms have been fixed at their geometrically ideal position and have been refined isotropically using the riding model. Geometric calculations have been carried out using PARST.642.5 Computational details
Electronic structure calculations were performed within the DFT framework. Geometry optimizations of the ground state (S0) and lowest triplet state (T1) were carried out with the B3LYP hybrid functional,65 the double-ζ quality basis set consisting of Hay and Wadt's effective core potential (LANL2DZ) for the iridium atom and the standard all-electron 6-31G(d) basis set for all other elements.66–68 Optimized geometries were identified as true local minima with no imaginary frequencies within the harmonic approximation. Vertical transition energies at the S0 and T1 geometries were computed via time-dependent DFT (TDDFT) with the same energy functional and basis set. The absorption spectra were simulated by convolution of Gaussian functions with a half-width at half maximum (HWHM) of 3500 cm−1. The vibrationally resolved emission spectra from T1 were simulated within the Franck–Condon approximation as described by Barone et al.69 and a HWHM of 650 cm−1 was used for the Gaussian convolution. All calculations were carried out considering isolated molecules in a vacuum, except for the optimization of the S0 and intra-ligand charge transfer (ILCT) singlet state of complex 4, for which the IEF-PCM method70 was employed in order to account for equilibrium solvation. In addition, the CAM-B3LYP functional71 was used for the optimization of the 1ILCT state of 4 to account for the strong charge-transfer nature of this transition, not properly described by the B3LYP functional.72 Interactions between ACN molecules and Ir(III) complexes in the crystal structure were analyzed using two molecular models: (i) two Ir(III) complexes and the central ACN molecule and (ii) one Ir(III) complex with two ACN molecules interacting via both PPh3 ligands. Interaction energies between ACN molecules and Ir(III) complexes were computed by applying the standard counterpoise correction for the basis set superposition error73 at the M06-2X/LANL2DZ,6-31G(d,p) level.74 The origin of the observed disorder for solvent molecules in the crystal structure was also investigated at the M06-2X/LANL2DZ,6-31G(d,p) level by carrying out a fixed rotation of the methyl group of one ACN molecule in the presence of the two neighboring interacting Ir(III) complexes with fixed geometries. Color coordinates for compounds 1–4 were calculated from the experimental emission spectra in the standard CIE (2°) 1931 color space following the conventional procedure75 (ESI†). All electronic structure calculations were performed using the Gaussian09 package.763. Results and discussion
3.1 Molecular and electronic structure
All synthesized complexes have a single Ir(III) metal center in a distorted octahedral coordination environment. Complexes 1–4 contain two triphenylphosphine ligands (PPh3) axially coordinated with the Ir(III) atom, since our previous studies suggested that these highly mobile groups could be responsible for the observation of an enhanced emission in aggregated phases.20,37,77 The four coordination positions on the equatorial plane are occupied by the N and C atoms of a Schiff base bidentate ligand, a chloride, and a hydride ligand. The differences between the four synthesized complexes are in the Schiff base ligand (Scheme 1). Concretely, complex 1 contains a pyrimidine ring, while complexes 2 and 3 have pyridine and methylpyridine moieties instead, respectively. Complex 4 corresponds to the substitution of the Schiff base with a diphenylamine donor group.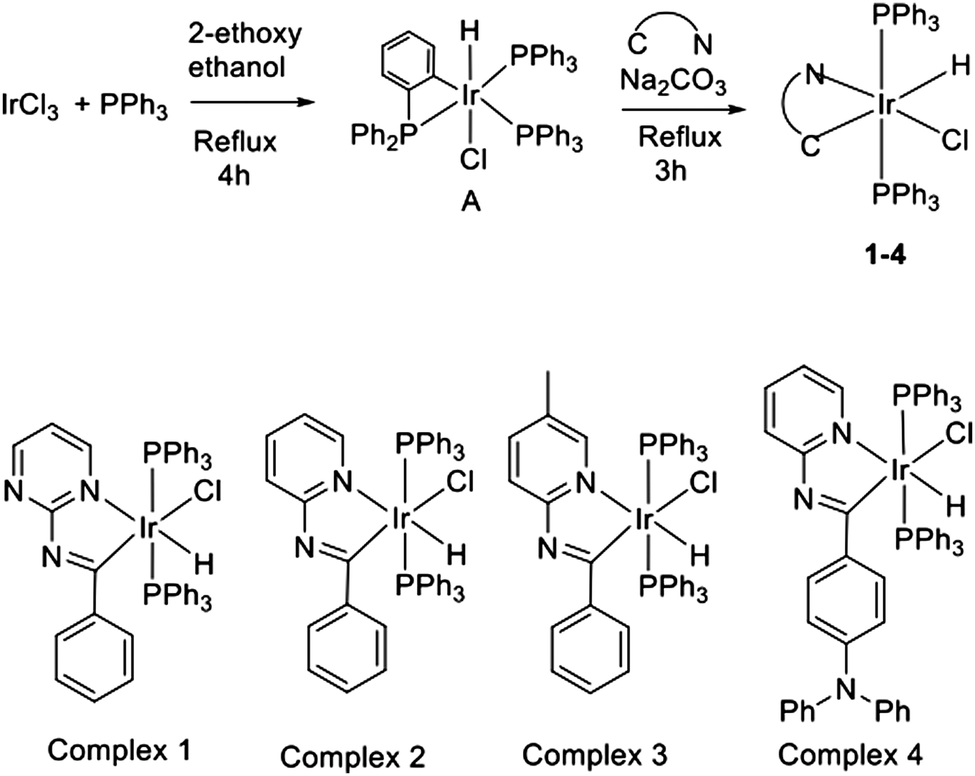 | ||
| Scheme 1 Synthesis of complexes 1–4. | ||
The principal geometrical parameters describing the distorted octahedral environment of the Ir(III) atom in complex 2 are gathered in Table S2 (ESI†). Optimized bond distances and angles for the ground state are in very good agreement with the X-ray molecular structure. A detailed discussion of the differences between the experimental and calculated geometries can be found in the ESI.†
Next we analyze in detail the electronic structure of the synthesized compounds by means of their frontier occupied and virtual molecular orbitals (MOs). In the following we refer to the z-axis as the one in the P–Ir–P direction and the x-axis defined by the C–Ir–Cl direction. Fig. 1 depicts the MO energy diagram for complex 2. Due to the heterogeneous ligand substitution pattern, all d-type orbitals from the Ir atom exhibit sizable mixings with ligand orbitals. In particular, Ir orbitals in the t2g-like set combine with occupied p-type orbitals from the chloride ligand, the lone pairs of the phosphorous atoms and π-type orbitals from the Schiff base ligand. The two highest occupied MOs, HOMO and HOMO−1, of complex 2 are practically degenerate combinations of metal dxy with chlorine py and metal dxz with chlorine pz AOs, respectively, with the HOMO−1 having an additional participation of π-type orbitals of the Schiff base ligand. The lowest unoccupied orbitals correspond mainly to π*-type orbitals of the Schiff base ligand. The lowest unoccupied MO (LUMO) has a small contribution of the Ir atomic orbitals, while the LUMO+3 shows participation of orbitals from two phenyl rings on the phosphine ligands. Further details regarding the MOs of 2 can be found in the ESI.†
 | ||
| Fig. 1 Molecular orbital diagram of 2 at the ground state geometry optimized at the B3LYP/LANL2DZ,6-31G(d) level. Participation of fragment orbitals to the whole system's MOs is indicated by the following color code: red for d-block orbitals of the iridium atom; light green for p orbitals of the chloride ligand; dark green for π and π* type orbitals of the phenyl rings of the phosphine ligands; blue for n, π and π*-type orbitals of the Schiff base ligand and orange for σ-type orbitals of the phosphorous's lone pairs. Hydrogen atoms are omitted from the picture for the sake of clarity. | ||
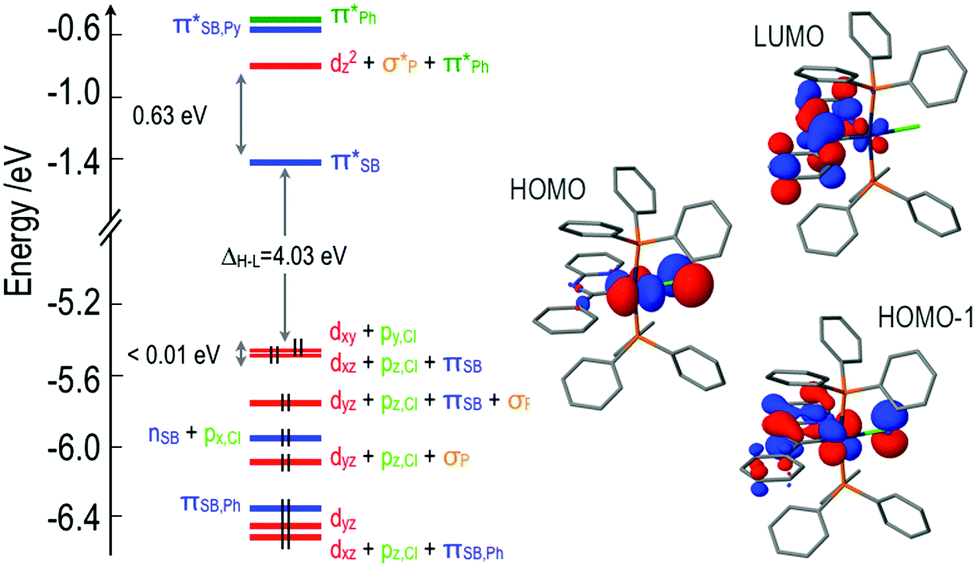
The MO diagrams for complexes 1 and 3 are qualitatively very similar to that of 2, except for subtle differences in the energies of the orbitals and the degree of mixing of fragment orbitals due to the differences in the Schiff bases. There is, though, an important difference between 4 and the other three complexes. Substitution of the Schiff base with an electron-donating diphenylamine group modifies dramatically its electronic structure. The HOMO is no longer an orbital centered on the iridium atom, but a pure π-type orbital of the donor amine unit (Fig. 2). The remaining frontier MOs of 4 are equivalent to those of 2.
 | ||
| Fig. 2 HOMO and LUMO of the optimized ground state of 4 at the B3LYP/LANL2DZ,6-31G(d) level. Hydrogen atoms have been omitted for the sake of clarity. | ||
3.2 Photophysical properties in solution and thin films
The lowest computed singlet and triplet states of complexes 1–3 correspond to excitations from the dxy and dxz type HOMOs to the Schiff base centered LUMO. Amplitude analysis of these excitations reveals that the corresponding excited states have important metal-to-ligand charge transfer (MLCT), ligand-to-ligand charge transfer (LLCT) from the chloride to the Schiff base, and ligand-centered (LC) π,π* character in the case of the excited state involving the dxz based occupied orbital. For simplicity, we will from now on refer to these excited states as MLLCTy and MLLCTz/LC, where y and z indicate the atomic p orbital of the chlorine atom involved in the occupied MO of the transition. The lowest excited singlet state for complexes 1–3 is MLLCTy, while the lowest triplet corresponds to MLLCTz/LC. In contrast, the lowest computed singlet and triplet states of complex 4 correspond to HOMO to LUMO transitions with ILCT character due to the donor nature of the diphenylamine substitution in the Schiff base. The computed vertical excitation energies to these states are gathered in Table 1.| State | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 3MLLCTz/LC | 456 | 472 | 479 | 420 |
| 3MLLCTy | 406 | 397 | 395 | 395 |
| 3ILCT | — | — | — | 528 |
| 1MLLCTz/LC | 370 (0.02) | 368 (0.04) | 368 (0.07) | 365 (0.01) |
| 1MLLCTy | 397 | 389 | 387 | 387 |
| 1ILCT | — | — | — | 416 (0.58) |
The UV-VIS absorption spectra in DCM solution for 1–4 are shown in Fig. 3. The moderately intense band at 300–450 nm is assigned by our calculations to the 1MLLCTz/LC state computed at ∼370 nm for all complexes. The absorption profiles of complexes 1–3 are very similar to each other, as predicted by the correspondence of their MO diagrams (Fig. 1). The absorption spectrum of complex 4 exhibits a rather intense band centered at ∼400 nm not present for the other three complexes, which is a signature of the ILCT transition in the Schiff base ligand. The vertical transition to 1ILCT in 4 is computed at 416 nm, in very good agreement with the experimental absorption maximum (404 nm). The simulated spectra of all complexes (ESI†) reproduce reasonably well the band positions and relative intensities of the experimental absorption spectra.
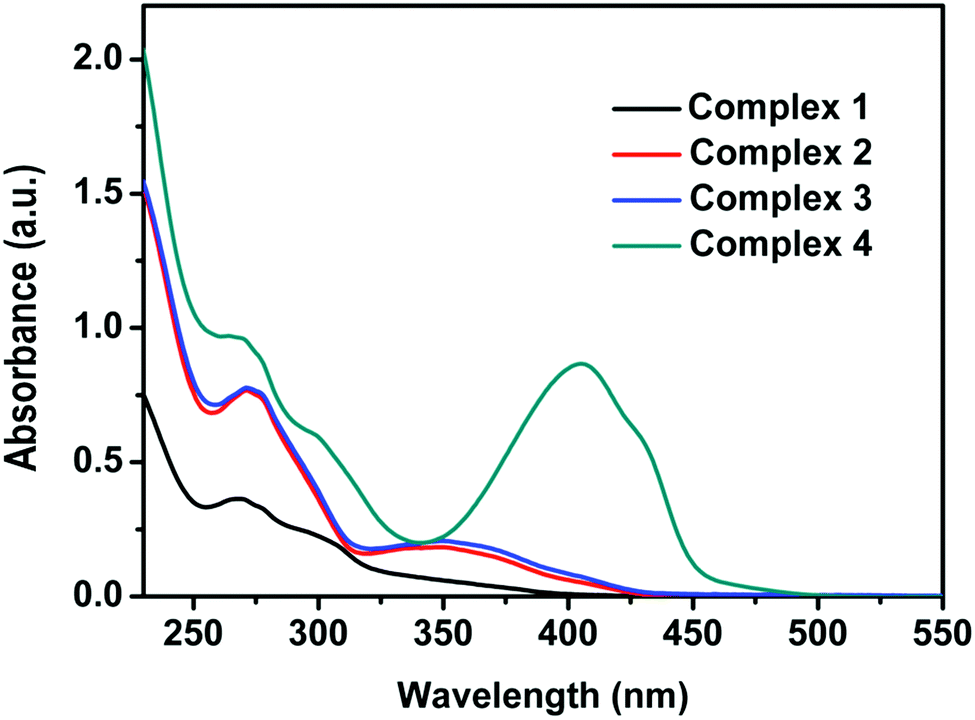 | ||
| Fig. 3 UV-VIS absorption spectra (in arbitrary units) of 1–4 (10−5 M) recorded in degassed DCM at room temperature. | ||
We have explored the solvent polarity dependence of the absorption spectra of the studied complexes. While the absorption spectra of 1–3 show no dependence on solvent polarity, the absorption spectrum of complex 4 shows a weak dependence with the solvent dielectric constant due to the strong absorption of the 1ILCT state and its CT character (Fig. S13, ESI†).
The normalized emission spectra of the synthesized complexes in DCM solution and in thin film are shown in Fig. 4. All complexes present structured emission profiles with a separation between the peaks on the order of 1300–1400 cm−1, which we assign to vibronic progressions.21,78,79 The position and relative intensity of the vibronic peaks in the thin film emission spectra are very close to their counterparts in solution. This matching is also reflected when comparing the emission colors in thin film with the emission colors in DCM solution shown in the chromaticity diagram (Fig. 5 and Table S4, ESI†), for which the same green to yellow transition is observed.
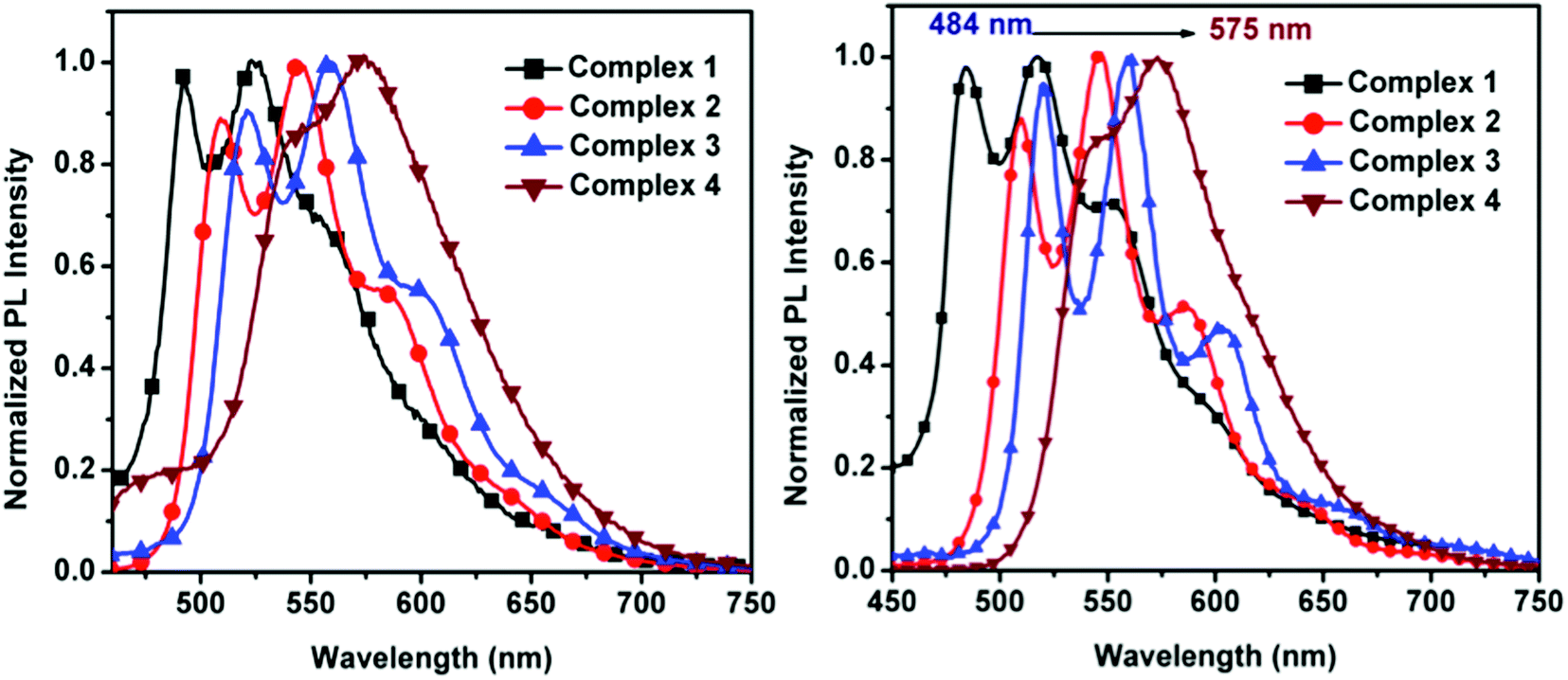 | ||
| Fig. 4 Normalized emission spectra of 1–4 in degassed DCM at room temperature (left) and thin film (right). | ||
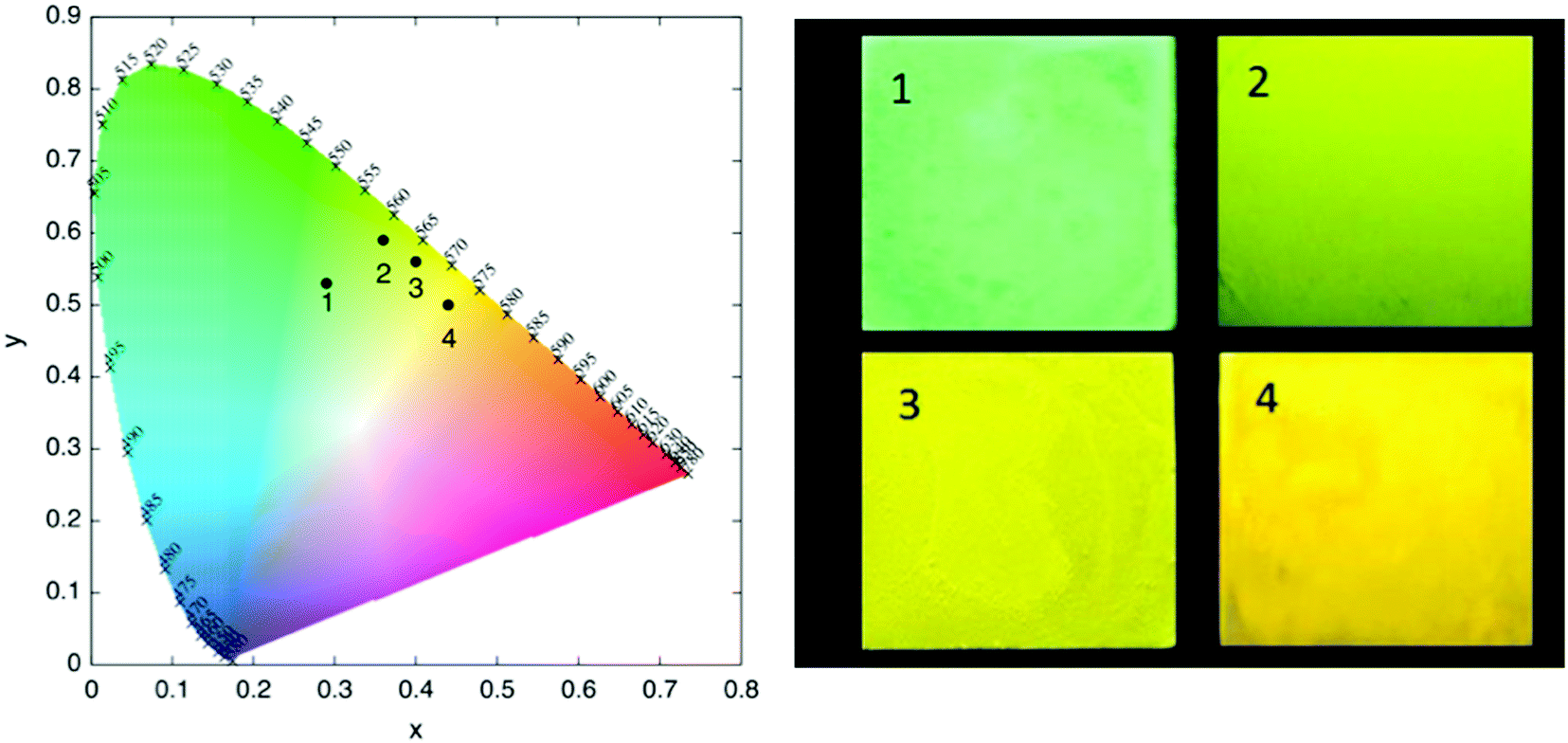 | ||
| Fig. 5 Chromaticity diagram for the emission of 1–4 in DCM (left) and their respective thin film emission color under 365 nm UV lamp (right). | ||
Our calculations predict the 3MLLCTz/LC state as the one responsible for the phosphorescence emission of complexes 1–3. For all three complexes there is a nice agreement between the experimental emission spectra and the simulated vibrationally resolved phosphorescence profiles (Fig. 6, Fig. S32 and S33, ESI†).
 | ||
| Fig. 6 Simulated (solid line) and experimental (dashed line) phosphorescence emission spectra of complex 2. Vertical sticks indicate the relative strength of vibronic transitions contributing to the simulated spectrum. Both spectra have been normalized and the calculated spectrum has been blue shifted by 0.12 eV to superimpose the most intense peaks. The 204 normal mode of vibration resulting in the vibrational structure corresponds to the CN stretching mode of the imine. | ||
For complexes 1–3, the main vibrational mode responsible for the structured emission is related to the stretching of the CN bond in the Schiff base, with a computed frequency of ∼1470–1450 cm−1 (Fig. S34, ESI†). The distortion associated with this mode is in fact the main geometrical modification along the molecular relaxation on the T1 state potential energy surface from the Franck–Condon region. Although this vibrational progression is the main contribution defining the overall shape of the emission spectra, there are other less intense normal modes also contributing to the emission profiles (Tables S5–S8, ESI†).
The emission spectrum of 4 in solution exhibits two clearly distinct bands (Fig. 7): (i) a solvatochromic band at 400–525 nm and (ii) a structured emission band in the 525–700 nm region, very similar to the emission band in 1–3, that is practically independent of the solvent's polarity (Fig. S14, ESI†). Due to this dual emissive behavior, the emission color of 4 may easily be tuned by varying the solvent's polarity. In particular, in low polarity solvents (benzene, 1,4-dioxane), the emission color of 4 is blue with an emission band maximum at 461 nm, whereas in more polar solvents (ethyl acetate, CHCl3, DCM and THF) the emission gradually shifts to longer wavelengths. In strongly polar solvents (DMSO) complex 4 shows yellow emission (Fig. 7).
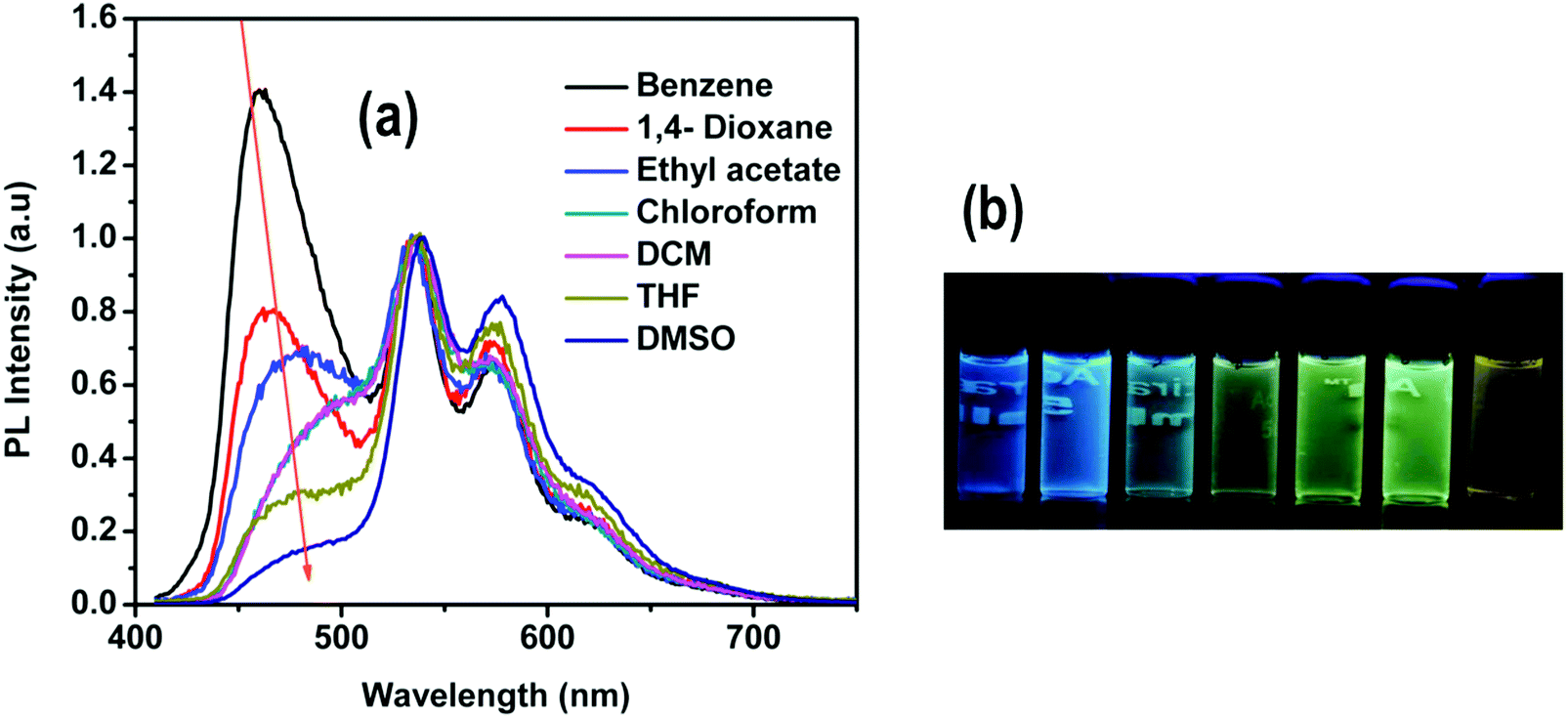 | ||
| Fig. 7 (a) Emission spectra of 4 in different solvents. (b) Photographs showing the emission colors of 4 under UV light in different solvents (from left to right): benzene, 1,4-dioxane, ethyl acetate, CHCl3, DCM, THF and DMSO. Concentration of 4 in all cases: 1 × 10−5 M. The spectra have been normalized to the peak at ∼535 nm. | ||
The similarities of the emission of 4 in the 525–700 nm region with the emission spectra of complexes 1–3 allow us to assign this band to the phosphorescence emission from the 3MLLCTz/LC state. On the other hand, our calculations indicate that the state responsible for the solvatochromic band below 500 nm corresponds to the singlet state with ILCT character. This assignment is also supported by time-resolved photoluminescence measurements by monitoring the emission maxima at 450 nm and 530 nm in different solvents (benzene, DCM, THF and DMSO). The lifetime data for the high-energy band is obtained within the 0.9–1.4 ns range, consistent with fluorescence emission (Fig. S15–S18 and Table S1, ESI†). On the other hand, the lifetime measurements of the band at 530 nm are in the microsecond range (from 1.4 to 1.7 μs), consistent with phosphorescence emission. The 1ILCT state of 4 was optimized in strongly polar and non-polar solvents, i.e. DMSO and benzene, for which the experimental emission maxima were measured at 480 and 459 nm, respectively. The calculated relative shift in the vertical emission energies of 4 in benzene and DMSO is 0.16 eV, in very good agreement with the shift observed in the experimental spectra (0.12 eV). As the polarity of the solvent increases, the LUMO is more stabilized than the HOMO (Table S12, ESI†), leading to the observed red-shift in the emission energy. Further calculations ruling out the possible formation of a twisted intramolecular charge transfer state (TICT) may be found in the ESI.†
The observed red-shifted emission of the fluorescent ILCT band in highly polar solvents of 4 is also accompanied by a decrease in its PL intensity. This is clearly seen in Fig. 8, where the emission spectra of 4 recorded with different fractions of benzene (fB) in THF are shown. The gradual addition of benzene to the THF solution results in a blue shifted emission with increasing PL intensity. The spectral data reveal that the intensity of the short wavelength band gradually increases with increasing benzene fractions.
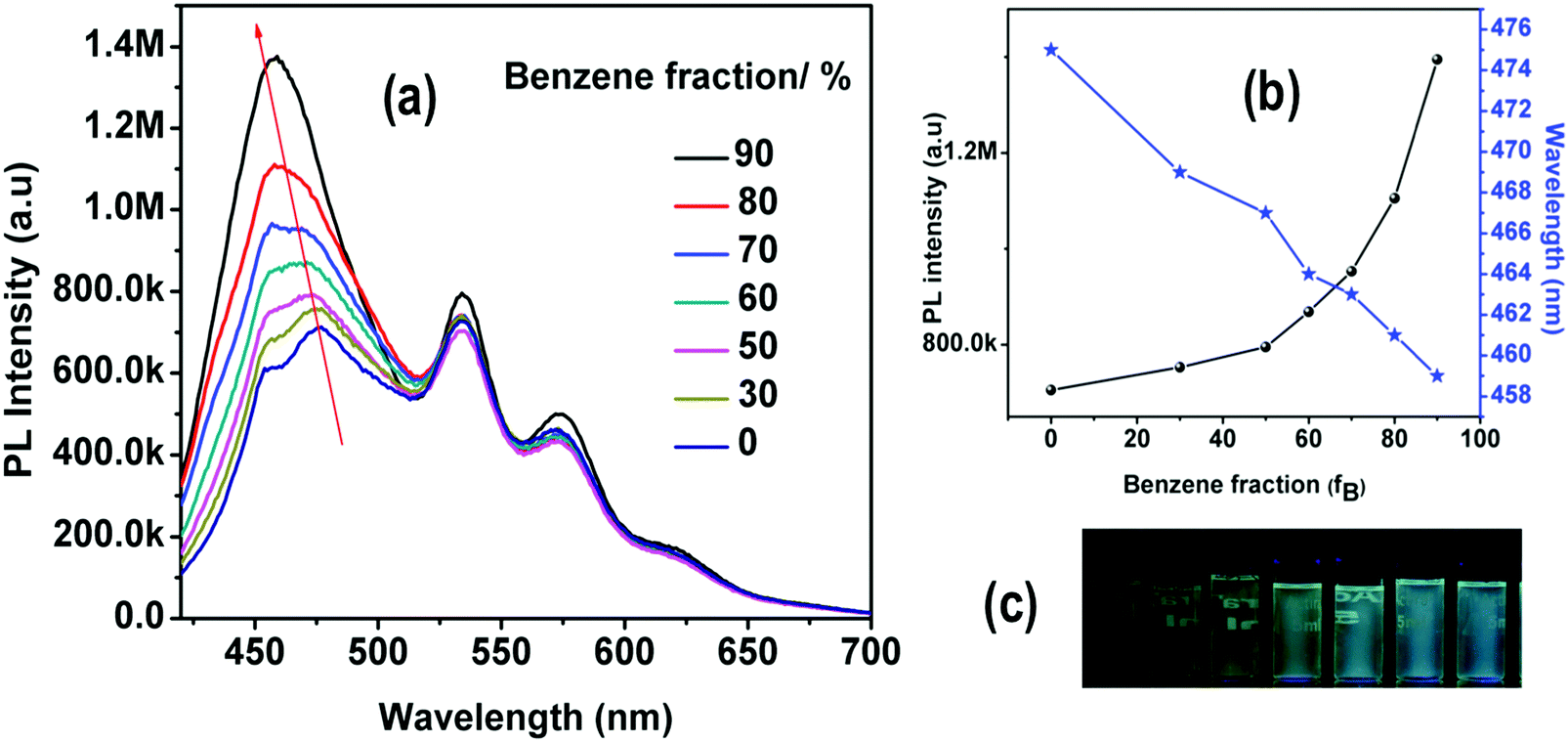 | ||
| Fig. 8 (a) Emission spectra of 4 in THF with different benzene fractions (0–90%). (b) Plots of maximum emission intensity and wavelength of 4versus benzene fraction (fB) in the THF/Benzene mixture. Solution concentration: 1 × 10−5 μM. (c) Photographs of 4 in THF/benzene mixtures with different fractions of benzene (fB) taken under 365 nm UV illumination. | ||
Frozen samples of 4 in benzene and 1,4-dioxane at −80 °C showed yellow emission under excitation at 365 nm (Fig. S19, ESI†), while at room temperature a blue emission was observed. Since the yellow emission of 4 has been attributed to the 3MLLCTz/LC state, this suggests that molecular relaxation of the 1ILCT state from the Franck–Condon geometry is slowed down in the frozen samples, allowing the population of the 3MLLCTz/LC state via intersystem crossing.
Finally, the excitation and emission spectra recorded in different solvents are in agreement with a dual emission from two different states, i.e.1ILCT and 3MLLCTz/LC (Fig. S20, ESI†).
3.3 Aggregation-induced enhanced emission
All four synthesized complexes exhibit weak emission in common organic solvents such as DCM, THF, MeOH, CHCl3, ACN, or DMSO, but they are highly emissive in the solid state. Such strong solid-state emission encouraged us to study the AIEE of these complexes. Complexes 1–3 are highly soluble in THF but insoluble in water, thus we explored the effects of aggregation on their PL properties in THF–water with a wide range of mixing ratios. The formation of aggregates when water is present was confirmed using a particle size analyzer, indicating the formation of nano-aggregates with diameters in the range of 124–674 nm (Fig. S11, ESI†). Solutions of 1–3 in pure THF emit weakly, while their emission gradually increases with the water fraction in THF–water mixtures. For mixtures with a high content of water (fw = 90%), the PL intensity of complexes 1, 2 and 3 is 45, 18, and 6 times stronger than that in pure THF solution (Fig. S21, ESI†), respectively.The AIEE property of 4 was also investigated in different solvents, namely THF, ACN and DMSO with gradual addition of water. The dilute solution of THF shows a faint green photoemission that intensifies and red shifts (from 535 nm to 578 nm) at fw = 10%. The addition of more water, (fw = 20%), leads again to a red shifted emission maximum (from 578 nm to 610 nm) and to an increase of the emission intensity. We attribute this spectral change (red shifted emission) to the stabilization of the charge transfer state with increasing solvent polarity.80 A further increase of the water fraction up to 70% does not alter the emission maximum and its intensity. At fw = 80% however, a sudden blue shifted structured emission (516 nm and 557 nm) appears along with a slight increase in the PL intensity. In contrast, the emission red shifts again (from 535 nm and 574 nm) at fw = 90%, but the structured profile remains and the PL intensity is 22 times higher than that in pure THF (Fig. 9). In this case, the observed emission enhancement at higher water content can also be safely attributed to aggregate formation (Fig. S11, ESI†). In addition, we argue that closely spaced aggregates could be responsible for creating a less polar environment, resulting in a blue shifted emission.81,82
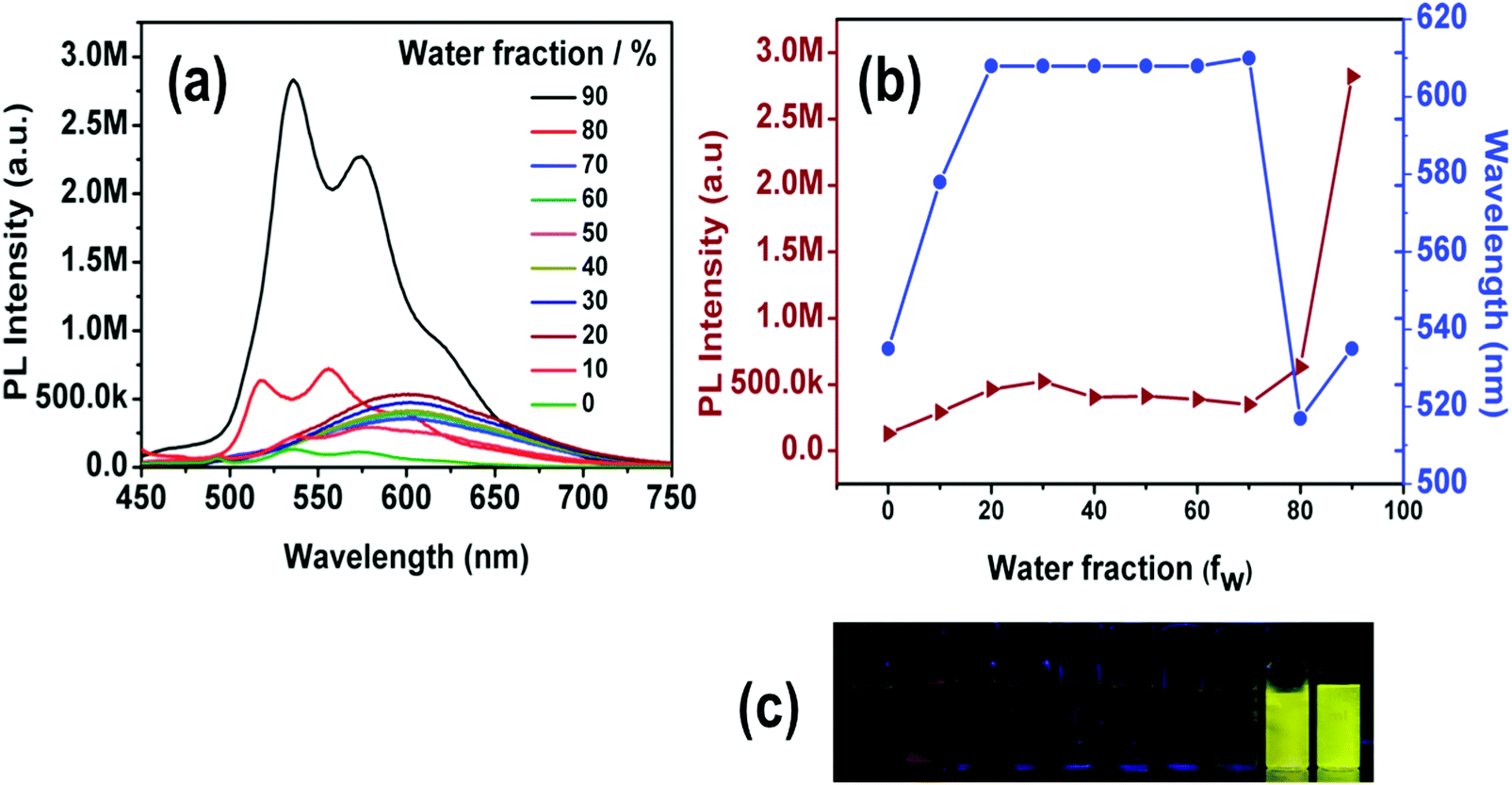 | ||
| Fig. 9 (a) Emission spectra of 4 in THF/water mixtures (0–90%). (b) Plot of maximum emission intensity and wavelength of 4versus water fraction. Concentration of 4: 1 × 10−5 M. (c) Photographs of 4 in THF/water mixtures taken under UV illumination. | ||
The emission behavior of 4 was also studied using mixtures with different ACN–water fractions (Fig. S22, ESI†). The emission intensity of 4 doubles at fw = 30% in comparison to pure ACN solution, while the emission maximum remains at 510 nm. At fw = 50% the PL intensity increases by 5 times with respect to pure ACN solution and the emission maximum slightly red-shifts to 523 nm. Further addition of water (fw = 70%) increases the PL intensity by 5 times in comparison to pure ACN solution, but now the emission band is no longer broad, displaying two distinct peaks at 517 nm and 552 nm, respectively. The structured band remains at fw = 90% with a significant red shift to 538 and 580 nm, however, the PL intensity weakens dramatically to only thrice that of the pure ACN solution.
The emission spectra of 4 in DMSO–water mixtures do not show any changes in the emission maximum, since there is already a highly polar environment in the pure organic solvent, but the PL intensity increases by 32 times as compared to the pure DMSO solution of the complex (Fig. 10).
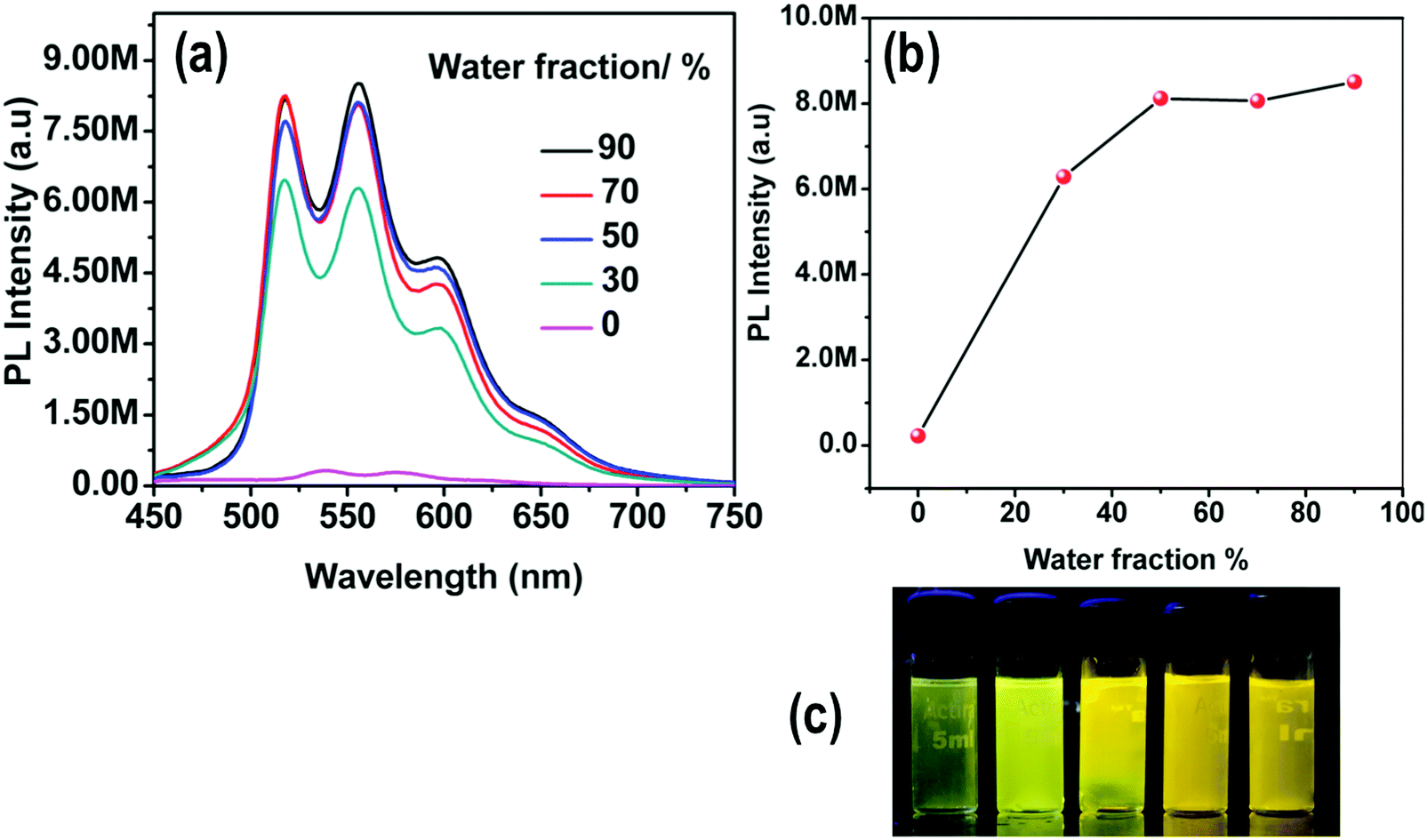 | ||
| Fig. 10 (a) Emission spectra and (b) plot of maximum emission intensity of 4 in DMSO/water mixtures (0–90%). Concentration of 4: 1 × 10−5 M. (c) Photographs of 4 in DMSO/water mixtures taken under UV illumination (excitation at 365 nm). | ||
The formation of nanoaggregates in the AIEE experiments of complex 4 was confirmed by the leveled-off tails81 in the visible region of the recorded UV-VIS spectra in different water fractions with DMSO, as well as with ACN (Fig. S23, ESI†).
The AIEE of these complexes was further studied by measuring the absolute emission quantum yields (QY) for solid samples, and comparing these data with those obtained in DCM solution. The QY of these complexes in solution is <0.1%, while the absolute QY in the solid state is 3–9% that is, about two orders of magnitude higher.
In order to explore the possibility of whether weak intermolecular interactions could be responsible for the restriction of intramolecular motions83,84 in the solid state and give rise to an enhancement of the PL intensity upon aggregation, the crystal packing of 2 was analyzed in detail considering that it might represent an acceptable structural model for the aggregates formed in the mixtures of water with organic solvents. The crystal structure of 2 has a unit cell with four equivalent Ir(III) complexes together with four cocrystallized ACN molecules exhibiting disorder in the position of their hydrogen atoms. Iridium complexes arrange in chains along the b-axis, with each chain being surrounded by six other chains in an approximate elongated hexagonal fashion as shown in Fig. 11. The molecules of the reference chain (highlighted in red) lie in the same ac-plane as the chains highlighted in grey. On the other hand, the chains shown in blue are displaced by half a unit cell along the b-axis with respect to the reference chain. Inspection of interatomic distances between molecules in the reference and grey chains indicates the absence of potentially important non-covalent interactions between them. The reference and blue chains, however, exhibit short interchain C–H⋯Cl distances (2.802 Å) between the chloride ligand and a hydrogen atom from an adjacent PPh3 ligand, indicative of non-negligible interchain interactions that represent a possible source of restriction of molecular motions in the solid state (Fig. S37, ESI†). Nevertheless, the most significant interactions are found along the chains themselves, where crystallized ACN solvent molecules alternate with iridium complexes, as shown in Fig. 12. An analysis of intermolecular distances hints to the presence of strong C–H⋯π interactions between each ACN molecule and two phenyl rings from PPh3 ligands of adjacent complexes. The computed interaction energy between one complex with the two neighboring ACN molecules was 9.1 kcal mol−1, confirming the presence of these C–H⋯π interactions.
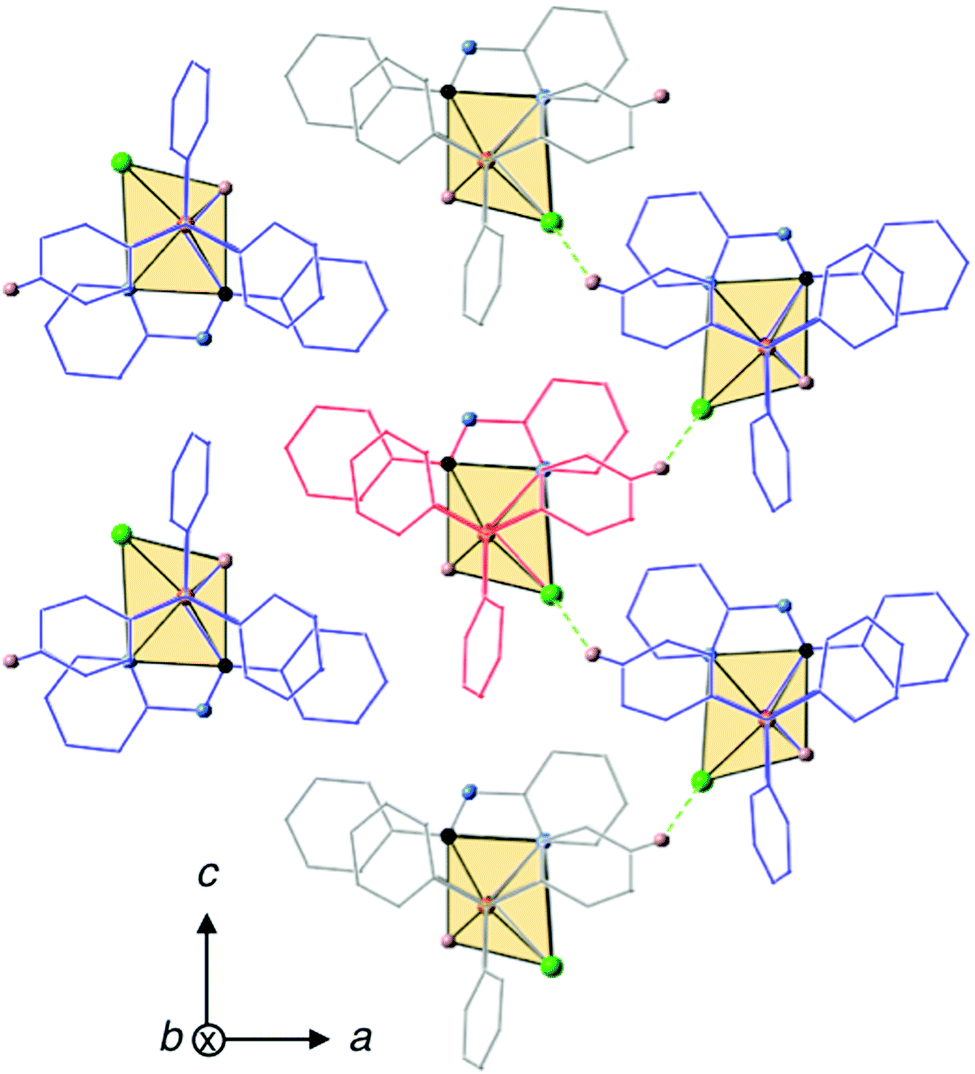 | ||
| Fig. 11 Crystal packing of 2 with a reference chain highlighted in red. C–H⋯Cl interactions (2.802 Å) between the chains are shown in green. Atom color code: nitrogen in blue, phosphorous in orange, chlorine in green, carbon in black and hydrogen in white. Hydrogen atoms not involved in intermolecular interactions are omitted for the sake of clarity. | ||
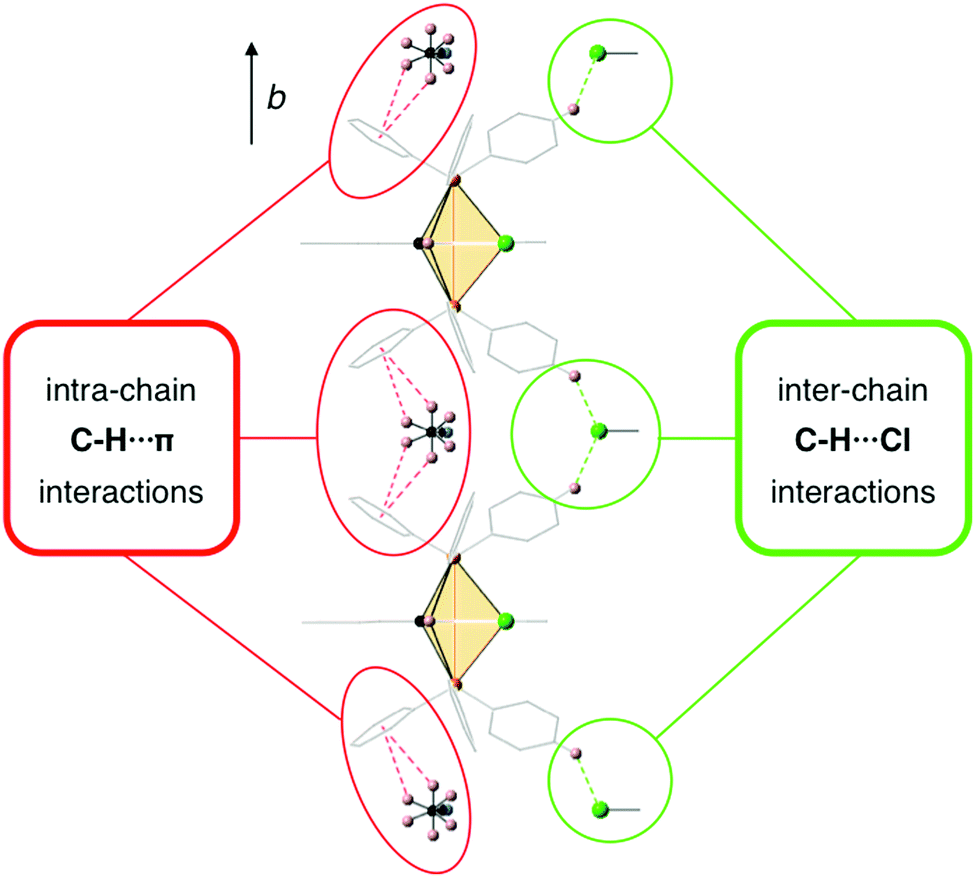 | ||
| Fig. 12 C–H⋯π interactions between ACN molecules and Ir(III) complexes along the crystal chains and inter-chain C–H⋯Cl interactions with the chains highlighted in blue in Fig. 11. ACN molecules present dynamic disorder. | ||
Further calculations were carried out to investigate the origin of the observed disorder for the hydrogen atoms of the ACN molecules in the crystal structure. Calculation of the energy cost for a rotation of the methyl group of the solvent molecule considering a fixed geometry was carried out in the presence of the two neighboring complexes (see Computational details). Two minima were obtained corresponding to the two hydrogen configurations found in the resolution of the crystal structure. The computed energy barrier (0.3 kcal mol−1) for the interconversion of the two conformations lies below the thermal energy kT at the temperature of crystal determination (0.4 kcal mol−1), suggesting that the origin of the observed disorder in the crystal structure is dynamic rather than static. These calculations show that in the crystal we should expect the methyl groups to rotate quite freely, establishing thus C–H⋯π interactions that restrict the motion of the PPh3 ligands and block non-radiative decay channels in the solid state with an increase of the emission intensity as a result. It has been recently shown that these types of interactions might play an important role in the solid state photophysics of iridium complexes.20
3.4 Reversible protonation–deprotonation
We also performed photophysical studies of complexes 1–4 in the presence of TFA/Et3N. Interestingly, addition of TFA triggers a considerable bathochromic shift in the thin film emission spectra of 1 and 4. The exposure of a thin film of 1 to TFA results in a broad emission band with a maximum at 539 nm responsible for a yellow color, which is highly reversible to its original green emission with maxima at 484 nm and 517 nm after exposure to Et3N (Fig. 13). The yellow emitting thin film of 4 becomes red emissive with an emission maximum at 617 nm upon exposure to TFA. Likewise, subsequent Et3N addition leads back to its original yellow emission. The thin films of complexes 2 and 3 also present reversible modification of the emission spectra under TFA/Et3N, although the emission color change is less pronounced (Fig. S24, ESI†).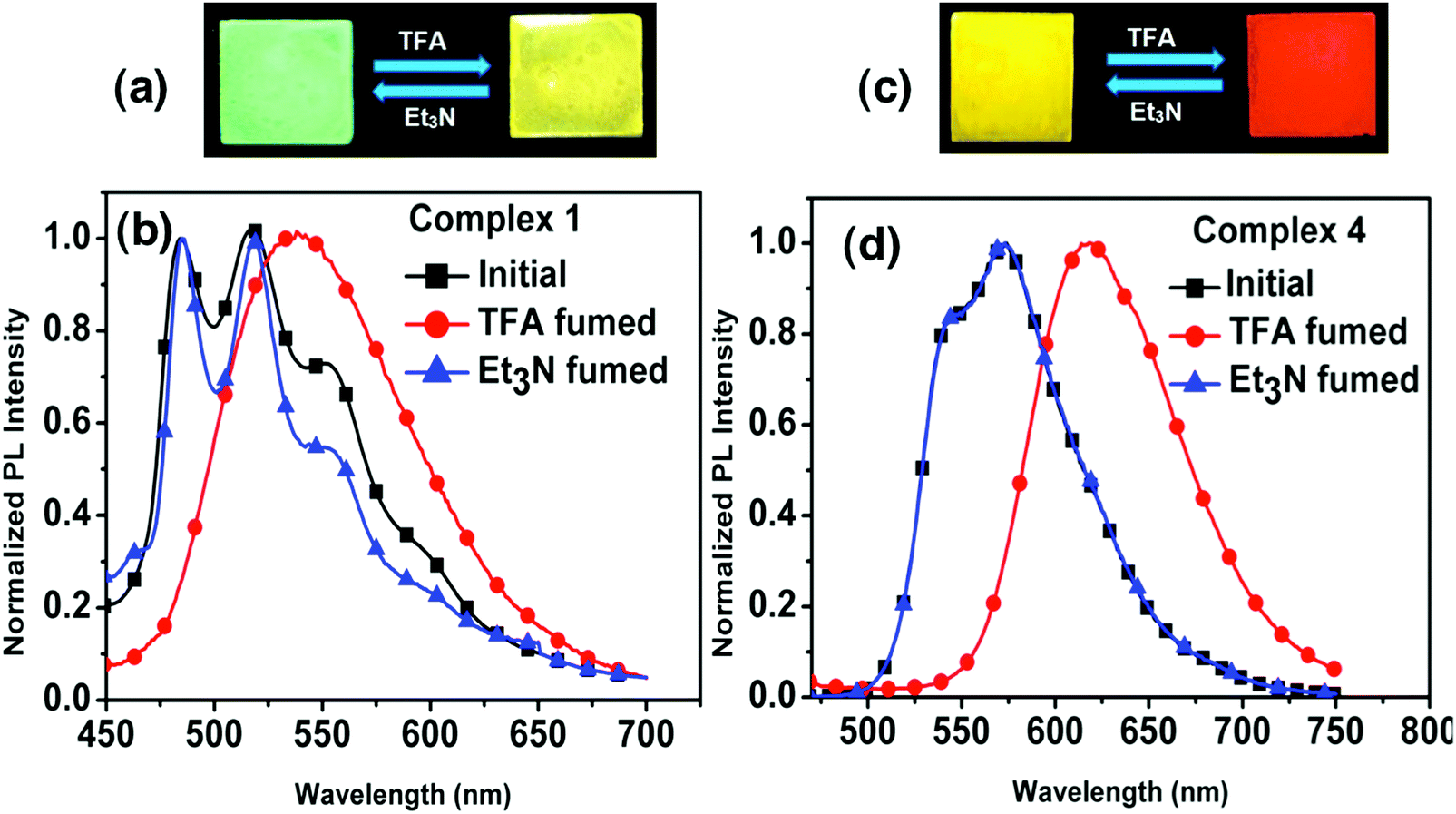 | ||
| Fig. 13 Thin film reversal of emission color upon TFA/Et3N exposure and emission spectra of complexes 1 (a and b) and 4 (c and d). The thin-film photographs and PL spectra were taken under an excitation wavelength of 365 nm and 400 nm, respectively. | ||
The reversibility of such protonation/deprotonation processes is confirmed by the 1H NMR spectra of the protonated complexes (1–4) recorded in CDCl3 solution. 1H NMR of complex 1 after exposure of the film to TFA shows a downfield signal at δ = 9.01 ppm, not present in the absence of TFA. Similarly 2, 3 and 4 display an even more downfield signal at δ = 13.56, 13.33, and 12.99 ppm, respectively. These 1H NMR spectra indicate the transformation of these complexes into a protonated form, where the downfield 1H signals may be attributed to the protonation of the nitrogen atom in the imine ligand unit (Fig. S12, ESI†). Electronic structure calculations also suggest that the energetically most favorable protonation occurs on the iminic nitrogen for all complexes (Table S9, ESI†). Computation of the protonated cationic form of complex 2 (labeled as 2H) indicates that the most important geometrical difference with respect to the neutral complex is the larger torsion angle of the phenyl ring of the Schiff base ligand due to steric hindrance between one of its hydrogen atoms and the protonating hydrogen (Fig. S38, ESI†). In the unprotonated complex, the Schiff base ligand is completely planar, while in the 2H form the phenyl ring experiences a torsion of 22° with respect to the complex's equatorial plane. Other structural differences of the coordination sphere of complex 2 upon protonation may be found in the ESI† (Table S10).
Protonation of the molecular species disrupts their electronic properties, resulting in changes in their photophysical behavior. Concretely, there is a reduction of the HOMO–LUMO gap upon protonation. Moreover, in the protonated form the electron density on the Schiff base ligand is strongly stabilized. As a result, the highest occupied orbitals with dxz and dyz participation do not show contributions from the conjugated systems of the bidentate ligand (Fig. S39, ESI†), and the computed excitation energies to the 3MLLCTy and 3MLLCTz states are very close to each other (Table S11, ESI†). These results point towards three factors responsible for the broad emission band with no vibrational structure observed upon exposure of the complexes to TFA: (i) the absence of LC character in the emitting triplet state, (ii) the loss of ligand planarity and (iii) the increase of molecular flexibility.
The dependence of the emission profiles for these complexes with pH suggests that these materials are potential candidates for H+ detection. Additional spectra of 1–4 in solution with the presence of TFA may be found in the ESI† (Fig. S25 and S26).
3.5 CO2 detection
The solubility of 4 was screened with different amines, e.g. pyridine, 1,8-diazabi-cyclo-[5,4,0]-undec-7-ene (DBU), piperidine and diethyl amine (DEA). Complex 4 was found to be completely soluble in DEA. The reaction between DEA and CO2 yields a viscous carbamate ionic liquid (CIL). In this case, the CIL was synthesized by a simple reaction condition where the DEA solution was purged by a large excess of CO2 gas for 10 minutes. The synthesized CIL was characterized by 1H NMR and 13C NMR spectroscopy and the results matched satisfactorily with earlier reports54 (Fig. S6, ESI†).The weak emission of complex 4 in DEA solution becomes gradually more intense after purging of CO2 bubbles. In fact, there is a linear relationship between the amount of CIL gradually added into the solution (fCIL) and the emission intensity of the sample. At fCIL = 90%, the PL intensity is 2.4 times stronger as compared to the intensity of the DEA solution (Fig. 14). This emission enhancement may be attributed to the increase in the viscosity of the medium that leads to a restriction of the motion of the phenyl rings in the PPh3 ligands possibly responsible for non-radiative decay. The increase of the PL intensity of complex 4 with the viscosity of the environment has been confirmed by comparing the emission spectra in PEG–THF mixtures (Fig. S27, ESI†). To further analyze the response of 4 in DEA solutions to CO2, additional experiments were carried out in which different fractions of CO2 were purged through a solution of 4 in DEA at a fixed rate and time. The emission of 4 was enhanced by gradual addition of CO2 in the system and resulted in a linear plot for the whole concentration range. These results indicate the potential use of DEA diluted complex 4 as a quantitative detector of CO2 (Fig. 15). Although the addition of CO2 facilitates the removal of O2 from the sample which might result in a larger emission intensity, it cannot account for the large PL intensity increase observed.
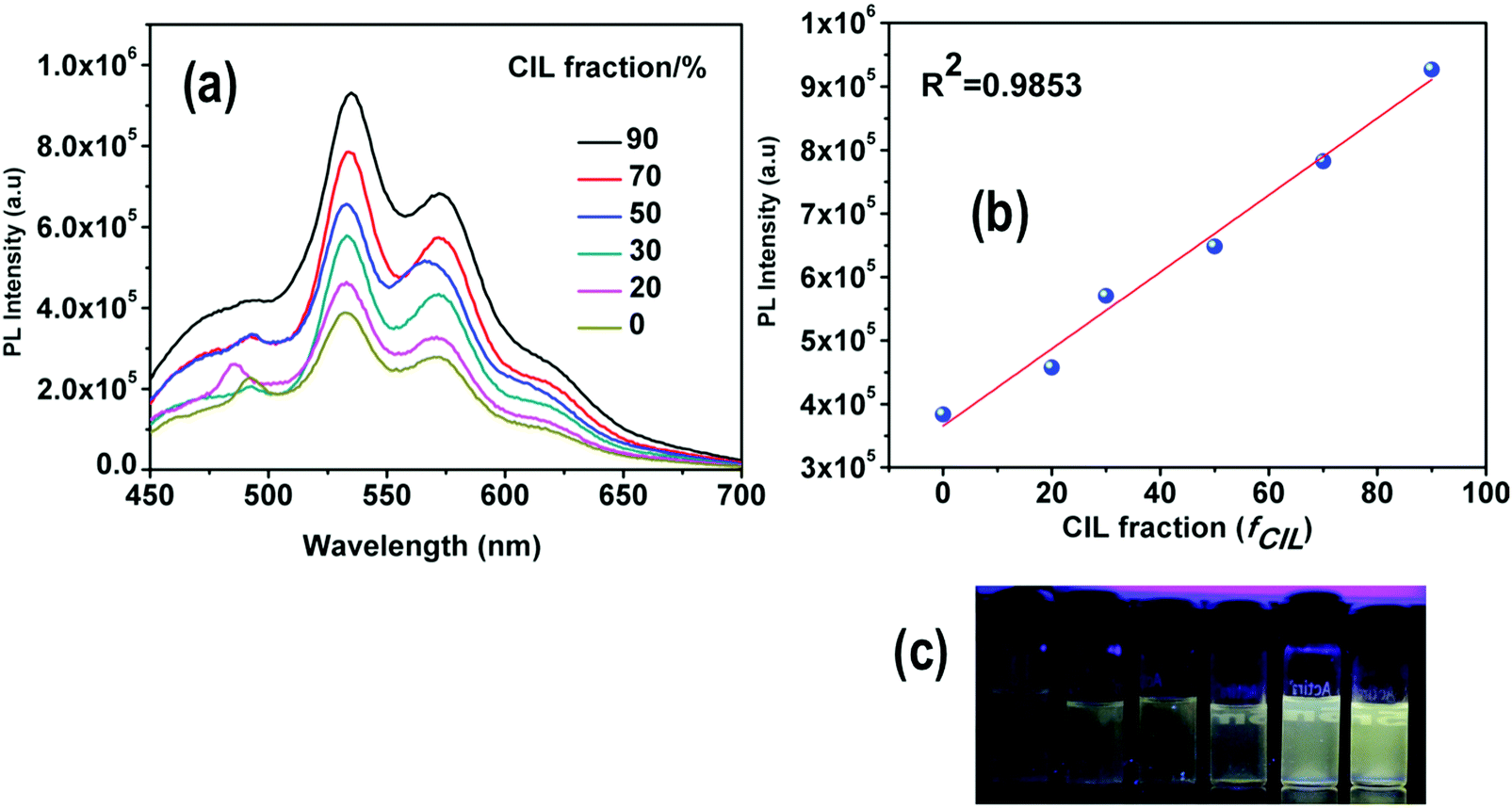 | ||
| Fig. 14 (a) Emission spectra of 4 in DEA with different fractions of CIL (0–90%). (b) Maximum emission intensity with respect to CIL. (c) Photographs of 4 in DEA/CIL mixtures taken under UV illumination. | ||
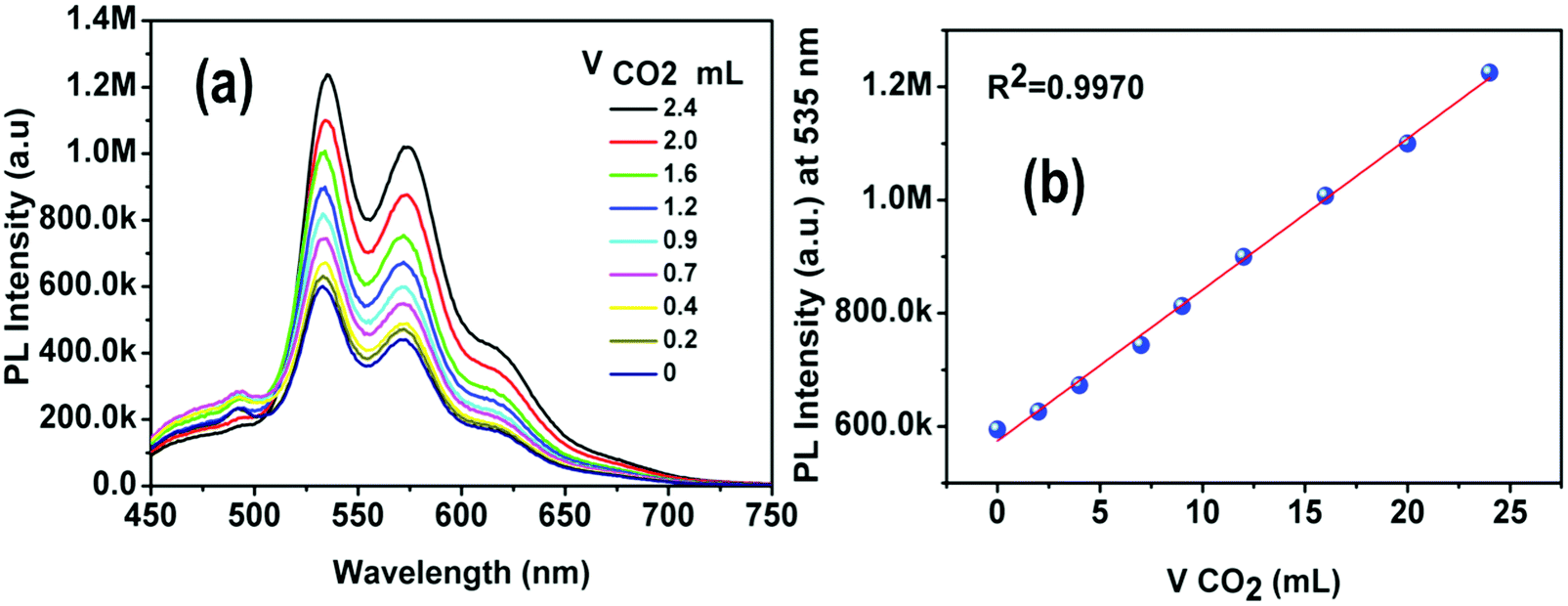 | ||
| Fig. 15 (a) PL spectra of 4 in DPA with different volumes of CO2 displaying a gradual enhancement of the PL intensity. (b) The PL intensity increased in a linear fashion (R2 = 0.9970) with gradual addition of CO2. | ||
4. Conclusions
In this study we have demonstrated a convenient route for the synthesis of a new series of ‘Aggregation-Induced Enhanced Emission’ (AIEE) active Ir(III) Schiff base complexes with facile color tuning. The calculations indicate that the triplet state responsible for the vibrationally structured phosphorescence band of the synthesized complexes is of mixed MLLCT and LC character of the Schiff base. The presence of a donor–acceptor unit in 4 is responsible for the additional fluorescence emission observed and its positive solvatochromism (increase in emission wavelength with increase in solvent polarity). The excited state responsible for this fluorescence has been characterized by our calculations as an ILCT state of the Schiff base ligand. Such dual emission present in 4 accounts for an outstanding color tuning from blue to yellow. In addition, 4 was found to be a probe molecule whose ILCT emission gradually increases with an increasing content of non-polar solvent (benzene) in polar media (THF). The crystal packing of 2 was analyzed revealing significant intra-chain C–H⋯π interactions (∼9 kcal mol−1) and inter-chain C–H⋯Cl interactions, which could be responsible for the hindering of internal motions of the PPh3 ligands, blocking the non-radiative decay channels and hence enhancing the emission in the solid state. Thin films of the synthesized complexes were successfully employed for reversible detection of acidic and basic vapors. Calculations and 1H NMR spectra hint at protonation of the iminic nitrogen as being responsible for the changes observed in the emission spectra of the thin films upon TFA exposure. An easy method for CO2 detection was also developed with 4 using a phosphorescence technique controlled by the viscosity of the medium. All the properties studied in this work point towards the potential application of the newly synthesized complexes as multi-responsive luminescent materials.Acknowledgements
This work was supported by the Basque Govt. (project IT588-13) and the Spanish Govt. (MINECO/FEDER projects CTQ2015-64579-C3-3-P and CTQ2016-80955). We are thankful to DST, Govt. of India, under a project (no. SB/S1/IC-13/2014) and CSIR, Govt. of India, (no. 01/2551/12/EMR-II) for financial assistance. The UGC-SAP and DST-FIST, Govt. of India, are acknowledged for the instrumental support to the Department of Chemistry, BITS Pilani. We thank Dr Neetu Singh, advanced instrumentation research facility, JNU, New Delhi (India), for providing technical help on time resolved fluorescence spectroscopy. IISER Mohali is acknowledged for providing the X-ray diffraction and NMR facilities. D.C. thanks IKERBASQUE, Basque Foundation for Science for financial support. C.C. is indebted to the Spanish “Ministerio de Economía y Competitividad” for a predoctoral FPI grant.References
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, Nearly 100% internal phosphorescence efficiency in an organic light-emitting device, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, High-efficiency fluorescent organic light-emitting devices using a phosphorescent sensitizer, Nature, 2000, 403, 750 CrossRef CAS PubMed.
- H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH, Weinheim, 2008 Search PubMed.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, Enantioselective α-Trifluoromethylation of Aldehydes via Photoredox Organocatalysis, J. Am. Chem. Soc., 2009, 131, 10875 CrossRef CAS PubMed.
- A. Kobayashi and M. Kato, Vapochromic Platinum(II) Complexes: Crystal Engineering toward Intelligent Sensing Devices, Eur. J. Inorg. Chem., 2014, 4469 CrossRef CAS.
- O. S. Wenger, Vapochromism in Organometallic and Coordination Complexes: Chemical Sensors for Volatile Organic Compounds, Chem. Rev., 2013, 113, 3686 CrossRef CAS PubMed.
- X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, Luminescence vapochromism in solid materials based on metal complexes for detection of volatile organic compounds (VOCs), J. Mater. Chem., 2012, 22, 11427 RSC.
- K. K.-W. Lo, C.-K. Chung, T. K.-M. Lee, L.-H. Lui, K. H.-K. Tsang and N. Zhu, New Luminescent Cyclometalated Iridium(III) Diimine Complexes as Biological Labeling Reagents, Inorg. Chem., 2003, 42, 6886 CrossRef CAS PubMed.
- K. K.-W. Lo, P.-K. Lee and J. S.-Y. Lau, Synthesis, Characterization, and Properties of Luminescent Organoiridium(III) Polypyridine Complexes Appended with an Alkyl Chain and Their Interactions with Lipid Bilayers, Surfactants, and Living Cells, Organometallics, 2008, 27, 2998 CrossRef CAS.
- K. K.-W. Lo, M.-W. Louie and K. Y. Zhang, Design of luminescent iridium(III) and rhenium(I) polypyridine complexes as in vitro and in vivo ion, molecular and biological probes, Coord. Chem. Rev., 2010, 254, 2603 CrossRef CAS.
- P. Alam, G. Kaur, C. Climent, S. Pasha, D. Casanova, P. Alemany, A. Roy Choudhury and I. R. Laskar, New aggregation induced emission (AIE) active cyclometalated iridium(III) based phosphorescent sensors: high sensitivity for mercury(II) ions, Dalton Trans., 2014, 43, 16431 RSC.
- P. Alam, G. Kaur, V. Kachwal, A. Gupta, A. Roy Choudhury and I. R. Laskar, Highly sensitive explosive sensing by aggregation induced phosphorescence active cyclometalated iridium(III) complexes, J. Mater. Chem. C, 2015, 3, 5450 RSC.
- P. Alam, G. Kaur, A. Sarmah, R. K. Roy, A. R. Choudhury and I. R. Laskar, Highly Selective Detection of H+ and OH− with a Single-Emissive Iridium(III) Complex: A Mild Approach to Conversion of Non-AIEE to AIEE Complex, Organometallics, 2015, 34, 4480 CrossRef CAS.
- J. Chen, B. Xu, X. Ouyang, B. Z. Tang and Y. Cao, Aggregation-Induced Emission of cis, cis-1,2,3,4-Tetraphenylbutadiene from Restricted Intramolecular Rotation, J. Phys. Chem. A, 2004, 108, 7522 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Aggregation-induced emission: phenomenon, mechanism and applications, Chem. Commun., 2009, 4332 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Aggregation-Induced Emission: The Whole Is More Brilliant than the Parts, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar!, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- V. Sathish, A. Ramdass, P. Thanasekaran, K.-L. Lu and S. Rajagopal, Aggregation-induced phosphorescence enhancement (AIPE) based on transition metal complexes—An overview, J. Photochem. Photobiol., C, 2015, 23, 25 CrossRef CAS.
- P. Alam, S. Dash, C. Climent, G. Kaur, A. R. Choudhury, D. Casanova, P. Alemany, R. Chowdhury and I. R. Laskar, ‘Aggregation induced emission’ active iridium(III) complexes with applications in mitochondrial staining, RSC Adv., 2017, 7, 5642 RSC.
- P. Alam, C. Climent, G. Kaur, D. Casanova, A. Roy Choudhury, A. Gupta, P. Alemany and I. R. Laskar, Exploring the Origin of Aggregation Induced Emission Activity and Crystallization Induced Emission in Organometallic Iridium(III) Cationic Complexes: Influence of Counterions, Cryst. Growth Des., 2016, 16, 5738 CAS.
- P. Alam, I. R. Laskar, C. Climent, D. Casanova, P. Alemany, M. Karanam, A. R. Choudhury and J. R. Butcher, Microwave-assisted facile and expeditive syntheses of phosphorescent cyclometallated iridium(III) complexes, Polyhedron, 2013, 53, 286 CrossRef CAS.
- Y. Jiang, G. Li, W. Che, Y. Liu, B. Xu, G. Shan, D. Zhu, Z. Su and M. R. Bryce, A neutral dinuclear Ir(III) complex for anti-counterfeiting and data encryption, Chem. Commun., 2017, 53, 3022 RSC.
- G. Li, W. Guan, S. Du, D. Zhu, G. Shan, X. Zhu, L. Yan, Z. Su, M. R. Bryce and A. P. Monkman, Anion-specific aggregation induced phosphorescence emission (AIPE) in an ionic iridium complex in aqueous media, Chem. Commun., 2015, 51, 16924 RSC.
- G. Li, X. Ren, G. Shan, W. Che, D. Zhu, L. Yan, Z. Su and M. R. Bryce, New AIE-active dinuclear Ir(III) complexes with reversible piezochromic phosphorescence behaviour, Chem. Commun., 2015, 51, 13036 RSC.
- G. Li, Y. Wu, G. Shan, W. Che, D. Zhu, B. Song, L. Yan, Z. Su and M. R. Bryce, New ionic dinuclear Ir(III) Schiff base complexes with aggregation-induced phosphorescent emission (AIPE), Chem. Commun., 2014, 50, 6977 RSC.
- G.-G. Shan, H.-B. Li, H.-Z. Sun, D.-X. Zhu, H.-T. Cao and Z.-M. Su, Controllable synthesis of iridium(III)-based aggregation-induced emission and/or piezochromic luminescence phosphors by simply adjusting the substitution on ancillary ligands, J. Mater. Chem. C, 2013, 1, 1440 RSC.
- Y. Wu, H.-Z. Sun, H.-T. Cao, H.-B. Li, G.-G. Shan, Y.-A. Duan, Y. Geng, Z.-M. Su and Y. Liao, Stepwise modulation of the electron-donating strength of ancillary ligands: understanding the AIE mechanism of cationic iridium(III) complexes, Chem. Commun., 2014, 50, 10986 RSC.
- K.-Y. Zhao, G.-G. Shan, Q. Fu and Z.-M. Su, Tuning Emission of AIE-Active Organometallic Ir(III) Complexes by Simple Modulation of Strength of Donor/Acceptor on Ancillary Ligands, Organometallics, 2016, 35, 3996 CrossRef CAS.
- A. J. Howarth, R. Patia, D. L. Davies, F. Lelj, M. O. Wolf and K. Singh, Elucidating the Origin of Enhanced Phosphorescence Emission in the Solid State (EPESS) in Cyclometallated Iridium Complexes, Eur. J. Inorg. Chem., 2014, 3657 CrossRef CAS.
- K. Huang, H. Wu, M. Shi, F. Li, T. Yi and C. Huang, Reply to comment on aggregation-induced phosphorescent emission (AIPE) of iridium(III) complexes: origin of the enhanced phosphorescence, Chem. Commun., 2009, 1243 RSC.
- Y. You, H. S. Huh, K. S. Kim, S. W. Lee, D. Kim and S. Y. Park, Comment on aggregation-induced phosphorescent emission (AIPE) of iridium(III) complexes: origin of the enhanced phosphorescence, Chem. Commun., 2008, 3998 RSC.
- Q. Zhao, L. Li, F. Li, M. Yu, Z. Liu, T. Yi and C. Huang, Aggregation-induced phosphorescent emission (AIPE) of iridium(III) complexes, Chem. Commun., 2008, 685 RSC.
- B. M. J. S. Paulose, D. K. Rayabarapu, J. P. Duan and C. H. Cheng, First Examples of Alkenyl Pyridines as Organic Ligands for Phosphorescent Iridium Complexes, Adv. Mater., 2004, 16, 2003 CrossRef CAS.
- D. K. Rayabarapu, B. M. J. S. Paulose, J. P. Duan and C. H. Cheng, New Iridium Complexes with Cyclometalated Alkenylquinoline Ligands as Highly Efficient Saturated Red-Light Emitters for Organic Light-Emitting Diodes, Adv. Mater., 2005, 17, 349 CrossRef CAS.
- C. S. Chin, M.-S. Eum, S. y. Kim, C. Kim and S. K. Kang, New Type of Photoluminescent Iridium Complex: Novel Synthetic Route for Cationic trans-Bis(2-phenylpyridinato)iridium(III) Complex, Eur. J. Inorg. Chem., 2006, 4979 CrossRef CAS.
- M.-S. Eum, C. S. Chin, S. y. Kim, C. Kim, S. K. Kang, N. H. Hur, J. H. Seo, G. Y. Kim and Y. K. Kim, Ligand Effects on Luminescence of New Type Blue Light-Emitting Mono(2-phenylpyridinato)iridium(III) Complexes, Inorg. Chem., 2008, 47, 6289 CrossRef CAS PubMed.
- P. Alam, P. Das, C. Climent, M. Karanam, D. Casanova, A. R. Choudhury, P. Alemany, N. R. Jana and I. R. Laskar, Facile tuning of the aggregation-induced emission wavelength in a common framework of a cyclometalated iridium(III) complex: micellar encapsulated probe in cellular imaging, J. Mater. Chem. C, 2014, 2, 5615 RSC.
- X. Yang, X. Xu, J. Zhao, J.-s. Dang, Z. Huang, X. Yan, G. Zhou and D. Wang, Phosphorescent Platinum(II) Complexes Bearing 2-Vinylpyridine-type Ligands: Synthesis, Electrochemical and Photophysical Properties, and Tuning of Electrophosphorescent Behavior by Main-Group Moieties, Inorg. Chem., 2014, 53, 12986 CrossRef CAS PubMed.
- K. K.-W. Lo, Luminescent Rhenium(I) and Iridium(III) Polypyridine Complexes as Biological Probes, Imaging Reagents, and Photocytotoxic Agents, Acc. Chem. Res., 2015, 48, 2985 CrossRef CAS PubMed.
- A. J. Howarth, D. L. Davies, F. Lelj, M. O. Wolf and B. O. Patrick, Tuning the Emission Lifetime in Bis-cyclometalated Iridium(III) Complexes Bearing Iminopyrene Ligands, Inorg. Chem., 2014, 53, 11882 CrossRef CAS PubMed.
- S. Kumar, Y. Hisamatsu, Y. Tamaki, O. Ishitani and S. Aoki, Design and Synthesis of Heteroleptic Cyclometalated Iridium(III) Complexes Containing Quinoline-Type Ligands that Exhibit Dual Phosphorescence, Inorg. Chem., 2016, 55, 3829 CrossRef CAS PubMed.
- S. Ladouceur, L. Donato, M. Romain, B. P. Mudraboyina, M. B. Johansen, J. A. Wisner and E. Zysman-Colman, A rare case of dual emission in a neutral heteroleptic iridium(III) complex, Dalton Trans., 2013, 42, 8838 RSC.
- K. K.-W. Lo, K. Y. Zhang, S.-K. Leung and M.-C. Tang, Exploitation of the Dual-emissive Properties of Cyclometalated Iridium(III)–Polypyridine Complexes in the Development of Luminescent Biological Probes, Angew. Chem., Int. Ed., 2008, 47, 2213 CrossRef CAS PubMed.
- Y.-S. Yeh, Y.-M. Cheng, P.-T. Chou, G.-H. Lee, C.-H. Yang, Y. Chi, C.-F. Shu and C.-H. Wang, A New Family of Homoleptic Ir(III) Complexes: Tris-Pyridyl Azolate Derivatives with Dual Phosphorescence, ChemPhysChem, 2006, 7, 2294 CrossRef CAS PubMed.
- Y. You, S. Cho and W. Nam, Cyclometalated Iridium(III) Complexes for Phosphorescence Sensing of Biological Metal Ions, Inorg. Chem., 2014, 53, 1804 CrossRef CAS PubMed.
- Y. You, Y. Han, Y.-M. Lee, S. Y. Park, W. Nam and S. J. Lippard, Phosphorescent Sensor for Robust Quantification of Copper(II) Ion, J. Am. Chem. Soc., 2011, 133, 11488 CrossRef CAS PubMed.
- K. Y. Zhang, H.-W. Liu, M.-C. Tang, A. W.-T. Choi, N. Zhu, X.-G. Wei, K.-C. Lau and K. K.-W. Lo, Dual-Emissive Cyclometalated Iridium(III) Polypyridine Complexes as Ratiometric Biological Probes and Organelle-Selective Bioimaging Reagents, Inorg. Chem., 2015, 54, 6582 CrossRef CAS PubMed.
- T. Guo, W. Zhong, J. Zou, L. Ying, W. Yang and J. Peng, Efficient binary white light-emitting polymers grafted with iridium complexes as side groups, RSC Adv., 2015, 5, 89888 RSC.
- H. Shi, H. Sun, H. Yang, S. Liu, G. Jenkins, W. Feng, F. Li, Q. Zhao, B. Liu and W. Huang, Cationic Polyfluorenes with Phosphorescent Iridium(III) Complexes for Time-Resolved Luminescent Biosensing and Fluorescence Lifetime Imaging, Adv. Funct. Mater., 2013, 23, 3268 CrossRef CAS.
- Z. Song, R. Liu, Y. Li, H. Shi, J. Hu, X. Cai and H. Zhu, AIE-active Ir(III) complexes with tunable emissions, mechanoluminescence and their application for data security protection, J. Mater. Chem. C, 2016, 4, 2553 RSC.
- T. Yoshihara, S. Murayama and S. Tobita, Ratiometric Molecular Probes Based on Dual Emission of a Blue Fluorescent Coumarin and a Red Phosphorescent Cationic Iridium(III) Complex for Intracellular Oxygen Sensing, Sensors, 2015, 15, 13503 CrossRef CAS PubMed.
- Z. Guo, N. R. Song, J. H. Moon, M. Kim, E. J. Jun, J. Choi, J. Y. Lee, C. W. Bielawski, J. L. Sessler and J. Yoon, A Benzobisimidazolium-Based Fluorescent and Colorimetric Chemosensor for CO2, J. Am. Chem. Soc., 2012, 134, 17846 CrossRef CAS PubMed.
- M. Ishida, P. Kim, J. Choi, J. Yoon, D. Kim and J. L. Sessler, Benzimidazole-embedded N-fused aza-indacenes: synthesis and deprotonation-assisted optical detection of carbon dioxide, Chem. Commun., 2013, 49, 6950 RSC.
- Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, Fluorescent Chemosensor for Detection and Quantitation of Carbon Dioxide Gas, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed.
- W. Lu, P. Xiao, J. Gu, J. Zhang, Y. Huang, Q. Huang and T. Chen, Aggregation-induced emission of tetraphenylethylene-modified polyethyleneimine for highly selective CO2 detection, Sens. Actuators, B, 2016, 228, 551 CrossRef CAS.
- Y. Ma, H. Xu, Y. Zeng, C.-L. Ho, C.-H. Chui, Q. Zhao, W. Huang and W.-Y. Wong, A charged iridophosphor for time-resolved luminescent CO2 gas identification, J. Mater. Chem. C, 2015, 3, 66 RSC.
- T. Tian, X. Chen, H. Li, Y. Wang, L. Guo and L. Jiang, Amidine-based fluorescent chemosensor with high applicability for detection of CO2: A facile way to see CO2, Analyst, 2013, 138, 991 RSC.
- Q. Xu, S. Lee, Y. Cho, M. H. Kim, J. Bouffard and J. Yoon, Polydiacetylene-Based Colorimetric and Fluorescent Chemosensor for the Detection of Carbon Dioxide, J. Am. Chem. Soc., 2013, 135, 17751 CrossRef CAS PubMed.
- S. Neethirajan, D. S. Jayas and S. Sadistap, Carbon Dioxide (CO2) Sensors for the Agri-food Industry—A Review, Food Bioprocess Technol., 2009, 2, 115 CrossRef CAS.
- A. Star, T. R. Han, V. Joshi, J. C. P. Gabriel and G. Grüner, Nanoelectronic Carbon Dioxide Sensors, Adv. Mater., 2004, 16, 2049 CrossRef CAS.
- CrystalClear 2.0, Rigaku Corporation, Tokyo, Japan, 2016.
- G. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- M. Nardelli, PARST95 – an update to PARST: a system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- A. D. Becke, Density functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- W. R. Wadt and P. J. Hay, Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- Vibrationally-resolved electronic spectra in Gaussian 09, http://dreamslab.sns.it/pdf/vibronic_spectra_G09-A02.pdf.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Failure of Time-Dependent Density Functional Theory for Long-Range Charge-Transfer Excited States: The Zincbacteriochlorin–Bacteriochlorin and Bacteriochlorophyll–Spheroidene Complexes, J. Am. Chem. Soc., 2004, 126, 4007 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- J. Schanda, Colourimetry: Understanding the CIE System, Wiley, Hoboken, New Jersey, 2007 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E 01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- P. Alam, G. Kaur, S. Chakraborty, A. Roy Choudhury and I. R. Laskar, Aggregation induced phosphorescence active rollover iridium(III) complex as a multi-stimuli-responsive luminescence material, Dalton Trans., 2015, 44, 6581 RSC.
- E. Baranoff, S. Fantacci, F. De Angelis, X. Zhang, R. Scopelliti, M. Grätzel and M. K. Nazeeruddin, Cyclometalated Iridium(III) Complexes Based on Phenyl-Imidazole Ligand, Inorg. Chem., 2011, 50, 451 CrossRef CAS PubMed.
- A. F. Rausch, H. H. H. Homeier and H. Yersin, in Photophysics of Organometallics, ed. A. J. Lees, Springer, Berlin, Heidelberg, 2010, p. 193 Search PubMed.
- Y. You and S. Y. Park, Inter-Ligand Energy Transfer and Related Emission Change in the Cyclometalated Heteroleptic Iridium Complex: Facile and Efficient Color Tuning over the Whole Visible Range by the Ancillary Ligand Structure, J. Am. Chem. Soc., 2005, 127, 12438 CrossRef CAS PubMed.
- J. Liang, Z. Chen, L. Xu, J. Wang, J. Yin, G.-A. Yu, Z.-N. Chen and S. H. Liu, Aggregation-induced emission-active gold(I) complexes with multi-stimuli luminescence switching, J. Mater. Chem. C, 2014, 2, 2243 RSC.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Efficient Solid Emitters with Aggregation-Induced Emission and Intramolecular Charge Transfer Characteristics: Molecular Design, Synthesis, Photophysical Behaviors, and OLED Application, Chem. Mater., 2012, 24, 1518 CrossRef CAS.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Restriction of Intramolecular Motions: The General Mechanism behind Aggregation-Induced Emission, Chem. – Eur. J., 2014, 20, 15349 CrossRef CAS PubMed.
- E. Quartapelle Procopio, M. Mauro, M. Panigati, D. Donghi, P. Mercandelli, A. Sironi, G. D'Alfonso and L. De Cola, Highly Emitting Concomitant Polymorphic Crystals of a Dinuclear Rhenium Complex, J. Am. Chem. Soc., 2010, 132, 14397 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc02250f |
| ‡ Both authors contributed equally to this work, C.C with the computational part and P.A with the experimental one. |
| This journal is © The Royal Society of Chemistry 2017 |