
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thieno[3,4-c]pyrrole-4,6-dione as novel building block for host materials for red PhOLEDs†
Paul
Kautny
 *a,
Chenyang
Zhao
b,
Dominik
Schopf
a,
Berthold
Stöger
c,
Ernst
Horkel
a,
Jiangshan
Chen
*b,
Dongge
Ma
b,
Johannes
Fröhlich
a and
Daniel
Lumpi
a
*a,
Chenyang
Zhao
b,
Dominik
Schopf
a,
Berthold
Stöger
c,
Ernst
Horkel
a,
Jiangshan
Chen
*b,
Dongge
Ma
b,
Johannes
Fröhlich
a and
Daniel
Lumpi
a
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, A-1060 Vienna, Austria. E-mail: paul.kautny@tuwien.ac.at
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: jschen@ciac.ac.cn
cInstitute of Chemical Technologies and Analytics, TU Wien, Getreidemarkt 9/164, A-1060 Vienna, Austria
First published on 31st January 2017
Abstract
In the presented work the electron accepting thieno[3,4-c]pyrrole-4,6-dione is introduced as a novel building block for donor–acceptor based host materials for Phosphorescent Organic Light Emitting Diodes (PhOLEDs). A series of three regioisomers consisting of the thieno[3,4-c]pyrrole-4,6-dione acceptor and carbazole donors linked via a phenylene linker was prepared and the impact of the phenylene substitution pattern on the molecular properties was analyzed. Regarding their applicability as host materials, the newly developed materials were investigated in red PhOLED devices achieving a high current efficiency of 30.6 cd A−1 corresponding to an external quantum efficiency of 17.7% and a high power efficiency of 23.8 lm W−1. Thus, we present the first successful application of the thieno[3,4-c]pyrrole-4,6-dione building block in host materials for PhOLEDs.
Introduction
Since the first reports on electroluminescence from organic materials,1,2 great efforts have been made in the development of organic light emitting diodes (OLEDs).3–8 The application of transition metal emitters in phosphorescent OLEDs (PhOLEDs)9,10 and of organic compounds capable of thermally activated delayed fluorescence (TADF)11 boosted the efficiency of OLED devices.12–16 Therefore, one major goal of research is the development of new host materials for these emitters.17–21 In particular, the application of bipolar host materials consisting of donor and acceptor subunits proved successful, owing to their balanced charge transport properties and thus simplified device structures and broad recombination zones.22–24The primary source of electron donating groups for optoelectronic materials are aromatic amines, such as triphenylamines,3,9,25–29 carbazoles,10,17,28,30–33 indolocarbazoles,34–37 phenoxazines15,38–40 and dihydroacridines.15,41 In contrast, the nature of the electron accepting moieties exhibits more variations. Among the most frequently used acceptors are oxadiazoles,27,33,38,42–44 phenantrolines,31,45 pyridines,32,46 benzimidazoles,26,28,47 1,3,5-triazines,39,48,49 1,2,4-triazoles,29,38,50 phosphine oxides,51–53 sulfones,15,54 ketones,40,41 carbolines55,56 and benzonitriles.11,57
The thieno[3,4-c]pyrrole-4,6-dione (TPD) (Scheme 1) has been successfully employed as electron accepting unit in donor–acceptor polymers for organic photovoltaics in combination with various donor units.58–62 Moreover, the application of a TPD based polymer in an organic field effect transistor has been reported.63 So far however, this particular unit has not been exploited as acceptor in materials for OLEDs.
 | ||
| Scheme 1 Molecular structure of thieno[3,4-c]pyrrole-4,6-dione (TPD). | ||
This circumstance prompted us to explore the possibility to utilize the TPD building block in donor–acceptor host materials for PhOLEDs. Hence, this study presents the first application of TPD in host materials in combination with the well-established carbazole donor. The variation of the linkage mode between the two molecular subunits allowed for a modification of the molecular properties of the derivatives. Suchlike developed compounds were employed as host materials in efficient red PhOLED devices (CEmax: 30.6 cd A−1, PEmax: 23.8 lm W−1, EQEmax: 17.7%), demonstrating the applicability of the TPD unit as electron withdrawing organic building block for optoelectronic applications.
Results and discussion
Synthesis
Three different TPD based host materials (p-PCzTPD, m-PCzTPD and o-PCzTPD), in which the TPD unit is connected to the carbazole donors by phenylene linkers, were prepared (Scheme 2). Thereby three different linkage modes (para, meta, ortho) were realized using the phenylene linker. The unsubstituted imide of the TPD unit was protected with an octyl chain, also facilitating the handling of the materials during the synthetic process. The key synthetic step in the preparation of the new materials was a Suzuki cross coupling reaction of dibrominated TPD 1 and the suitable substituted boronic acid esters 2a–c (Scheme 2). Products p-PCzTPD and m-PCzTPD were obtained with yields of 67% and 70%, respectively. In contrast the reaction towards o-PCzTPD proceeded significantly slower and with a low yield of 17% due to the high steric demand resulting from the ortho linkage mode. All materials were characterized by 1H and 13C NMR and high resolution mass spectrometry.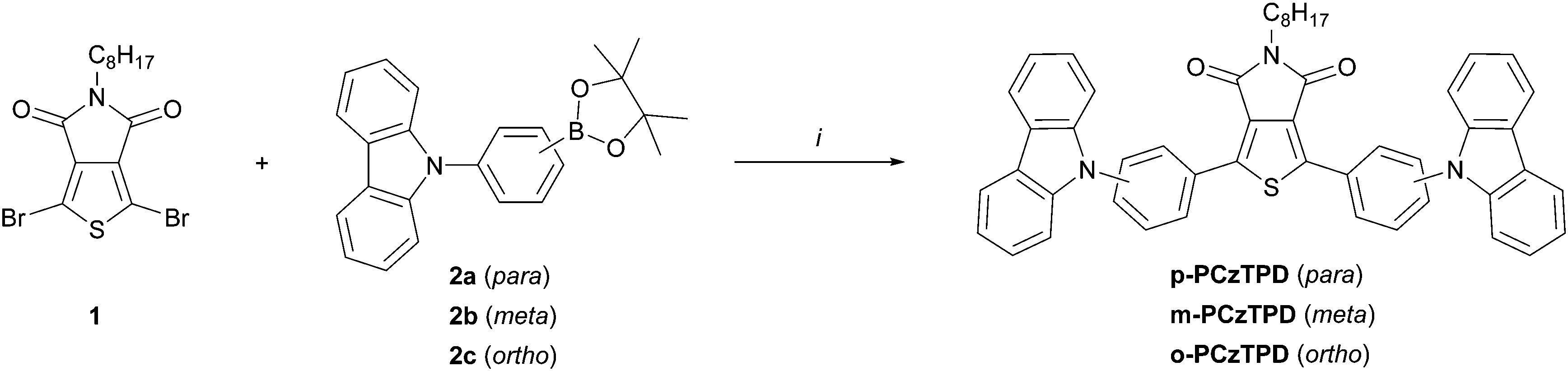 | ||
| Scheme 2 Synthesis of TPD based host materials; (i) 1 (1.0 eq., 25 mM), 2a–c (2.5 eq.), Pd(PPh3)4 (0.05 eq.), K2CO3 (5.0 eq., 2 M aqueous solution), THF, reflux. | ||
In the case of p-PCzTPD the molecular structure was unequivocally confirmed by X-ray crystal analysis (Fig. 1). Both phenylene linkers are close to coplanar with the TPD core [angles between the least square (LS) planes: Φ2 = 15.04(11)° and Φ3 = 18.08(11)°] and thus fully conjugated with the central aromatic system. In contrast, the carbazole moieties and the phenylene linkers are more pronouncedly inclined [angles between the LS planes: Φ1 = 54.53(12)° and Φ4 = 61.68(11)°] due to the steric interaction of the ortho hydrogen atoms.
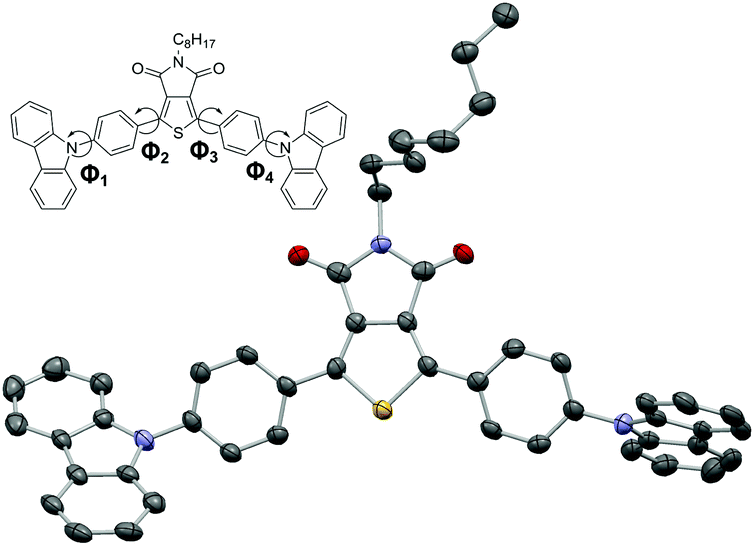 | ||
| Fig. 1 Molecular structure of p-PCzTPD. C, N, O and S atoms are represented by gray, blue, red and yellow ellipsoids drawn at 70% probability levels. H atoms are omitted for clarity. | ||
Thermal properties
The thermal behaviour of the developed materials was studied to evaluate their applicability as host materials for PhOLEDs (Table 1). m-PCzTPD and p-PCzTPD exhibited high decomposition temperatures (Td – corresponding to 5% mass loss) of 438 °C and 448 °C during thermogravimetric analysis (Fig. S7, ESI†). In contrast, the Td of o-PCzTPD was lower (369 °C). The glass transition temperatures (Tg) of the compounds were determined by differential scanning calorimetry, to estimate the morphological stability of thin films of the materials. Whereas o-PCzTPD exhibited a rather low Tg of 62 °C, the Tgs increased with increasingly linear shape of the molecules to 84 °C for m-PCzTPD and 102 °C for p-PCzTPD (Fig. S8, ESI†).| T g/Tm/Tda [°C] | Opt. BGb,c [eV] | λ PL,max [nm] | HOMO/LUMO [eV] | E T [eV] | |||
|---|---|---|---|---|---|---|---|
| Exp.d | Cal.e | Exp.f | Cal.g | ||||
a Determined by thermogravimetric analysis and differential scanning calorimetry. Tm: melting temperature.
b Estimated from the absorption onset.
c Measurements were carried out in CH2Cl2 (5 μM) at room temperature.
d HOMO levels were determined from the onset of the oxidation peak obtained during cyclic voltammetric measurements (0.5 mM solution in anhydrous CH2Cl2 with Bu4NBF4 (0.1 M) as supporting electrolyte); LUMO levels were calculated from the HOMO levels and the optical bandgap.
e Calculated applying density functional theory level (B3LYP/6-311+G*).
f Estimated from the highest energy vibronic transition in solid solution of toluene/EtOH (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 77 K.
g Calculated applying time dependent density functional theory level (B3LYP/6-311+G*). :1) at 77 K.
g Calculated applying time dependent density functional theory level (B3LYP/6-311+G*).
|
|||||||
| p-PCzTPD | 102/208/448 | 2.84 | 476 | −5.68/−2.84 | −5.68/−2.57 | 2.09 | 2.11 |
| m-PCzTPD | 84/195/438 | 3.18 | 506 | −5.70/−2.52 | −5.71/−2.56 | 2.21 | 2.31 |
| o-PCzTPD | 62/133/369 | 3.34 | 485 | −5.65/−2.31 | −5.75/−2.13 | 2.33 | 2.62 |
Photophysical properties
UV/Vis absorption, room temperature photoluminescence and low temperature phosphorescence spectra were recorded to investigate the impact of the substitution pattern on the photophysical properties of the materials (Fig. 2). The key photophysical data is summarized in Table 1. All derivatives exhibit a distinct absorption peak at 292 nm, which can be attributed to a transition centered on the carbazole moieties.31 Moreover, weaker transitions at approximately 325 nm and 340 nm, which are typical for phenylcarbazole derivatives,64 are found for all compounds. Fully conjugated p-PCzTPD features an additional broad absorption peak with a peak maximum at 386 nm. This peak can be attributed to an intramolecular charge transfer transition between the electron rich carbazoles and the TPD acceptor unit. Owing to the decreased effective conjugation resulting from the ortho and meta linkages,33 this transition is absent in o-PCzTPD and m-PCzTPD. In anology, the absorption onsets are shifted towards lower energy from o-PCzTPD (3.34 eV) to m-PCzTPD (3.18 eV) and p-PCzTPD (2.84 eV).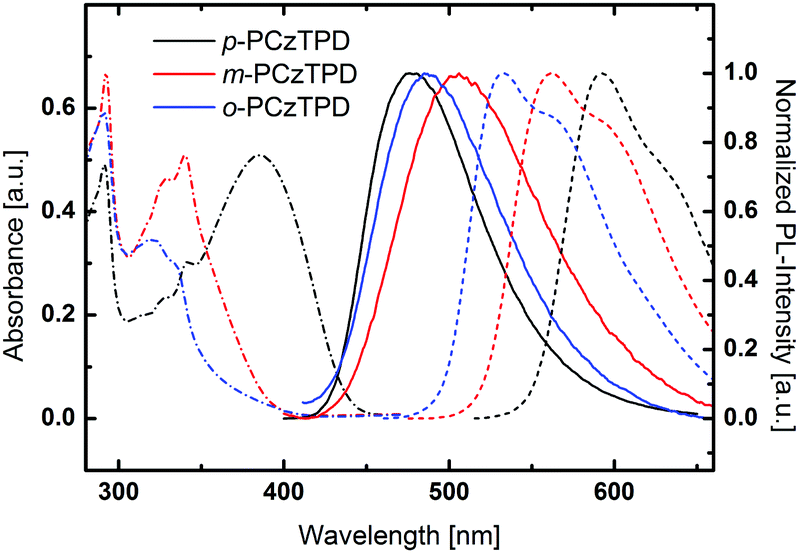 | ||
| Fig. 2 UV-Vis absorption (dashed and dotted lines), normalized fluorescence at room temperature (solid lines) and normalized phosphorescence spectra at 77 K (dashed lines) of the materials under investigation. | ||
The fluorescence of the materials is distinctly red shifted compared to the absorption, and emission maxima are located at 476 nm (p-PCzTPD), 506 nm (m-PCzTPD) and 485 nm (o-PCzTPD).
To determine the triplet energy (ET) of the developed materials low temperature phosphorescence was recorded. The order of the emission maxima follows the absorption onset. Thus, o-PCzTPD features the highest ET of 2.33 eV, whereas the ET of m-PCzTPD is slightly lower (2.21 eV) and p-PCzTPD exhibits the lowest ET of 2.09 eV. In p-PCzTPD, the pronounced donor–acceptor interaction between the carbazoles and the TPD, which is also evident in the absorption behaviour of the material, lowers the ETs via intramolecular charge transfer.22 The theoretically calculated ET display the same trend, the absolute values for m-PCzTPD and o-PCzTPD are, however, higher than the experimentally determined energies. A closer inspection of the S0 → T1 transition reveals that in case of m-PCzTPD and o-PCzTPD transitions from low lying molecular orbitals contribute significantly to the lowest excited triplet state. These transitions may be slightly overestimated in the applied model, leading to the prediction of too high ETs of the two derivatives. Ultimately, the application of different linkage modes allowed to control the degree of the overall conjugation in m-PCzTPD and o-PCzTPD. Therefore, the intramolecular charge transfer could be decreased, resulting in higher ETs. Notably, in this series the ortho derivative o-PCzTPD exhibits a higher ET than meta linked m-PCzTPD, which is unlike the behaviour of related host materials based on 1,3,4-oxadiazole acceptor units.33,65
Electrochemical properties
The levels of the Highest Occupied Molecular Orbitals (HOMOs) of the materials were determined by cyclic voltammetry (CV). All derivatives exhibited irreversible oxidation, as commonly observed for carbazole based materials, owing to the instability of the formed cations.66 The HOMO levels were calculated from the onset of the oxidation peaks relative to the oxidation of ferrocene and span a narrow range between −5.65 eV and −5.70 eV (Fig. S9, ESI†), indicating no significant barrier for hole injection from the adjacent layers. The Lowest Unoccupied Molecular Orbital (LUMO) levels of the materials were estimated from the HOMO levels and the optical bandgaps and are located between −2.84 eV and −2.31 eV, whereby p-PCzTPD features the lowest value owing to the lower bandgap of this compound.Computational investigations
Density functional theory was applied to obtain a deeper understanding of the electronic layouts of the investigated materials on the molecular level. In general, the calculated HOMO and LUMO energy values are in good agreement with the experimental results (Table 1). Moreover, the energetic order of the triplet energies of the developed host materials was reliably reproduced by the theoretical calculations, albeit slightly higher energies were predicted for m-PCzTPD and o-PCzTPD compared to the experimental values.The spatial distributions of the HOMOs and LUMOs are depicted in Fig. 3. In the case of the fully conjugated p-PCzTPD, the HOMO level expands over the whole aromatic backbone of the molecule with a distinctive localization of electron density on the central TPD unit. The LUMO of p-PCzTPD is more localized with major contributions coming from the TPD acceptor and the adjacent phenyl moieties. Looking at m-PCzTPD, the morphology of the HOMO is distinctly different. The electron density of the orbital is solely located on one of the carbazole units and the neighbouring phenyl ring owing to the decreased overall conjugation. In contrast, the shape of the LUMO is essentially unaffected by the altered linkage mode of the donor units. Consequently, the decreased spatial overlap of the two molecular orbitals leads to a lower degree of intramolecular charge transfer in m-PCzTPD, as reflected by the absence of a distinct charge transfer transition in the absorption spectra and also an increased ET.22
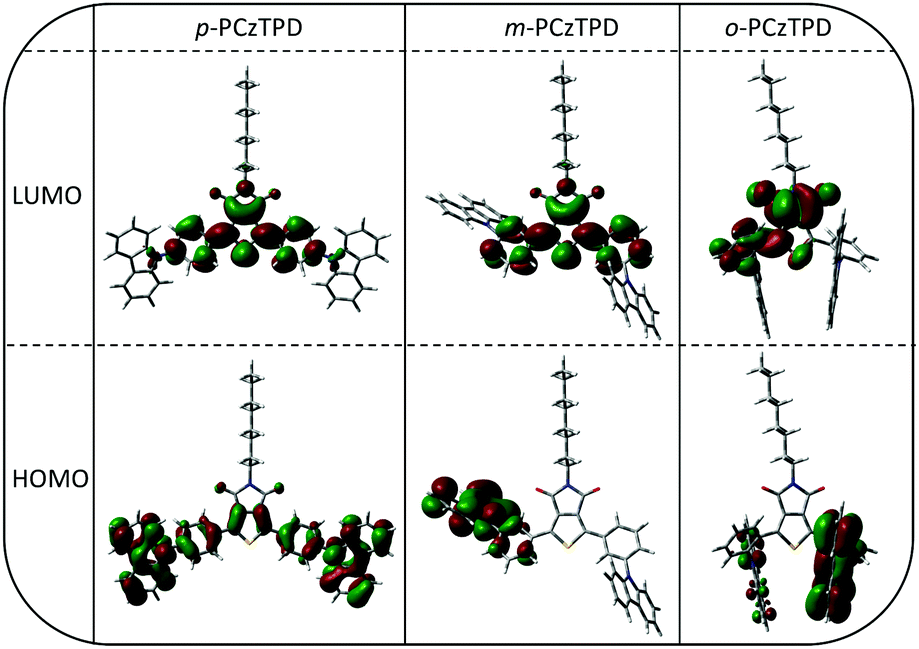 | ||
| Fig. 3 Spatial distribution of the HOMOs and LUMOs of p-PCzTPD, m-PCzTPD and o-PCzTPD. | ||
In the case of o-PCzTPD, however, the overall situation changed as a result of the geometric structure of the molecule. The calculated shape of the aromatic part of p-PCzTPD is in excellent agreement with the molecular structure determined by X-ray diffraction. Accordingly, the optimized torsion angles between the aromatic subunits deviate only slightly from the experimentally determined values (Table 2). Similar torsion angles were observed for m-PCzTPD. In contrast the molecular structure of o-PCzTPD is distinctly different. Both carbazole units are oriented away from the TPD, owing to the steric demand of the carbonyl oxygen atoms (Fig. 3). This leads to a rotation of the phenylene linkers to avoid a close proximity of the carbazoles. Therefore, the torsion angles Φ2 and Φ3 between the TPD and the adjacent phenyl rings are distinctly enlarged to 49.43° and 53.46° and also the torsion angles between the phenylene linkers and the carbazole are larger than in the other two derivatives (Table 2). The highly twisted structure of o-PCzTPD restricts the HOMO to one of the carbazoles with only minor electron density on the neighbouring phenylene linker as well as on the opposite carbazole. Notably, also the distribution of the LUMO is altered compared to the other derivatives. The LUMO is exclusively located on the TPD acceptor and the phenylene linker opposite of the carbazole that is hosting the HOMO. The increased spatial separation of the two molecular orbitals impedes their electronic exchange and thus explains the increased ET of o-PCzTPD compared to m-PCzTPD. Thus, the ortho linkage effect33 is further enhanced for these particular class of materials, owing to the steric demand of the TPD, which leads to an increased electronic separation of the molecular subunits, even compared to the corresponding meta derivatives.
| Φ 1 [°] (Cz1–Ph1) | Φ 2 [°] (Ph1–TPD) | Φ 3 [°] (TPD–Ph2) | Φ 4 [°] (Ph2–Cz2) | |
|---|---|---|---|---|
| a Determined by single-crystal X-ray diffraction. b Calculated applying density functional theory level (B3LYP/6-311+G*). | ||||
| p-PCzTPD | 54.53(12) | 15.04(11) | 18.08(11) | 61.68(11) |
| p-PCzTPD | 55.67 | 19.14 | 20.30 | 55.52 |
| m-CzTPD | 59.65 | 25.75 | 23.47 | 61.64 |
| o-CzTPD | 67.16 | 49.43 | 53.46 | 67.91 |
Electroluminescent properties
The title compounds were employed in PhOLED devices, to investigate their practical applicability as host materials. Owing to the higer ET of the three materials compared to red emitting Ir(MDQ)2(acac) (2.0 eV),67 Ir(MDQ)2(acac) was chosen as phosphorescent emitter for this purpose. PhOLED devices were fabricated with the structure of ITO/MoO3 (8 nm)/TAPC (75 nm)/EML (12 nm)/BPhen (70 nm)/LiF/Al (Fig. S10, ESI†). The EMLs consisted of host materials p-PCzTPD (R1), m-PCzTPD (R2) or o-PCzTPD (R3) doped with Ir(MDQ)2(acac). A dopant concentration of 5% was applied without further optimization. TAPC was employed as hole transporting layer and BPhen was utilized as electron transporting and hole blocking layer. Current density–voltage–luminance and current efficiency–luminance–power efficiency curves of the three devices are depicted in Fig. 4 and the key electroluminescent properties are summarized in Table 3. | ||
| Fig. 4 Current density–voltage–luminance (left; luminance: full symbols, current density: hollow symbols) and current efficiency–luminance–power efficiency (right; current efficiency: full symbols, power efficiency: hollow symbols) curves of devices R1–R3. | ||
All devices exhibited electroluminescent emission exclusively from the Ir(MDQ)2(acac) dopant (Fig. S11, ESI†), indicating that exciton transfer from the host to the dopant or direct charge trapping on the phosphorescent emitter was effective. Moreover, all devices exhibited turn-on voltages between 3.2 V and 4.0 V. The slightly lower turn on voltage of 3.2 V of R1 can be attributed to the lower bangap and in particular the low lying LUMO level of p-PCzTPD compared to its regioisomers.
Among the three devices, m-PCzTPD based R2 exhibited the best performance with a high maximum current efficiency (CE) of 30.8 cd A−1 corresponding to a satisfying maximum external quantum efficiency (EQE) of 17.7% and a maximum power efficiency (PE) of 23.8 lm W−1. Compared to R2 devices R1 and R3 displayed somewhat lower efficiencies with a CEmax of 18.5 cd A−1 and 22.2 cd A−1 and a PEmax of 11.8 lm W−1 and 12.9 lm W−1, respectively. The lower efficiency of R1 may be attributed to the fact that the ET of p-PCzTPD is rather close to the ET of the red (MDQ)2(acac) dopant (2.0 eV),67 which is also in accordance with the higher efficiency roll-off of R1 compared to R2 and R3. Although the performance of R3 is slightly better compared to R1, it does not reach the efficiency values of R2 despite a higher ET of the host material. Nonetheless, the efficiency roll-off above a brightness of 1000 cd m−2 is less pronounced in R3.
Conclusions
In this study we incorporated the electron accepting thieno[3,4-c]pyrrole-4,6-dione building block in donor–acceptor host materials for PhOLEDs and prepared three isomeric derivatives for this purpose. The newly developed materials displayed favourable molecular properties for the application in red PhOLED devices and prototype devices employing Ir(MDQ)2(acac) as phosphorescent emitter featured satisfying current and power efficiencies. Thus, we have successfully demonstrated the applicability of the thieno[3,4-c]pyrrole-4,6-dione as acceptor in host materials for PhOLEDs and introduced this particular molecular unit as novel building block for functional organic materials for OLEDs.Experimental section
General information
All reagents and solvents were purchased from commercial suppliers and used without further purification. Column chromatography was performed on silica 60 (Merck, 40–63 μm). NMR spectra were recorded on a Bruker Avance DRX-400 Spectrometer. A Thermo Scientific LTQ Orbitrap XL hybrid FTMS (Fourier Transform Mass Spectrometer) equipped with a Thermo Scientific MALDI source was used for high resolution mass spectrometry. The thermal analysis was carried out with a heating rate of 10 K min−1 in a flowing argon atmosphere (25 mL min−1). TG measurements were performed on a Netzsch TG 209 F9 Tarsus system with open aluminium oxide crucibles. DSC measurements were performed on a Netzsch DSC 200 F3 Maia, working with aluminium pans with pierced lids. UV/Vis absorption and fluorescence emission spectra were recorded in DCM solutions (5 μM) with a Perkin Elmer Lambda 750 spectrometer and an Edinburgh FLS920, respectively. The time resolved low temperature phosphorescence spectra were recorded in solid solutions (1 mg mL−1; toluene:EtOH = 9:1) at 77 K with a Perkin Elmer Instruments LS 50B luminance spectrometer. Cyclic voltammetry was performed using a three electrode configuration consisting of a Pt working electrode, a Pt counter electrode and an Ag/AgCl reference electrode and a PGSTAT128N, ADC164, DAC164, External, DI048 potentiostat provided by Metrohm Autolab B.V. Measurements were carried out in a 0.5 mM solution in anhydrous DCM with Bu4NBF4 (0.1 M) as supporting electrolyte. The solutions were purged with nitrogen for 15 minutes prior to measurement. HOMO energy levels were calculated from the onset of the oxidation peaks, which was determined from the intersection of two tangents drawn at the background and the rising of the oxidation peaks.
Synthetic details
General procedure for the synthesis of the host materials: Host materials p-PCzTPD, m-PCzTPD and o-PCzTPD were prepared using a Suzuki cross coupling reaction. A three necked flask was flushed with argon and charged with dibromide 1 (1.0 eq.) and boronic acid esters 2a–c (2.5 eq.) before degassed THF (2.5 mM) and aqueous K2CO3 (5.0 eq., 2 M) were added. Subsequently, Pd(PPh3)4 (0.05 eq.) was added and the reaction mixture was heated to reflux until full conversion (TLC, 72 h). After cooling to room temperature the reaction mixture was poured on water and extracted with CH2Cl2 three times. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification of the crude product was accomplished by column chromatography.Computational details
The (TD)DFT computations were performed using the Gaussian 09 package, revision D.01.68 Density functional theory (DFT) and time-dependent (TD)DFT calculations were performed using the Becke three parameters hybrid functional with Lee–Yang–Perdew correlation (B3LYP),69,70 in combination with Pople basis sets (6-31G*, 6-311+G*).71 The geometry optimizations were performed in gas phase and without symmetry constraints. For the calculation of HOMO/LUMO levels, the ground state (S0) geometries were optimized applying the 6-311+G* basis set. The determination of the triplet energy was achieved by the calculation of the T1 excitation energy applying TDDFT level and the 6-311+G* basis to a S0 geometry optimized at DFT level using the 6-31G* basis set. Orbital plots were generated using GaussView.72Single crystal diffraction
A crystal of p-PCzTPD suitable for single crystal diffraction was selected under a polarizing microscope, embedded in perfluorinated oil and attached to Kapton® micromounts. Intensity data were collected in a dry stream of nitrogen at 100 K on a Bruker KAPPA APEX II diffractometer system. Data were reduced using SAINT-Plus73 and an empirical absorption correction using the multi-scan approach implemented in SADABS73 was applied. The crystal structures were solved by charge-flipping implemented in SUPERFLIP74 and refined against F with the JANA200675 software package. The non-H atoms were refined with anisotropic displacement parameters. The H atoms were placed at calculated positions and refined as riding on the parent C atoms.Device fabrication and measurement
All synthesized materials were additionally purified by crystallization (light petrol with minor amounts of toluene) prior to device fabrication. OLED fabrication and testing: the fabricated devices were grown on clean glass substrates pre-coated with ITO with a sheet resistance of 10 Ω sq−1. After cleaning with detergent and deionized water, they were dried in an oven at 120 °C for 30 minutes and then treated with UV-ozone for 15 minutes. All layers were grown in succession by thermal evaporation without breaking the vacuum (∼10−4 Pa). The evaporation rates for the organic layer were in a range of 1–2 Å s−1, LiF and Al were evaporated with a rate of approximately 0.1 and 5.0 Å s−1, respectively. All measurements were carried out in ambient atmosphere. The current–voltage–brightness characteristics were recorded using a Keithley source measurement unit (Keithley 2400 and Keithley 2000) with a calibrated silicon photodiode. The electroluminescence (EL) spectra were measured with a Spectrascan PR650 spectrophotometer.Acknowledgements
This work was supported in part by the TU Wien “Innovative Projects” research funds and the Austrian Federal Ministry of Science, Research and Economy. The X-ray centre of the TU Wien is acknowledged for providing access to the single-crystal diffractometer. P. K. and J. F. gratefully acknowledge financial support by the Austrian Science Fund (FWF) (Grant No. I 2589-N34). J. C. and D. M. gratefully acknowledge financial support by the National Natural Science Foundation of China (Grant No. 11661131001).Notes and references
- M. Pope, P. Magnante and H. P. Kallmann, J. Chem. Phys., 1963, 38, 2042–2043 CrossRef CAS.
- W. Helfrich and W. G. Schneider, Phys. Rev. Lett., 1965, 14, 229–231 CrossRef CAS.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- D. Braun and A. J. Heeger, Appl. Phys. Lett., 1991, 58, 1982–1984 CrossRef CAS.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- M. C. Gather, A. Köhnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef CAS PubMed.
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, Rev. Mod. Phys., 2013, 85, 1245–1293 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908–912 CrossRef CAS PubMed.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lussem and K. Leo, Nature, 2009, 459, 234–238 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. H. Li, R. F. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- W.-C. Chen, C.-S. Lee and Q.-X. Tong, J. Mater. Chem. C, 2015, 3, 10957–10963 RSC.
- R. J. Holmes, S. R. Forrest, Y.-J. Tung, R. C. Kwong, J. J. Brown, S. Garon and M. E. Thompson, Appl. Phys. Lett., 2003, 82, 2422–2424 CrossRef CAS.
- S. Tokito, T. Iijima, Y. Suzuri, H. Kita, T. Tsuzuki and F. Sato, Appl. Phys. Lett., 2003, 83, 569–571 CrossRef CAS.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169–3190 CrossRef CAS PubMed.
- H. Sasabe and J. Kido, Eur. J. Org. Chem., 2013, 7653–7663 CrossRef CAS.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed.
- S. O. Jeon and J. Y. Lee, J. Mater. Chem., 2012, 22, 4233–4243 RSC.
- B. E. Koene, D. E. Loy and M. E. Thompson, Chem. Mater., 1998, 10, 2235–2250 CrossRef CAS.
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M.-a. Kakimoto, Adv. Funct. Mater., 2008, 18, 584–590 CrossRef CAS.
- Y. Tao, Q. Wang, Y. Shang, C. Yang, L. Ao, J. Qin, D. Ma and Z. Shuai, Chem. Commun., 2009, 77–79 RSC.
- C. H. Chen, W. S. Huang, M. Y. Lai, W. C. Tsao, J. T. Lin, Y. H. Wu, T. H. Ke, L. Y. Chen and C. C. Wu, Adv. Funct. Mater., 2009, 19, 2661–2670 CrossRef CAS.
- Y. Tao, Q. Wang, L. Ao, C. Zhong, C. Yang, J. Qin and D. Ma, J. Phys. Chem. C, 2010, 114, 601–609 CAS.
- Y. Kuwabara, H. Ogawa, H. Inada, N. Noma and Y. Shirota, Adv. Mater., 1994, 6, 677–679 CrossRef CAS.
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M.-a. Kakimoto, Org. Lett., 2008, 10, 421–424 CrossRef CAS PubMed.
- S.-J. Su, H. Sasabe, T. Takeda and J. Kido, Chem. Mater., 2008, 20, 1691–1693 CrossRef CAS.
- Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 304–311 CrossRef CAS.
- H.-C. Ting, Y.-M. Chen, H.-W. You, W.-Y. Hung, S.-H. Lin, A. Chaskar, S.-H. Chou, Y. Chi, R.-H. Liu and K.-T. Wong, J. Mater. Chem., 2012, 22, 8399–8407 RSC.
- P. Kautny, D. Lumpi, Y. Wang, A. Tissot, J. Bintinger, E. Horkel, B. Stöger, C. Hametner, H. Hagemann, D. Ma and J. Fröhlich, J. Mater. Chem. C, 2014, 2, 2069–2081 RSC.
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050–5055 CrossRef CAS PubMed.
- P. Kautny, Z. Wu, J. Eichelter, E. Horkel, B. Stöger, J. Chen, D. Ma, J. Fröhlich and D. Lumpi, Org. Electron., 2016, 34, 237–245 CrossRef CAS.
- J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599–4604 RSC.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Chem. Mater., 2013, 25, 3766–3771 CrossRef CAS.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 126, 6520–6524 CrossRef.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- Y. Hamada, C. Adachi, T. Tsutsui and S. Saito, Jpn. J. Appl. Phys., Part 1, 1992, 31, 1812–1816 CrossRef CAS.
- C. Adachi, T. Tsutsui and S. Saito, Appl. Phys. Lett., 1989, 55, 1489–1491 CrossRef CAS.
- P. Kautny, Z. Wu, B. Stöger, A. Tissot, E. Horkel, J. Chen, D. Ma, H. Hagemann, J. Fröhlich and D. Lumpi, Org. Electron., 2015, 17, 216–228 CrossRef CAS.
- D. F. O'Brien, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 74, 442–444 CrossRef.
- S.-J. Su, Y. Takahashi, T. Chiba, T. Takeda and J. Kido, Adv. Funct. Mater., 2009, 19, 1260–1267 CrossRef CAS.
- Z. Gao, C. S. Lee, I. Bello, S. T. Lee, R.-M. Chen, T.-Y. Luh, J. Shi and C. W. Tang, Appl. Phys. Lett., 1999, 74, 865–867 CrossRef CAS.
- H. Inomata, K. Goushi, T. Masuko, T. Konno, T. Imai, H. Sasabe, J. J. Brown and C. Adachi, Chem. Mater., 2004, 16, 1285–1291 CrossRef CAS.
- D. R. Lee, M. Kim, S. K. Jeon, S.-H. Hwang, C. W. Lee and J. Y. Lee, Adv. Mater., 2015, 27, 5861–5867 CrossRef CAS PubMed.
- J. Kido, C. Ohtaki, K. Hongawa, K. Okuyama and K. Nagai, Jpn. J. Appl. Phys., Part 2, 1993, 32, L917–L920 CrossRef CAS.
- P. E. Burrows, A. B. Padmaperuma, L. S. Sapochak, P. Djurovich and M. E. Thompson, Appl. Phys. Lett., 2006, 88, 183503 CrossRef.
- S. O. Jeon, K. S. Yook, C. W. Joo and J. Y. Lee, Adv. Funct. Mater., 2009, 19, 3644–3649 CrossRef CAS.
- C. Han, G. Xie, H. Xu, Z. Zhang, L. Xie, Y. Zhao, S. Liu and W. Huang, Adv. Mater., 2011, 23, 2491–2496 CrossRef CAS PubMed.
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi and P. Wang, Adv. Mater., 2014, 26, 5198–5204 CrossRef CAS PubMed.
- C. W. Lee and J. Y. Lee, Adv. Mater., 2013, 25, 5450–5454 CrossRef CAS PubMed.
- Y. Im and J. Y. Lee, Chem. Commun., 2013, 49, 5948–5950 RSC.
- T. Nakagawa, S.-Y. Ku, K.-T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS PubMed.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed.
- C. B. Nielsen, R. S. Ashraf, B. C. Schroeder, P. D'Angelo, S. E. Watkins, K. Song, T. D. Anthopoulos and I. McCulloch, Chem. Commun., 2012, 48, 5832–5834 RSC.
- M.-C. Yuan, M.-Y. Chiu, S.-P. Liu, C.-M. Chen and K.-H. Wei, Macromolecules, 2010, 43, 6936–6938 CrossRef CAS.
- X. Guo, N. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Nat. Photonics, 2013, 7, 825–833 CrossRef CAS.
- X. Guo, R. P. Ortiz, Y. Zheng, M.-G. Kim, S. Zhang, Y. Hu, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 13685–13697 CrossRef CAS PubMed.
- H. Li, Y. Wang, K. Yuan, Y. Tao, R. Chen, C. Zheng, X. Zhou, J. Li and W. Huang, Chem. Commun., 2014, 50, 15760–15763 RSC.
- Y. Tao, Q. Wang, L. Ao, C. Zhong, J. Qin, C. Yang and D. Ma, J. Mater. Chem., 2010, 20, 1759–1765 RSC.
- E. Mondal, W.-Y. Hung, H.-C. Dai and K.-T. Wong, Adv. Funct. Mater., 2013, 23, 3096–3105 CrossRef CAS.
- G. Schwartz, S. Reineke, T. Rosenow, K. Walzer and K. Leo, Adv. Funct. Mater., 2009, 19, 1319–1333 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revission D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem, Inc., Shawnee Mission, KS, 2009 Search PubMed.
- Bruker computer programs: APEX2, SAINT and SADABS, Bruker AXS Inc., Madison, WI, 2015 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. – Cryst. Mater., 2014, 229, 345–352 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, DSC, TGA and CV analysis and crystallographic information. CCDC 1519402. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6tc05248g |
| This journal is © The Royal Society of Chemistry 2017 |