
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards enhancing spin states in doped arylamine compounds through extended planarity of the spin coupling moieties†
Łukasz
Skórka
a,
Jean-Marie
Mouesca
bc,
Jacek B.
Gosk
d,
Roman
Puźniak
 e,
Jacques
Pécaut
bc,
Vincent
Maurel
*bc and
Irena
Kulszewicz-Bajer
*a
e,
Jacques
Pécaut
bc,
Vincent
Maurel
*bc and
Irena
Kulszewicz-Bajer
*a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: ikulsz@ch.pw.edu.pl
bINAC, SyMMES Université Grenoble Alpes, F-38000 Grenoble, France
cINAC, SyMMES, CEA Grenoble, F-38054 Grenoble, France
dFaculty of Physics, Warsaw University of Technology, Koszykowa 75, 00-662 Warsaw, Poland
eInstitute of Physics, Polish Academy of Science, Al. Lotników 32/46, 02-668 Warsaw, Poland
First published on 19th June 2017
Abstract
Arylamine moieties oxidized to radical cations are promising spin bearers for organic high-spin compounds. However, successive oxidations of next-neighbour sites as well as the resulting ferromagnetic exchange interactions depend strongly on the charge and spin distributions over the molecule. We report here that the use of rigid segments composed of m-phenylene spin couplers linked to arylamine spin bearers in a co-planar way facilitates successive oxidations, enhances spin exchange interactions and doubles the observed spin state (S = 2) in a polymer (PQA) when compared to the chemically equivalent (but locally free rotating) PA2 polymer (S = 1). Such a quintet spin state is observed for the first time for a linear polyarylamine with an m-phenylene coupler. DFT calculations of the dimer QA reproduce the experimental J value and indicate that oxidation of these co-planar compounds leads indeed to well localized radical cations, a fact of crucial importance for the preparation of arylamine-type high-spin materials.
Introduction
Organic high-spin dimers, oligomers and polymers have been the object of intense research efforts for the last twenty years,1–5 with the aim of obtaining materials useful for spintronics,6 MRI contrast agents7 or purely organic magnetic materials.8 The most popular and versatile strategy to obtain high-spin organic materials is to connect π-conjugated free radicals acting as “spin bearers” via π-conjugated “spin couplers”, the most documented example of which is the m-phenylene group.1,2 Such spin couplers mediate ferromagnetic spin coupling following the theory of alternant hydrocarbons.9 High-spin oligomers and polymers based on the m-phenylene spin coupler and arylamine radical cation spin bearers have been therefore extensively studied, due to the high stability of these spin bearers (up to room temperature).10,11 However, the highest spin state obtained to date is S = 9/2 for highly branched and poorly processable polymers.12 For the best linear polymer13 the highest spin state was found to be S = 1 with the exchange coupling constant, J/kB = 18 K (PA2, see Scheme 1).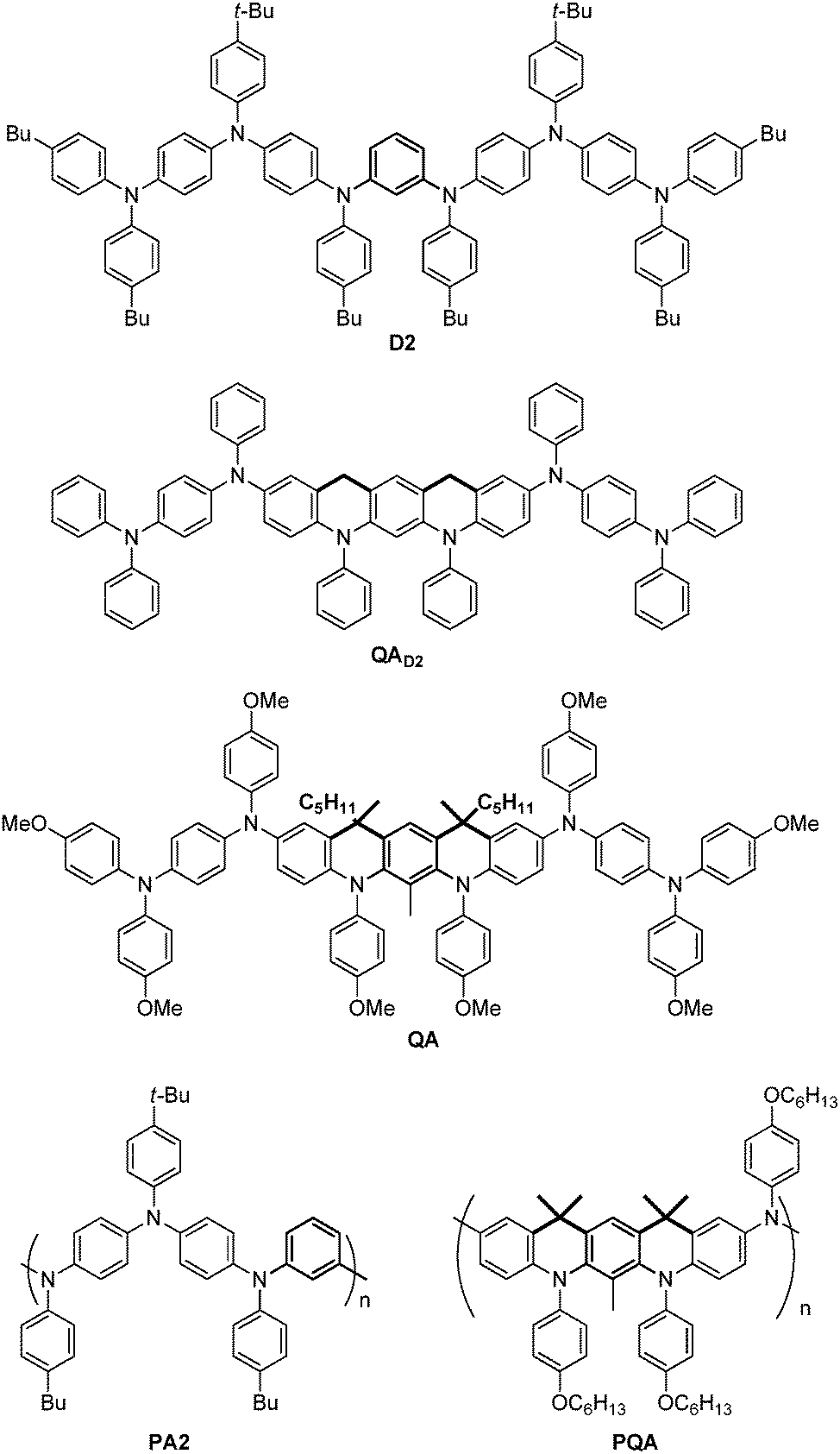 | ||
| Scheme 1 The chemical structures of QA and PQA with a rigid coupler and their flexible analogues D2 and PA2. | ||
An important factor that prevents the achievement of higher S and J values is that in most systems reported to date (like in D2 and PA2, Scheme 1 and ref. 13), the phenyl rings of the spin couplers and spin bearers are not coplanar and therefore do not behave strictly as alternant hydrocarbons. A solution to this problem would be to prevent the rotation of the spin bearers around the Ccoupler–Nbearer bond (i.e. the C–N bonds in the meta positions of the central m-phenyl group in D2 or QA) by the insertion of an additional covalent link. Rajca and co-workers demonstrated the benefit of this last approach synthesizing fully rigid ladders of m-aniline oligomers4,14 which were converted into neutral oligoaminyl radicals with high estimated J values and high spin up to S = 2 in the tetramer.14
Herein we report the synthesis and the characterization of partially rigid compounds QA and PQA, formally derived from the more “flexible” D2 and PA2 analogues (Scheme 1). This study aimed at examining the effect of such a local co-planarity between the spin coupler ring and the spin bearers on the redox properties, the maximum spin S observed and the exchange coupling constant. Notably, here are the main results: (i) the synthetic strategy of such semi-rigid compounds could be extended to the polymeric system like PQA, (ii) rather unexpectedly, the electrochemical properties of the semi-rigid QA and PQA compounds were more favourable than those of the flexible analogues, (iii) the magnetic properties of QA and PQA oxidized to radical cations (spin S value achieved, the magnetic exchange coupling constant for PQA) were significantly improved compared with those of D2 and PA2, demonstrating the interest of introducing local co-planarity.
Results and discussion
Synthetic strategy
The model compound QA and its polymeric analogue, PQA, were synthesized in similar ways. The synthetic routes incorporated palladium catalysed Buchwald–Hartwig aminations combined with Sonogashira coupling and several classical organic transformations. As presented in Scheme 1, the main structural unit unifying QA and PQA is the 5,7,12,14-tetrahydroquinolino[3,2-b]acridine core, which provides the planarization of the molecule and increases its rigidity. The complete synthetic pathway can be divided into two parts: the synthesis of intermediate bromides 5a and 5b (Scheme 2) and their further transformation into the title QA and PQA (Scheme 3). Thus bromides 1 (for the synthesis see the ESI†) were coupled with 2,6-diaminotoluene (used to avoid the unwanted cyclization direction in the next step) in Buchwald–Hartwig amination, catalysed by Pd2(dba)3/P(t-Bu)3 (2a) or Pd(OAc)2/P(t-Bu)3 (2b). Then, both derivatives were subjected to cyclization, performed in a mixture of acetic acid and phosphoric acid. For the compound 3b however, two possible diastereomers were possible to form: one meso (12R,14S) and two enantiomers: (12S,14S) and (12R,14R). Without subsequent separation of the diastereomers, any further step became significantly difficult due to small differences in chemical shifts and therefore lack of reliable product identification. Diastereomers could be separated by chromatography and then used in pure isomeric form (the respective, superimposed 1H NMR spectra of diastereomers of 3b are presented in Fig. S1, ESI†). The derivatives 3 containing secondary amine sites were transformed into tertiary amines via Buchwald–Hartwig amination giving compounds 4a and 4b, which upon treatment with NBS afforded the bromides 5a and 5b. One of the diastereomers of 5b (the meso form) formed single crystals suitable for X-ray study. Based on the crystallographic data (Fig. S2, ESI†) we were able to assign the absolute configurations and differentiate the NMR spectra from Fig. S1 (ESI†) corresponding to the adequate diastereomeric form.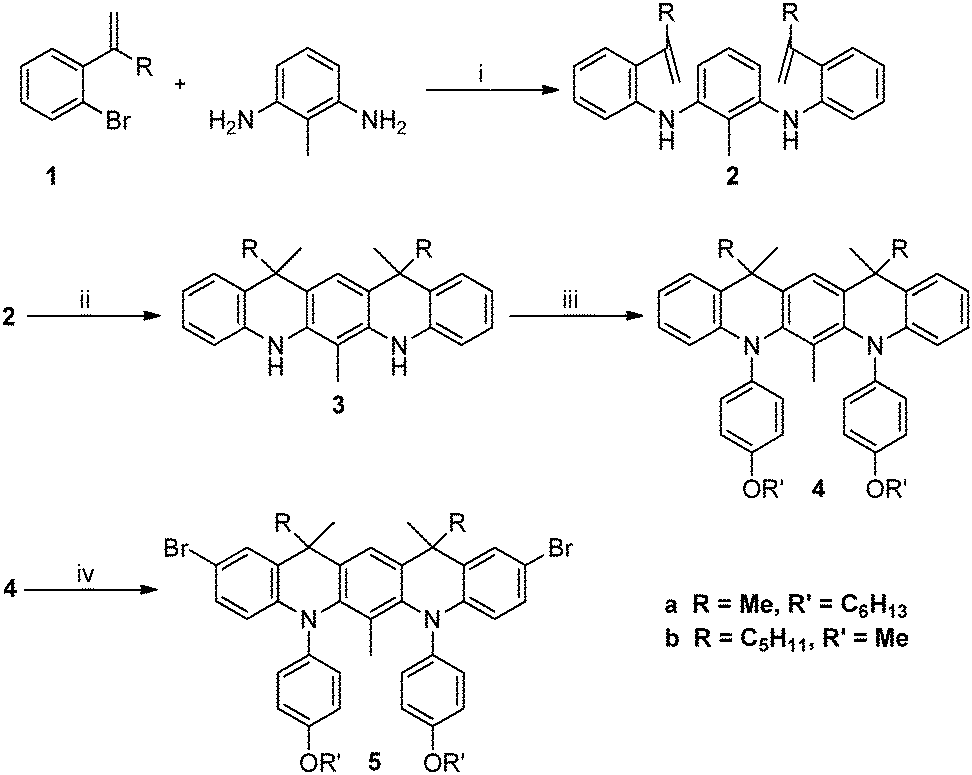 | ||
| Scheme 2 Synthesis of intermediate bromides. Conditions: (i) 5a 3% [Pd2(dba)3], P(t-Bu)3, t-BuONa, Ar, 110 °C, 5b 3% [Pd(OAc)2], P(t-Bu)3, t-BuONa, Ar, 120 °C; (ii) 85% H3PO4, MeCO2H, Ar, 100 °C; (iii) 1-(hexyloxy)-4-iodobenzene (7a) or 4-iodoanisole (7b) 3–5% [Pd(OAc)2], P(t-Bu)3, t-BuONa, Ar, 110 °C; (iv) NBS, DMF, Ar, 0 °C. | ||
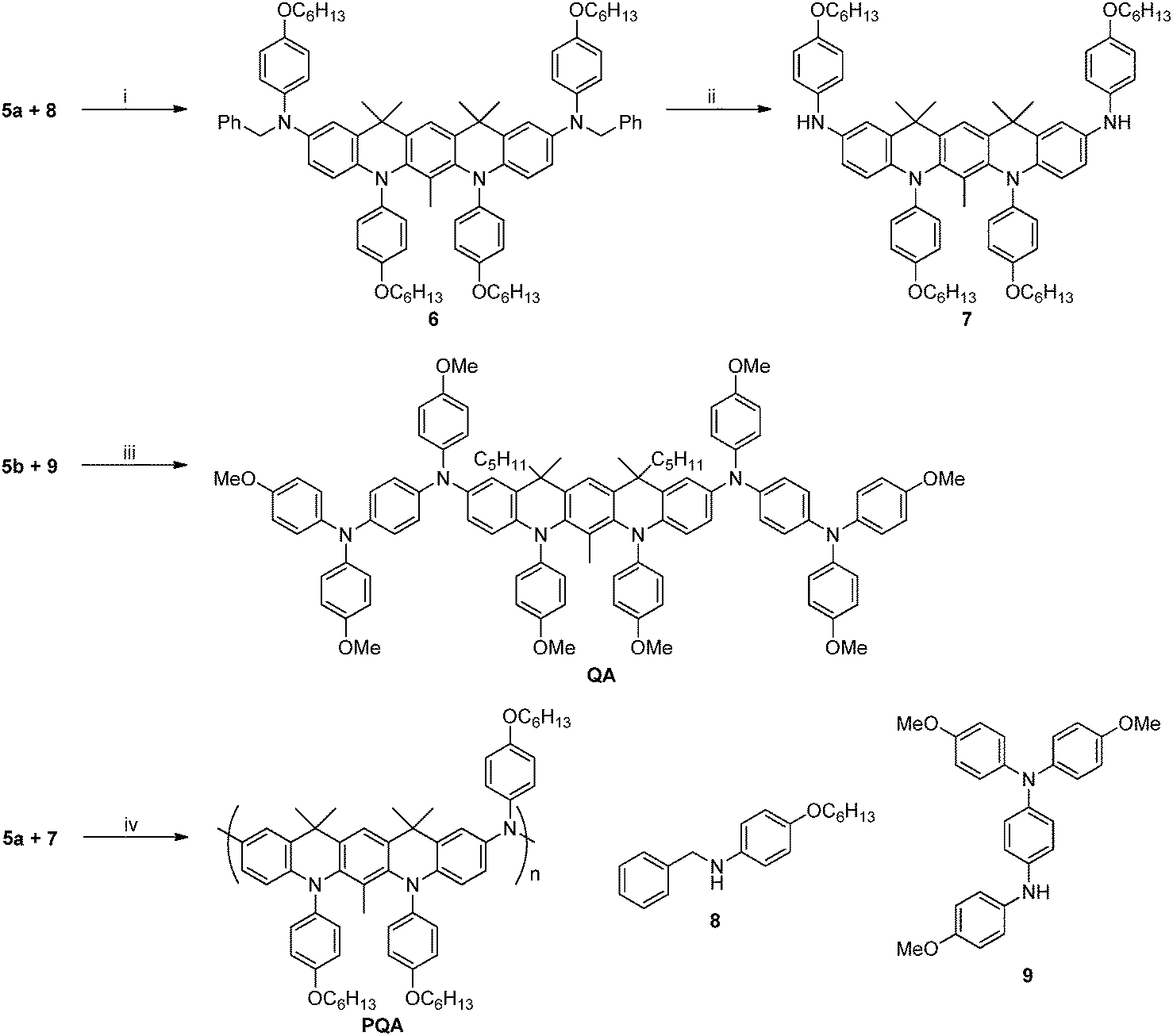 | ||
| Scheme 3 Synthesis of the target dimer (QA) and polymer (PQA). Conditions: (i) 3% [Pd(OAc)2], P(t-Bu)3, t-BuONa, Ar, 110 °C; (ii) HCO2NH4, 10% Pd/C, MeOH, Ar, reflux; (iii) 3% [Pd(OAc)2], P(t-Bu)3, t-BuONa, Ar, 110 °C; (iv) 3% [Pd(OAc)2], P(t-Bu)3, t-BuONa, Ar, 110 °C. | ||
The model compound QA and corresponding PQA were synthesized as depicted in Scheme 3. QA was obtained via coupling of 5b with amine 9 (the synthesis of 8 and 9 is shown in ESI,† Scheme S2) using the Pd(OAc)2/P(t-Bu)3 catalyst. The synthesis of PQA required the functionalization of 5a with amine 8, which upon reduction with ammonium formate resulted in the formation of amine 7. The polycondensation of dibromoderivative 5a with diamine 7 in the presence of Pd(OAc)2/P(t-Bu)3 yielded the polymer PQA. The polymer exhibited a weight-averaged molecular weight of 161 kDa and a number-averaged molecular weight of 497 kDa, Mw/Mn = 3.08.
Electrochemical studies
The electrochemical oxidation of the dimer QA and the polymer PQA were studied by cyclic voltammetry and differential pulse voltammetry and their voltammograms are compared with previously reported voltammograms for D2 and PA2 in Fig. 1 (for the results of chemical oxidation followed by UV-vis-NIR spectroscopy see the ESI,† Section VI).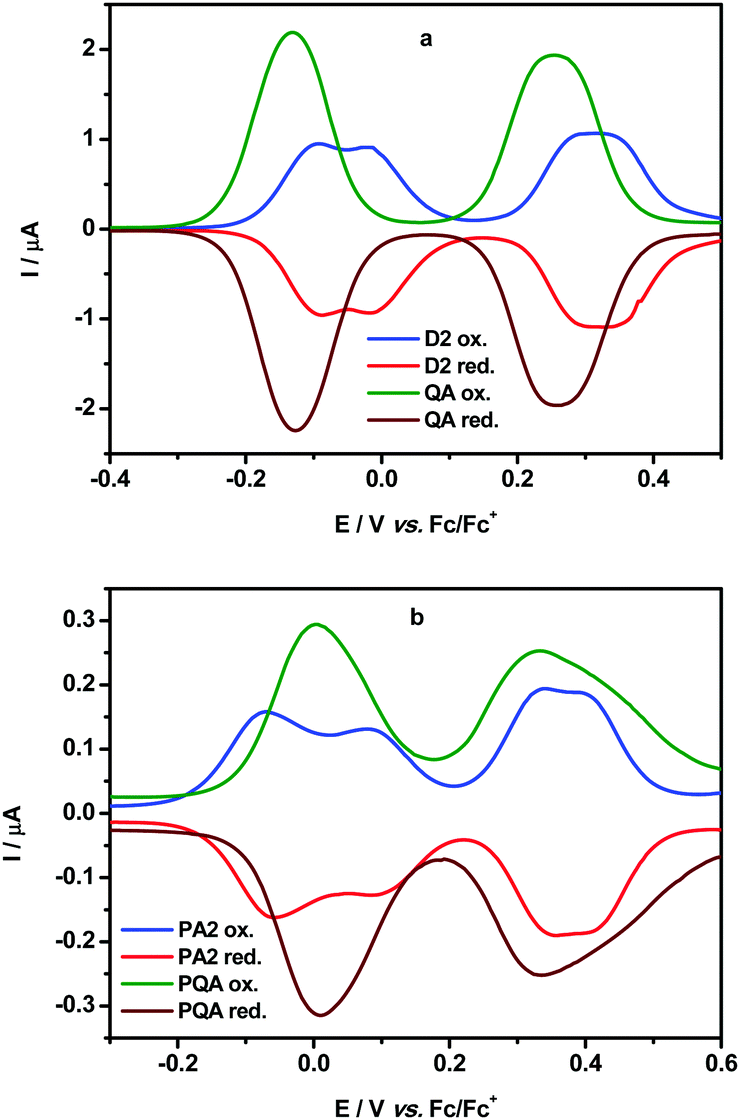 | ||
| Fig. 1 Differential pulse voltammograms of dimers QA and D2 (a) and of polymers PQA and PA2 (b) in CH2Cl2 solution (c = 1 × 10−3 M) containing an electrolyte – 0.1 M Bu4NBF4 (modulation time 50 ms, modulation amplitude 10 mV, step potential 5 mV). | ||
The voltammogram of QA showed two reversible waves at −0.125 V and 0.26 V (vs. Fc/Fc+) independently of diastereisomeric forms (Fig. 1a). Both peaks were rather broad with Eox − Ered = 100 mV and 120 mV, respectively, suggesting that multi-electron oxidation processes took place in both cases. The first peak can be related to the formation of one radical cation within each arylamine conjugated segment separated via the m-phenylene coupler. By contrast, the oxidation of D2 to one radical cation in each arylamine segment proceeded at higher potentials and in two steps (−0.09 V and −0.005 V vs. Fc/Fc+) separated by a 85 mV shift. The second oxidation wave of QA can be attributed to the formation of a second radical cation within each conjugated segment.
Similar patterns were observed in the cyclic voltammogram of the polymer PQA, namely two broad reversible oxidation peaks were observed at 0.005 V and 0.34 V (vs. Fc/Fc+, Eox − Ered = 140 mV and 190 mV, respectively) (Fig. 1b). Similar to the oxidation of QA these peaks correspond to the formation of one and then two radical cations per polymer unit. By contrast, the peaks corresponding to the first and the second one-electron oxidations in PA2 were observed with a shift of 140 mV (E = −0.065 and 0.085 V) and correspond to the formation of radical cations in two adjacent mers (see ref. 13).
Normally, the redox potential increase can impede the creation of radical cations in adjacent units and this was identified as the main cause of unsuccessful attempts to obtain high-spin polyradical cations from oligo(m-aniline) derivatives.15 Remarkably here, in terms of redox potentials, the use of the planar 5,7,12,14-tetrahydroquinolino[3,2-b]acridine core for both QA and PQA almost cancels the cost of oxidizing two arylamines connected by the m-phenylene spin coupler. This feature appears to be crucial for obtaining high-spin polymers with radical cation spin bearers.
This behaviour could be related to (i) the intrinsic favourable localization of the cationic charge after the first oxidation, and/or (ii) energetic compensation upon second oxidation (via local geometry constraint relaxation and/or rearrangement of the counter-anions and/or various interactions with the solvent, see ref. 16 for a review of such effects).
DFT calculations
As a preliminary way of exploring these possibilities, and for the sake of comparison, we first performed DFT calculations in vacuo on the mono-oxidized states of both D2 and a QA model (QAD2) derived from D2 only by adding two connecting CH2 groups (see Scheme 1 above; the same procedure was then applied to a more suitable QA model: see below). Within an isotropic environment (i.e. without a counter-anion), the unpaired electron is artificially fully delocalized by DFT in both D2 and QAD2 models (Fig. S12, ESI†). However, when placing sideways a negative charge mimicking the counter-anion, the topological difference between D2 and QAD2 is manifested (see Fig. 2 and the ESI,† Fig. S13 for details). In QAD2, the alternant (conjugated) hydrocarbon theory is enforced by local planarity of the coupler and adjacent bearers. This results in complete lateralization of the cationic hole on one side of the dimer. By contrast, placing the same negative charge sideways, lateralization is only partly realized in D2 because of non-planarity around the coupler leading to charge leakage between both sides (see Fig. 2 and Fig. S13, ESI†). The better localization of the charge in QAD2 compared with D2 can explain, at least partially, the more favourable redox properties of QA reported above. To further validate these calculations, the meta-phenylene coupler in QAD2 was changed into a para-phenylene coupler. Consistent with the alternant hydrocarbon theory, no localization of the charge was observed in spite of the introduction of the same lateral negative charge (Fig. S14, ESI†). Finally, the same procedure was applied to a quasi-QA dimer, with its OMe moieties and >CMe2 bridges (QACMe), encompassing all the main features of QA. In the presence of the same lateral charge, its HOMO becomes strongly lateralized, as already observed in the case of QAD2 (Fig. 2, bottom left). This is more remarkable as QACMe (and therefore QA) is actually slightly bent: the relative angle defined by the planes of the central (coupler) phenyl ring and of the adjacent (bearer) phenyl ring is 15°; the intersection of these planes occurs precisely at the level of the >CMe bridges. Such a difference between ideal planarity (D2 and QAD2) and the actual dimer (QACMe) has here negligible (as observed experimentally) consequences for the redox properties.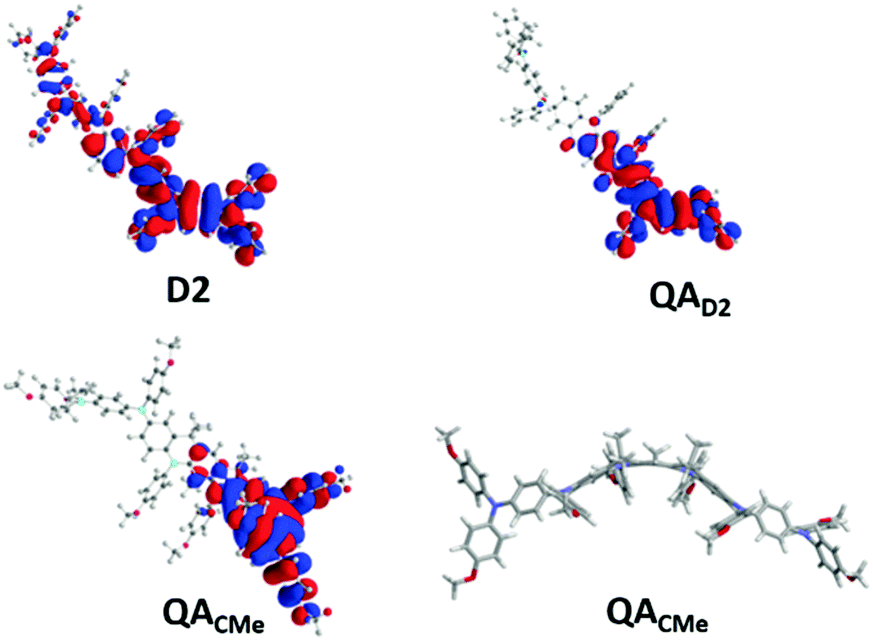 | ||
| Fig. 2 HOMOs for the mono-oxidized redox state of D2 (top left), QAD2 (top right) and QACMe (bottom left) in the presence of a lateral negative charge (see main text and the ESI† for details). Isodensity value: 0.01 a.u. Bottom right: Lateral view of QAOMe showing bending of the molecular structure around the central coupler. | ||
EPR nutation and SQUID magnetometry studies
The magnetic interaction between the created spins was studied with pulsed EPR nutation spectroscopy (Fig. 3 and the ESI,† Section VII) and SQUID magnetometry (Fig. 4, 5 and ESI,† Section VIII). When QA was chemically oxidized to one radical cation per arylamine unit (using tris(4-bromophenyl) amminium hexachloroantimonate, TBA, see ESI,† Section IV), the EPR nutation spectrum of the frozen solution exhibited a very dominant signal at the S = 1 nutation frequency with a very small residual signal at the S = 1/2 frequency (Fig. 3). The magnetization data obtained by SQUID magnetometry (at T = 2 K) for samples oxidized to the same level, but after removal of the solvent, can be very well fitted with the effective Brillouin function for S = 1 including mean-field correction (θ = −0.92 K) accounting for intermolecular antiferromagnetic interactions (the ESI,† Section VIII, Fig. S8). From this fit, it was also shown that 78% of QA dimers were in the triplet state expected for this oxidation stoichiometry. The product of magnetic susceptibility and temperature versus the temperature curve of the QA sample was dominated by intermolecular antiferromagnetic interaction (ESI,† Fig. S9), which impeded the measurement of the J exchange coupling constant within the diradical dication. To decrease such antiferromagnetic interactions, the QA dimer was diluted in a polystyrene matrix. The magnetization data versus magnetic field at T = 2 K were similar to the former one and confirmed the presence of S = 1 (Fig. 4a). The shape of χT vs. T (Fig. 4b) is now mainly due to the ferromagnetic interaction between both radical cations; the intramolecular coupling constant J/kB was estimated to be ca. 50 K (versus the value of 35 K for D2).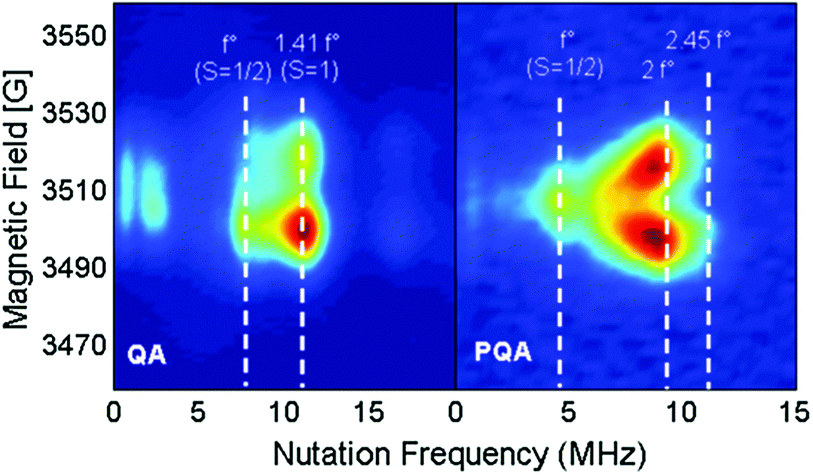 | ||
| Fig. 3 EPR nutation spectra of QA and PQA (T = 10 K) oxidized with 1 eq. per arylamine unit. The microwave power was set so f°(S = 1/2) = 7.7 and 4.4 MHz for QA and PQA (respectively). | ||
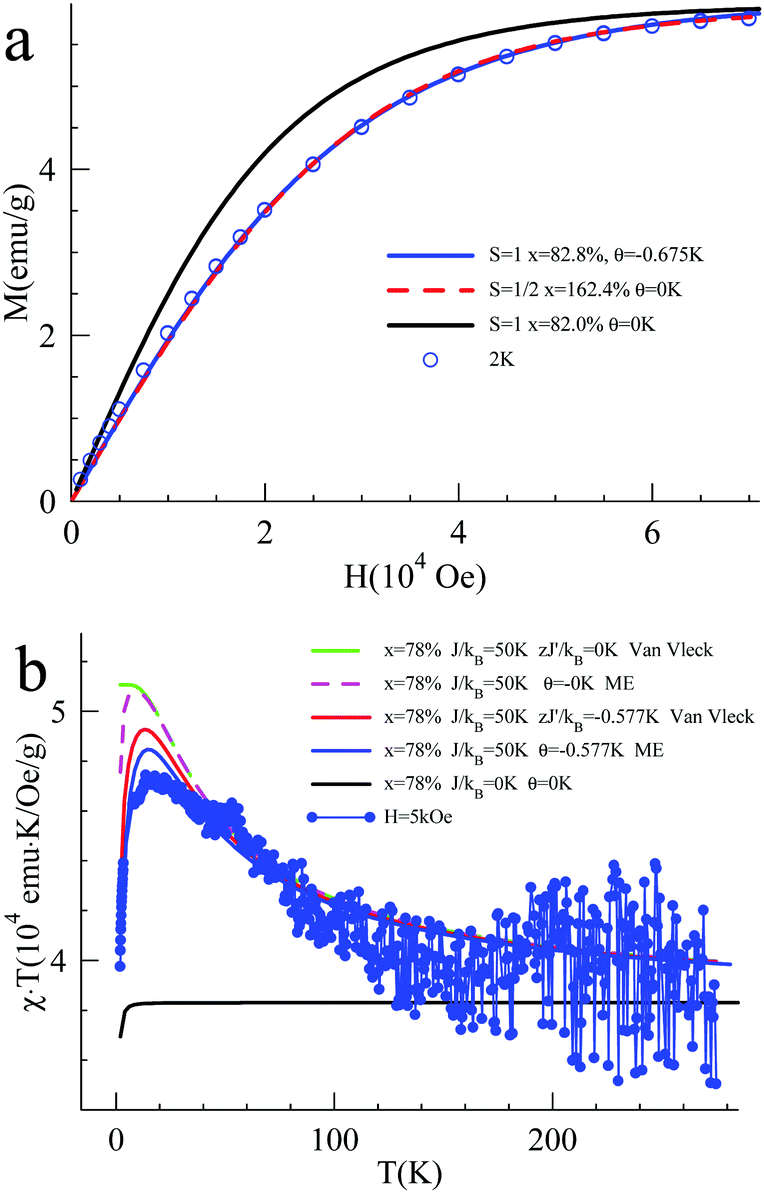 | ||
| Fig. 4 (a) Magnetization of QA diluted in a polystyrene matrix (0.58%mol) versus magnetic field at T = 2 K. (b) Product of magnetic susceptibility and temperature (χT) vs. temperature at H = 5000 Oe obtained for the QA sample diluted in a polystyrene matrix (see the ESI,† Section VIII for expression used for the Van Vleck eqn (S3) and the magnetization eqn (S4), derived from the Heisenberg exchange Hamiltonian). | ||
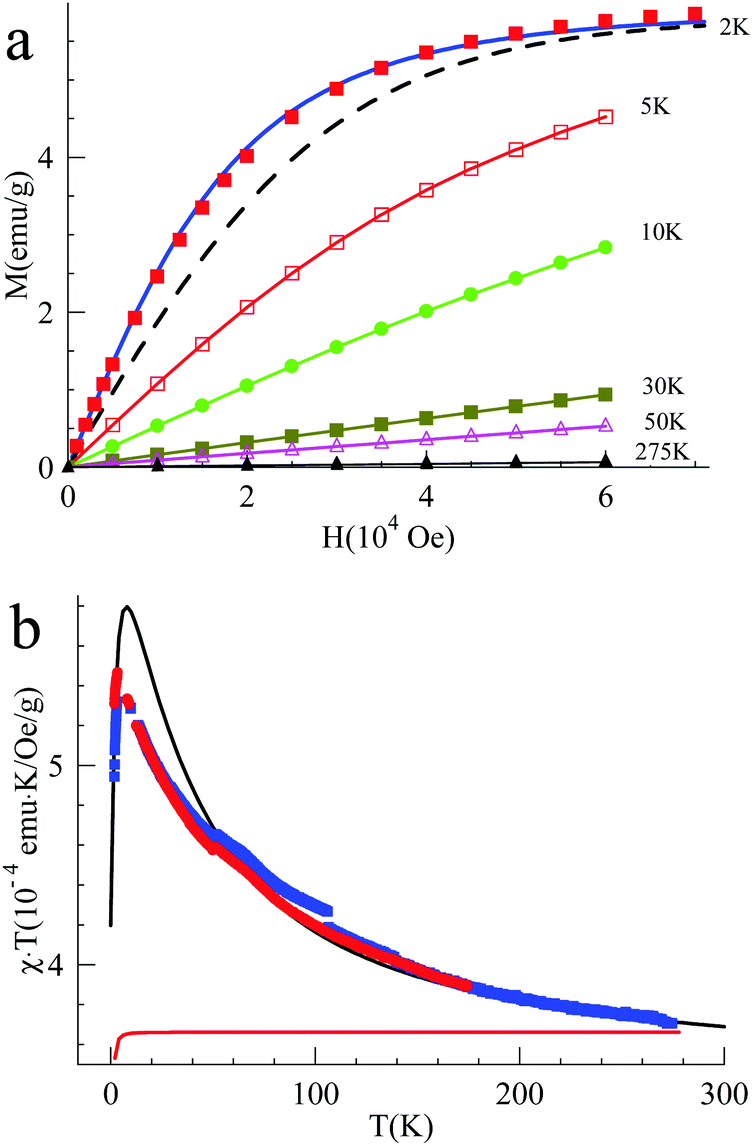 | ||
| Fig. 5 (a) Magnetization of PQAvs. magnetic field (solid blue line – for S = 2, x = 23.5%, θ = −0.85 K; dashed black line – for S = 1/2, θ = 0 K). (b) The product of magnetic susceptibility and temperature vs. temperature at H = 0.5 × 104 Oe (red points) and at 104 Oe (blue points) for PQA. The black line corresponds to the numerical calculations of the eqn (S5) for x = 20%, θ = −1 K, J/kB = 50 K (the four FM spin interaction/quintet state) while the red line represents non-interacting spins described by the standard Brillouin function with S = 1/2, x = 87.5%. | ||
When PQA was oxidized chemically to one radical cation per mer, the EPR nutation spectrum of the frozen solution exhibited a dominant signal at a nutation frequency close to 2·f° (f° being the S = 1/2 nutation frequency). Moreover, a smaller but clear signal can be observed up to √6·f° (see Fig. 3). Such a spectrum is typical of S = 2 spin states11,17,18 (see the ESI† for details) and demonstrates that S = 2 states were dominant at this oxidation stoichiometry. This spin multiplicity was thus twice that detected for the equivalent but flexible polymer PA2. Lower and higher oxidation stoichiometries lead to lower S values (see the ESI†). The magnetization measured as a function of magnetic field by SQUID magnetometry (at T = 2 K) for PQA samples oxidized in the best stoichiometry was very well fitted with the effective Brillouin function for S = 2 including mean-field correction (θ = −0.85 K) which reflects interchain antiferromagnetic interactions (Fig. 5a). From this fit it appears that ca. 90% mers of the PQA polymer contribute to the S = 2 state observed both by SQUID and by pulsed EPR nutation experiments. The χT vs. T plot rises up for decreasing temperature from 275 to 6 K and then decreases for temperatures approaching 2 K (Fig. 5b). The experimental curve reveals ferromagnetic interactions between spins along the polymer chain and antiferromagnetic ones between chains, which dominate at low temperatures. It should be emphasized that the polymer chains cannot be so closely packed as in the case of QA molecules, due to different local conformations, thus the χT curve allows for the detection of the ferromagnetic interaction even for solid state samples. The experimental data were approximated using the numerical solution of the Hamiltonian (S5) (see the ESI,† Section VIII) which leads to the exchange coupling constant J/kB = 50 K, to be compared to a value of 18 K for the equivalent PA2 polymer. The magnetic studies unequivocally confirm the formation of a quintet ground state in PQA, thus four spins in adjacent mers of the polymer chain can be effectively coupled in a ferromagnetic fashion.
DFT calculations of exchange coupling constants
To compare the magnetic properties determined experimentally to those theoretically predicted by DFT, we considered the exchange coupling constant J/kB for the various dimers (D2, QAD2 and QACMe) already introduced above (see Scheme 1 and Fig. 2). For QAD2, we obtained a J/kB value of 116 K, about three times larger than 35 K (both measured and computed) for D2 (see Table 1). The incorporation of the methoxy OMe groups on the phenyl rings (compared to the simple QAD2) as well as the central (donor) methyl group to the m-phenylene spin coupler decreases however the value of J/kB down to 42 K for QACMe, close to the experimental value (50 K), also determined experimentally for PQA. Though somewhat disappointing, this last value is still three times higher than that measured for the analogous D2 polymer (i.e.PA2): 18 K.13 At this stage, the main reason that can be invoked to explain the J/kB values' difference between QAD2 and QA(CMe) has to do with the fact that the former one is strictly planar whereas the latter one is slightly bent (cf. the discussion about the redox properties). Following these observations, a more detailed DFT study of both substituent electronic effects and structural features on the magnitude of J coupling constants is currently underway and will be the subject of a specific theoretical publication.| Molecules | D2 | PA2 | QAD2 | QACMe | PQA |
|---|---|---|---|---|---|
| J/kB (DFT) (K) | 35 | — | 116 | 42 | — |
| J/kB (exp.) (K) | 35 | 18 | — | ∼50 | ∼50 |
Finally, it should be now emphasized that the doping efficiencies of QA and PQA (ca. 80–85% and ca. 90%, respectively) were distinctly higher than those of D2 and PA2 (65% and 66%, respectively). The high doping efficiencies of rigid compounds can be related to the formation of localized radical cations as evidenced by electrochemical and DFT studies. As a result, it increases the radical cation concentration.
Both effects (doping and redox) are important to minimize the number of spin defects which have a negative impact on spin coupling, especially in polymer chains. However, the most important question for us was to know how the insertion of additional bonds (i.e. via bridges) to prevent the relative rotation between spin bearers and a spin coupler would affect the spin multiplicity of PQA compared with the very limited spin multiplicity (S = 1) already reported for PA2. The EPR and magnetization studies showed that, for PQA, an almost pure quintet state (S = 2) was obtained with high doping efficiency. The presence of rigid, co-planar segments (m-phenylene coupler linked to adjacent spin bearers) most probably increases the overall stiffness of the chain, thus resulting in extended coil conformations. As it turns out, the ferromagnetic spin interaction can now be spread along the polymer chain up to four neighbouring units, related to the fact that a twist of the geometry can now only take place at the central nitrogen atom of the spin bearers (a PQA mer possesses 2 – confined in the bearer – free bonds out of 6 bonds, whereas all 6 successive bonds are potentially free in a PA2 mer). This constitutes progress as, to date, the presence of quintet spin states in linear polyarylamines was only detected for linear polymers with 3,4′-biphenyl couplers.17 Remarkably, the coupling of four spins in PQA involves 8 free bonds (two per mer), whereas the coupling of four spins in the linear polymers PB2 and PB3 (see the ESI,† p. 53) with 3,4′-biphenyl couplers involves a very similar number of 9 free bonds (three per spin coupling unit). Similar trends were observed by Oka et al. for the polynitroxides 1, 2 and 3 (Scheme 4).19
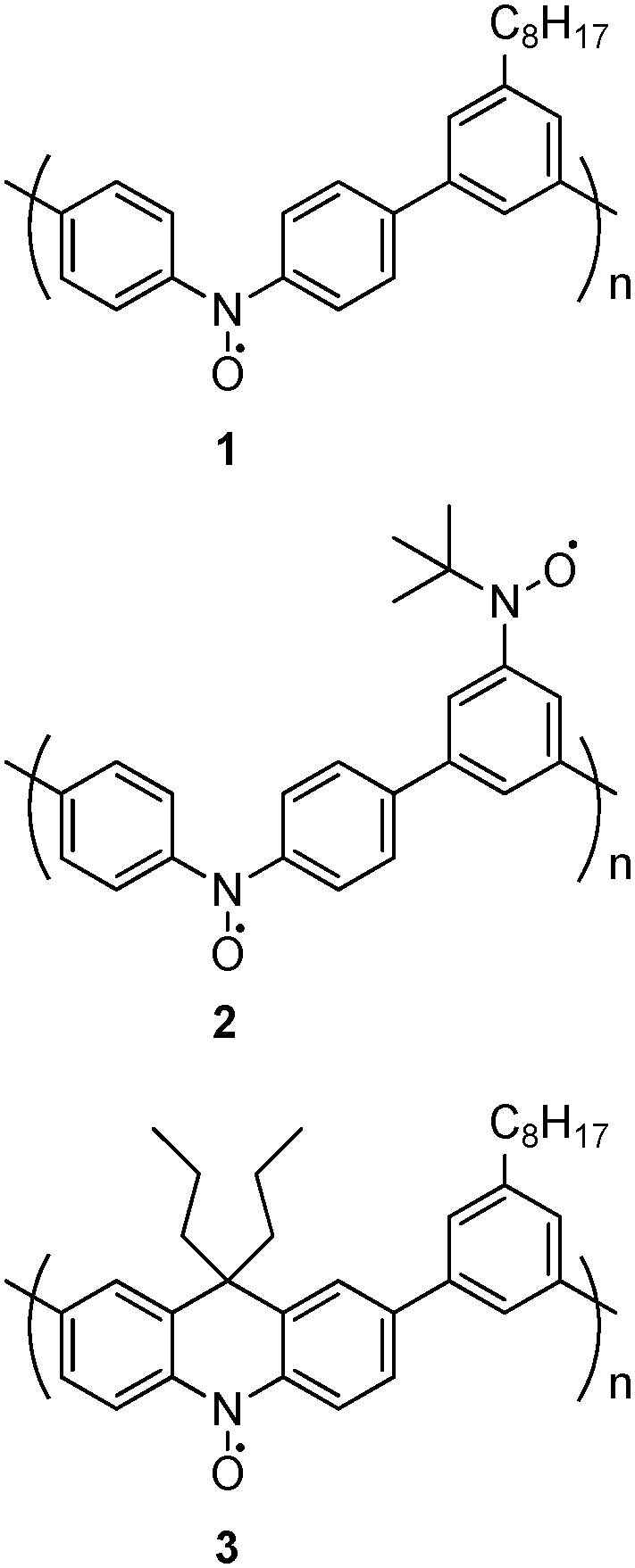 | ||
| Scheme 4 The chemical structures of polynitroxides studied by Oka et al.19 | ||
In the polynitroxide 1 each mer contains 4 bonds with free rotations around the nitroxide spin bearing unit, resulting in S = 1 spin state. In 3 one >CPr2 bridge per mer is introduced and removes the free rotation of 2 bonds, yielding a S = 3/2 state. In the case of 1 and 3 polynitroxide, the increase of S (from 1 to 3/2) is probably less pronounced than that from PA2 to PQA (from 1 to 2), because only 2 bonds out of 4 are immobilized for the polynitroxide instead of 4 bonds out of 6 for PQA. Moreover in the case of PQA all bonds around the meta-phenyl spin coupling moiety are blocked, which is more efficient than blocking only the bonds around the spin bearing NO° moiety in 3.
Experimental section
For any experimental details on the chemical sources, synthesis, pulsed EPR and SQUID measurements as well as DFT calculations see the ESI.†Conclusions
In summary, we demonstrated the efficient synthesis of the model compound (dimer) QA and of the polymer PQA with semi co-planar structures. The first two successive oxidations were observed without significant increase of the redox potential, which is crucial for obtaining high-spin arylamine materials and this can be rationalized by the formation of well localized radical cations. Though the presence of a co-planar structure around the spin couplers potentially leads to a significant increase of the ferromagnetic exchange coupling constant (cf.D2versusQAD2: Table 1), the slightly bent structure of QA (and, accordingly, of PQA) as well as the presence of the pending donor groups to stabilize the molecules upon oxidation, resulted in a relatively “small” magnitude of J/kB. Still, this enabled an increase of the spin multiplicity in polymeric compounds: a quintet spin state (S = 2) was observed for the first time in the linear polymer PQA with an m-phenylene coupler. Moreover, the exchange coupling constant determined for PQA (∼50 K) from magnetization measurements still showed a significant increase of the J/kB value (ca. three times) in comparison to that estimated for the flexible analogue, PA2 (18 K).Acknowledgements
IKB and LS wish to acknowledge financial support from the National Science Centre in Poland (NCN, Grant No. 2015/17/B/ST5/00179). We acknowledge prof. Andrzej Twardowski and Anita Gardias for preliminary SQUID studies.References
- (a) P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus and A. Pron, Chem. Soc. Rev., 2013, 42, 8895–8999 RSC; (b) A. Ito, D. Sakamaki, H. Ino, A. Taniguchi, Y. Hirao, K. Tanaka, K. Kanemoto and T. Kato, Eur. J. Org. Chem., 2009, 4441–4450 CrossRef CAS; (c) Y. Hirao, H. Ino, A. Ito, K. Tanaka and T. Kato, J. Phys. Chem. A, 2006, 110, 4866–4872 CrossRef CAS PubMed; (d) A. Ito, Y. Yamagishi, K. Fukui, S. Inoue, Y. Hirao, K. Furukawa, T. Kato and K. Tanaka, Chem. Commun., 2008, 6573–6575 RSC; (e) A. Ito, K. Ota, K. Tanaka, T. Yamabe and K. Yoshizawa, Macromolecules, 1995, 28, 5618–5625 CrossRef CAS.
- (a) J. A. Crayston, J. N. Devine and J. C. Walton, Tetrahedron, 2000, 56, 7829–7857 CrossRef CAS; (b) D. A. Dougherty, Acc. Chem. Res., 1991, 24, 88–94 CrossRef CAS; (c) T. Sugawara, S. Murrata, K. Kimura, Y. Sugawara, H. Iwasaki and H. Iwamura, J. Am. Chem. Soc., 1985, 107, 5293–5394 CrossRef CAS; (d) I. Fujita, Y. Teki, T. Takui, T. Kinoshita, K. Itoh, F. Miko, Y. Sawaki, H. Iwamura, A. Izuoka and T. Sugawara, J. Am. Chem. Soc., 1990, 112, 4074–4075 CrossRef CAS.
- (a) T. Aoki, T. Kaneko and M. Teraguchi, Polymer, 2006, 47, 4867–4892 CrossRef CAS; (b) T. Michinobu, J. Inui and H. Nishide, Polym. J., 2010, 42, 575–582 CrossRef CAS; (c) D. Sakamaki, A. Ito, K. Tanaka, K. Furukawa, T. Kato and M. Shiro, Angew. Chem., Int. Ed., 2012, 51, 8281–8285 CrossRef CAS PubMed; (d) D. Sakamaki, A. Ito, A. Furukawa, T. Kato, M. Shiro and K. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 12776–12781 CrossRef CAS PubMed; (e) Y. Yokoyama, D. Sakamaki, A. Ito, K. Tanaka and M. Shiro, Angew. Chem., Int. Ed., 2012, 51, 9403–9406 CrossRef CAS PubMed.
- (a) N. M. Gallagher, A. Olankitwanit and A. Rajca, J. Org. Chem., 2015, 80, 1291–1298 CrossRef CAS PubMed; (b) A. Rajca, K. Shiraishi, M. Pink and S. Rajca, J. Am. Chem. Soc., 2007, 129, 7232–7233 CrossRef CAS PubMed; (c) A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS; (d) A. Rajca, S. Rajca and J. Wongsriratanakul, J. Am. Chem. Soc., 1999, 121, 6308–6309 CrossRef CAS; (e) A. Rajca, J. Wongsriratanakul and S. Rajca, J. Am. Chem. Soc., 2004, 126, 6608–6626 CrossRef CAS PubMed; (f) S. Rajca, A. Rajca, J. Wongsriratanakul, P. Butler and S. M. Choi, J. Am. Chem. Soc., 2004, 126, 6972–6986 CrossRef CAS PubMed.
- A. Rajca, Chem. – Eur. J., 2002, 8, 4834–4841 CrossRef CAS.
- T. Sugawara, H. Komatsu and K. Suzuki, Chem. Soc. Rev., 2011, 40, 3105–3118 RSC.
- A. Rajca, Y. Wang, M. Boska, J. T. Paletta, A. Olankitwanit, M. A. Swanson, D. G. Mitchell, S. S. Eaton, G. R. Eaton and S. Rajca, J. Am. Chem. Soc., 2012, 134, 15724–15727 CrossRef CAS PubMed.
- A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503–1505 CrossRef CAS PubMed.
- (a) A. A. Ovchinnikov, Theor. Chim. Acta, 1978, 47, 297–304 CrossRef CAS; (b) R. J. Bushby, D. R. McGill, K. M. Ng and N. Taylor, J. Chem. Soc., Perkin Trans. 2, 1997, 1405–1414 RSC; (c) R. J. Bushby, N. Taylor and R. A. Williams, J. Mater. Chem., 2007, 17, 955–964 RSC; (d) T. D. Selby, K. R. Stickley and S. C. Blackstock, Org. Lett., 2000, 2, 171–174 CrossRef CAS PubMed; (e) E. Fukuzaki and H. Nishide, Org. Lett., 2006, 8, 1835–1838 CrossRef CAS PubMed.
- R. J. Bushby, D. Gooding and M. E. Vale, Philos. Trans. R. Soc., A, 1999, 357, 2939–2957 CrossRef CAS.
- E. Dobrzynska, M. Jouni, P. Gawrys, S. Gambarelli, J. M. Mouesca, D. Djurado, L. Dubois, I. Wielgus, V. Maurel and I. Kulszewicz-Bajer, J. Phys. Chem. B, 2012, 116, 14968–14978 CrossRef CAS PubMed.
- T. Michinobu, J. Inui and H. Nishide, Org. Lett., 2003, 5, 2165–2168 CrossRef CAS PubMed.
- V. Maurel, M. Jouni, P. Baran, N. Onofrio, S. Gambarelli, J. M. Mouesca, D. Djurado, L. Dubois, J. F. Jacquot, G. Desfonds and I. Kulszewicz-Bajer, Phys. Chem. Chem. Phys., 2012, 14, 1399–1407 RSC.
- A. Rajca, A. Olankitwanit, Y. Wang, P. J. Boratynski, M. Pink and S. Rajca, J. Am. Chem. Soc., 2013, 135, 18205–18215 CrossRef CAS PubMed.
- R. J. Bushby, C. A. Kilner, N. Taylor and M. E. Vale, Tetrahedron, 2007, 63, 11458–11466 CrossRef CAS.
- R. F. Winter, Organometallics, 2014, 33, 4517–4536 CrossRef CAS.
- L. Skorka, J. M. Mouesca, L. Dubois, E. Szewczyk, I. Wielgus, V. Maurel and I. Kulszewicz-Bajer, J. Phys. Chem. B, 2015, 119, 13462–13471 CrossRef CAS PubMed.
- A. Schweiger and G. Jeschke, Principles of Pulsed Electron Paramagnetic Resonance, Oxford University Press, Oxford, 2001 Search PubMed.
- (a) H. Oka, Y. Kiyohara, H. Kouno and H. Tanaka, Polyhedron, 2007, 26, 2059–2064 CrossRef CAS; (b) H. Oka, H. Kouno and H. Tanaka, J. Mater. Chem., 2007, 17, 1209–1215 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1545978. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7tc01932g |
| This journal is © The Royal Society of Chemistry 2017 |