
Stabilization of Ca-deficient hydroxyapatite in biphasic calcium phosphate ceramics by adding alginate to enhance their biological performances†
Xiangfeng
Li
,
Yanglong
Deng
,
Menglu
Wang
,
Xuening
Chen
*,
Yumei
Xiao
 * and
Xingdong
Zhang
* and
Xingdong
Zhang
National Engineering Research Center for Biomaterials, Sichuan University, Chengdu 610064, China. E-mail: xchen6@scu.edu.cn; xymzl2000@126.com
First published on 23rd November 2017
Abstract
It is of significance to further improve the bioactivity of existing calcium phosphate (Ca–P) biomaterials to satisfy the needs of regenerative medicine. Due to its compositional similarity to natural bone mineral, calcium-deficient hydroxyapatite (CDHA) is supposed to possess excellent bioactivity. However, it is difficult to fabricate Ca–P ceramics with a high amount of CDHA, as CDHA is easy to decompose during the sintering process. The present study introduced an effective approach to stabilize CDHA in biphasic calcium phosphate (BCP) ceramics by adding alginate, and investigated the roles of CDHA in their biological performances. The characterization of the phase composition, crystal structure, and functional group demonstrated that the addition of alginate could obviously inhibit CDHA decomposition to attain novel BCP ceramics with a high CDHA content (BCP-A), which could better mimic the inorganic composition of natural bones as compared with the conventional BCP ones (BCP-C). In vitro studies suggested BCP-A showed better bioactivity and osteoinductive capacity than BCP-C, as evidenced by the increased serum protein adsorption, better bone-like apatite formation and cell spreading, and promoted osteogenic differentiation. In vivo intramuscular implantation further confirmed that BCP-A could induce more ectopic bone formation than BCP-C, suggesting BCP-A had a stronger osteoinductivity. Altogether, this study demonstrates that the stabilization of CDHA in BCP ceramics by adding alginate offers a promising principle for designing regenerative biomaterials to process superior biological performances.
1. Introduction
It is becoming a promising research area in regenerative medicine to take advantage of the body's natural ability, especially in endowing biomaterials with biomimetic properties to attain excellent biological functions and induce tissue regeneration.1–3 Calcium phosphate (Ca–P) ceramics are extensively considered as a kind of excellent bone regenerative graft due to their similarity to the bone mineral composition, as well as their good biocompatibility and bioactivity.4–8 Among them, the biphasic calcium phosphate (BCP) ceramic seems one of the best choices, due to its suitable solubility and high osteoinductivity.6,9–11 However, the bioactivity of conventional BCP ceramics still cannot match the natural bone, despite partly mimicking the bony composition and porous structure.5,8,12 In order to bridge this gap, further biomimicking the composition and micro–nano structures of natural bone might be an alternative. It is speculated that as the chemical composition and crystal structure of Ca–P ceramics are designed to be further closer to natural bone, their biological performances should be improved evidently.It is known that bone mineral is a non-stoichiometric apatite (Ca/P ratio < 1.67) with a poor crystalline structure.13–15 To the best of our knowledge, compared with other kinds of synthetic Ca–P materials, calcium-deficient hydroxyapatite (CDHA) is more similar to natural bone mineral with respect to chemical composition and crystal structure.14–17 Thus it is supposed that CDHA should show better bioactivity than other conventional Ca–P ceramics, which has been confirmed by extensive previous studies.18–21 For instance, it was reported that CDHA elicited an immediate precipitation of biologically equivalent apatite on its surface when immersed in a simulated physiological fluid, whereas the precipitation rate on stoichiometric HA was relatively low.18 Kasten P et al.19,20 compared the effects of CDHA and β-TCP on the behaviors of human bone marrow stromal cells (BMSCs), suggesting that CDHA showed more favorable properties than β-TCP regarding reproducibility of the seeding efficacy. Liu CS et al.21 introduced CDHA in calcium phosphate cement, and observed the good biocompatibility and osteogenicity of the CDHA scaffolds. On the basis of the biomimetic point of view, it was hypothesized that BCP ceramics with a high content of CDHA phase should exhibit evidently improved biological performances.
However, it is generally difficult to introduce the CDHA phase in BCP ceramics, as CDHA, which is generally considered as a low-temperature Ca–P phase, is easy to decompose into β-TCP and HA during the high-temperature (above 700 °C) sintering process.22,23 Until now, little attention has been paid to the stabilization of CDHA in the sintering process of BCP ceramics, and thus little is known about the effects of the CDHA phase on the biological performances of BCP ceramics. Recently, alginate has received extensive studies due to its capability of forming a relatively stable calcium alginate hydrogel through ionotropic gelation in the presence of Ca2+.24–27 In our previous work,6,28 alginate gelatinizing technology was employed to prepare porous spherical Ca–P bioceramic granules, observing that the burning of calcium alginate had some influence on their chemical composition.
Herein, we proposed a strategy to stabilize the CDHA phase in BCP ceramics by adding alginate, and further evaluated their biological performances (Scheme 1). First of all, the effects of the decomposition of calcium alginate on the phase composition, crystal structure and functional group of BCP ceramics were investigated. Then, the in vitro biological performances of BCP ceramics with a high content of CDHA (BCP-A) were evaluated, including Tris–HCl buffer immersion, bone-like apatite formation, serum protein adsorption, as well as the cell viability and osteoblastic differentiation of BMSCs. Finally, the osteoinductive capacity of BCP-A ceramics was further investigated via the in vivo canine intramuscular implantation.
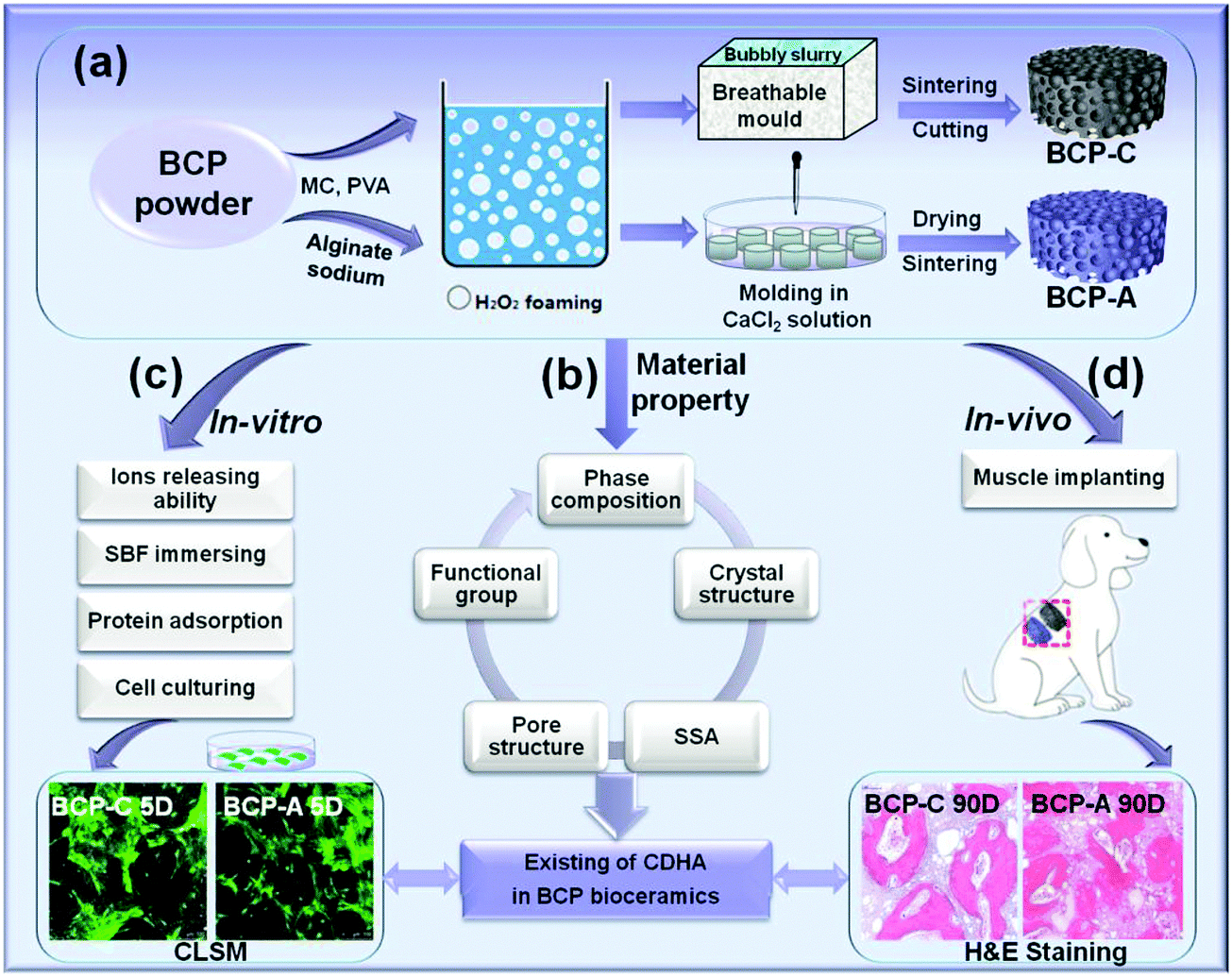 | ||
| Scheme 1 Schematic description of the experimental design: (a) preparation of BCP-C and BCP-A scaffolds (Φ8 × 2 mm), (b) material characterization, (c) in vitro biological evaluation and (d) in vivo intramuscular implantation of BCP-C and BCP-A. | ||
2. Materials and methods
2.1 Material preparation and characterization
BCP powder (HA/β-TCP = 20/80) were synthesized by a wet chemical method in the National Engineering Research Center for Biomaterials of Sichuan University, China.5,6 Firstly, BCP powder (10 g) was uniformly dispersed in 25 mL sodium alginate solution (6 wt%). Then the bubbly slurry foaming by the H2O2 gas-foaming method was dripped into a plastic mold (Φ16 mm × 5 mm) and gelled in CaCl2 solution (0.45 mol L−1). Subsequently, these gelled discs were hardened in CaCl2 solution, washed with deionized water, and then dried at 60 °C. Finally, the discs were sintered at 1100 °C for 2 h to obtain BCP ceramics (BCP-A, Φ8 mm × 2 mm). For comparison, the conventional BCP porous ceramics (BCP-C, Φ8 mm × 2 mm) were also fabricated via H2O2 foaming and sintering at 1100 °C for 2 h, by following a similar method as mentioned before.5,29,30 Furthermore, the pre-sintered BCP-A and BCP-C were also sintered at 600, 700, 800, 900 and 1000 °C for 2 h, respectively, to investigate the influence of calcium alginate on the sintering behaviors of BCP ceramics.The microstructures of the samples were visualized by scanning electron microscopy (SEM, S-4800, Hitachi, Japan). Phase composition was analyzed by X-ray diffractometry (XRD, Philips X’Pert 1 X-ray diffractometer), and the obtained peaks were compared with the standard references for HA (09-0432) and β-TCP (09-0169). The phase ratios of the HA phase and the β-TCP phase in the samples were calculated by the indexes of R211/(211+0210) and R0210/(211+0210), respectively, where R211 was the intensity of the HA(211) peak at 2θ = 31.8° and R0210 was the intensity of the β-TCP(0210) peak at 2θ = 31.03°.22,31 The lattice parameters and crystal sizes of the HA phase and the β-TCP phase in the samples were separately calculated with Jade 5.0 software. The functional groups of the samples were identified by FT-IR (Perkin-Elmer Spectrum one B, USA) and scanned from 4000 cm−1 to 500 cm−1. The pH values and Ca/P ratios were tested by using a pH meter (METTLER TOLEDO EF20 Plus) and an inductively coupled plasma optical emission spectrometer (ICP-OES, SPECTRO ARCOS). The specific surface area (SSA) and pore structure were tested by using a surface area analyzer (Gemini VII 2390t, Micromeritics) and mercury intrusion porosimetry (MIP, AutoPore IV 9500, Micromeritics), respectively.
2.2 In vitro experiments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) for 0.5, 1, 3, 5 and 7 days, respectively. At each time point, 0.5 mL immersed solution was taken out to test the concentration of Ca2+ and PO43−, meanwhile, the same amount of fresh Tris–HCl buffer solution was also added.
000 rpm for 15 min. Following the manufacturer's instruction, the intracellular ALP activity in the lysate was tested colorimetrically by means of Alkaline Phosphatase Assay Kit (Beyotime, China), based on the conversion of p-nitrophenyl phosphate (pNPP) to p-nitrophenol after incubation at 37 °C for 30 min. The results were normalized to the total protein content measured by using the Pierce® BCA protein assay kit (Themofisher Scientific, USA). Meanwhile, the amount of OCN produced by BMSCs cultured on each group (n = 3) was also quantified by using the Enzyme-linked Immunosorbent Assay (ELISA) Kit for OCN (Cloud-clone Corp, USA) and normalized to the total protein content.
:100) for 0.5, 1, 3, 5 and 7 days, respectively. At each time point, 0.5 mL immersed solution was taken out to test the concentration of Ca2+ and PO43−, meanwhile, the same amount of fresh Tris–HCl buffer solution was also added.
000 rpm for 15 min. Following the manufacturer's instruction, the intracellular ALP activity in the lysate was tested colorimetrically by means of Alkaline Phosphatase Assay Kit (Beyotime, China), based on the conversion of p-nitrophenyl phosphate (pNPP) to p-nitrophenol after incubation at 37 °C for 30 min. The results were normalized to the total protein content measured by using the Pierce® BCA protein assay kit (Themofisher Scientific, USA). Meanwhile, the amount of OCN produced by BMSCs cultured on each group (n = 3) was also quantified by using the Enzyme-linked Immunosorbent Assay (ELISA) Kit for OCN (Cloud-clone Corp, USA) and normalized to the total protein content.
2.3 In vivo experiments
000 U per dog was administered by ectopic muscle injection once a day over the first postoperative 3 days. After being implanted for 45 and 90 days, the dogs were sacrificed with an overdose of pentobarbital sodium and saturated potassium chloride, and the samples plus their surrounding tissue were retrieved. All the samples were immediately fixed by 4% paraformaldehyde solution buffered in 0.1 mol L−1 phosphate solution (pH = 7.2) for 5 days before further analysis.
2.4 Statistical analysis
All quantitative results were obtained from at least triplicate measurements. Statistical analysis of the data was performed using one-way analysis of variance (ANOVA) by using SPSS 11.0 software (SPSS Inc., USA). Statistical significance was assumed at p < 0.05.3. Results
3.1 Material properties
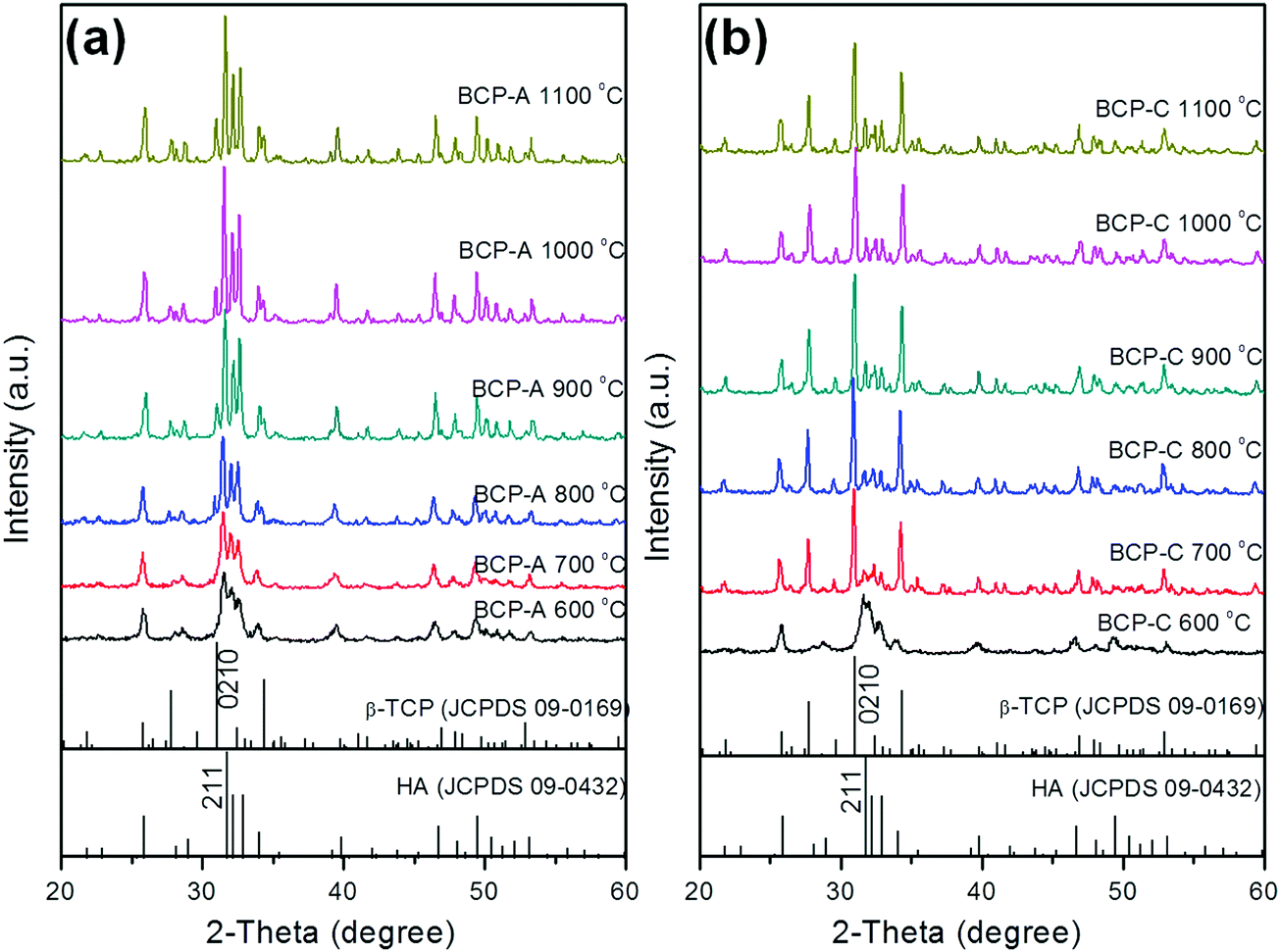 | ||
| Fig. 1 XRD patterns of BCP-A (a) and BCP-C (b) sintered with different temperatures (600, 700, 800, 900, 1000 and 1100 °C). | ||
| T (°C) | Phase | BCP-A | BCP-C | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ratio (%) | a = b (nm) | c (nm) | V (nm3) | XSa (nm) | Ratio (%) | a = b (nm) | c (nm) | V (nm3) | XSa (nm) | ||
| a XS represents the crystal size calculated from the HA main peak (211) or the β-TCP main peak (0210). | |||||||||||
| 600 | HA | 100 | 0.9503 | 0.6885 | 0.5379 | 10.4 | 100 | 0.9471 | 0.6906 | 0.5365 | 9.7 |
| β-TCP | — | — | — | — | — | — | — | — | — | — | |
| 700 | HA | 100 | 0.9523 | 0.6890 | 0.5415 | 16.3 | 19.6 | 0.9436 | 0.6811 | 0.5250 | 17.5 |
| β-TCP | — | — | — | — | — | 80.4 | 1.0466 | 3.7483 | 3.5554 | 34.1 | |
| 800 | HA | 74.6 | 0.9422 | 0.6899 | 0.5305 | 26.5 | 17.4 | 0.9437 | 0.6821 | 0.5261 | 28.0 |
| β-TCP | 25.4 | 1.0498 | 3.7358 | 3.5659 | 25.2 | 82.6 | 1.0472 | 3.7522 | 3.5633 | 32.5 | |
| 900 | HA | 77.6 | 0.9490 | 0.6860 | 0.5340 | 31.2 | 22.1 | 0.9429 | 0.6791 | 0.5229 | 42.6 |
| β-TCP | 22.4 | 1.0474 | 3.7603 | 3.5728 | 26.3 | 77.9 | 1.0426 | 3.7425 | 3.5228 | 33.4 | |
| 1000 | HA | 80.8 | 0.9504 | 0.6866 | 0.5371 | 34.0 | 19.3 | 0.9416 | 0.6777 | 0.5203 | 38.3 |
| β-TCP | 19.2 | 1.0460 | 3.7289 | 3.5333 | 30.5 | 80.7 | 1.0426 | 3.7425 | 3.5228 | 35.7 | |
| 1100 | HA | 83.7 | 0.9494 | 0.6862 | 0.5356 | 33.9 | 19.2 | 0.9417 | 0.6830 | 0.5294 | 41.7 |
| β-TCP | 16.3 | 1.0490 | 3.7614 | 3.5843 | 34.0 | 80.8 | 1.0463 | 3.7381 | 3.5445 | 35.9 | |
| Standard | HA | 0.9418 | 0.6884 | 0.5288 | β-TCP | 1.0429 | 3.7380 | 3.5209 | — | ||
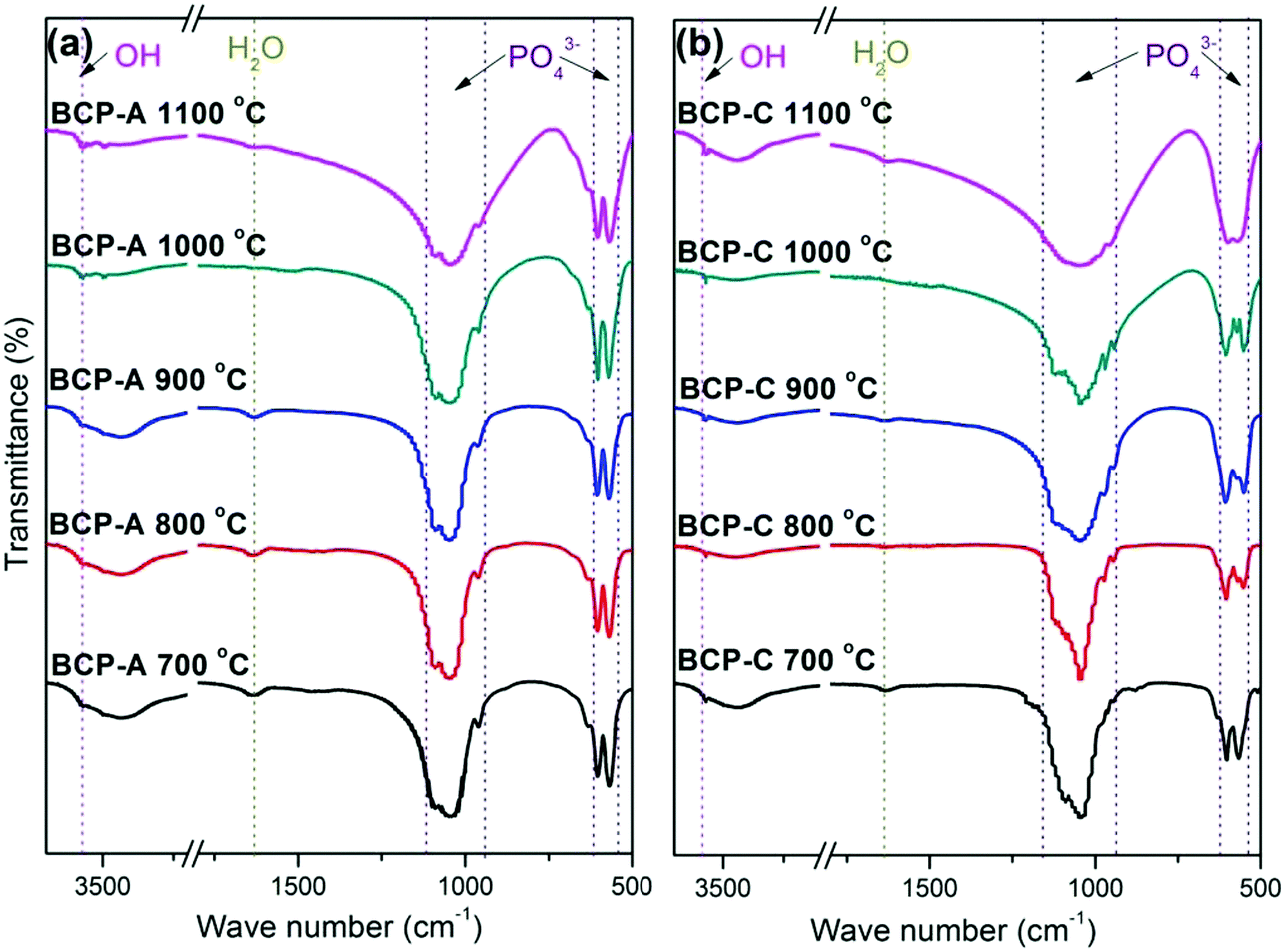 | ||
| Fig. 2 FT-IR spectra of BCP-A (a) and BCP-C (b) sintered with different temperatures (600, 700, 800, 900, 1000 and 1100 °C). | ||
Fig. 3 shows the enlarged characteristic peaks ranging from 800–400 cm−1 in the FT-IR spectra of BCP-A and BCP-C sintered at different temperatures. By comparison, it was found that the bending vibration peak of the OH group at 630 cm−1 in BCP-A was stronger than the one in BCP-C. In particular, the bending vibration peaks of OH group were not observed in BCP-C sintered at 900 and 1100 °C. Moreover, the splitting degree of the bending vibration peaks of the PO43− group (v4, at about 550 and 600 cm−1) in BCP-A was higher than that in BCP-C.
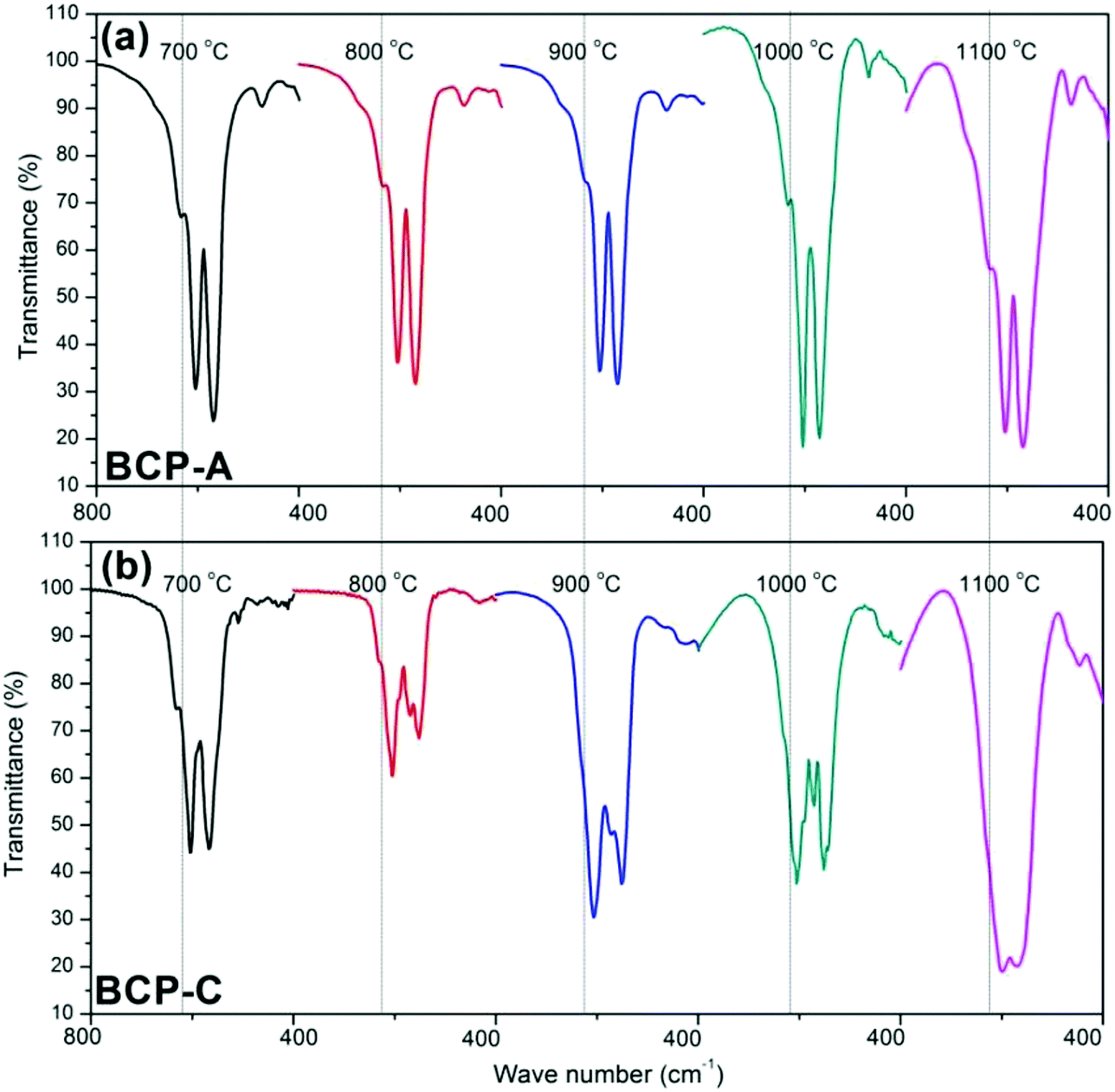 | ||
| Fig. 3 Comparisons of the enlarged characteristic peaks ranging from 800–400 cm−1 in the FT-IR spectra of BCP-A (a) and BCP-C (b) sintered with different temperatures (600, 700, 800, 900, 1000 and 1100 °C). | ||
:10), the same test was also done for BCP-C. The pH values of the solution immersed by BCP-A and BCP-C were both neutral (7.17 and 7.18, respectively) as listed in Table 2, indicating that there was no CaO residue in BCP-A. Moreover, the Ca/P ratioa (listed in Table 2) of BCP-A (1.577, dissolved in nitric acid) was higher than that of BCP-C (1.534), suggesting that some calcium sources pyrolyzing from calcium alginate had entered into BCP-A.
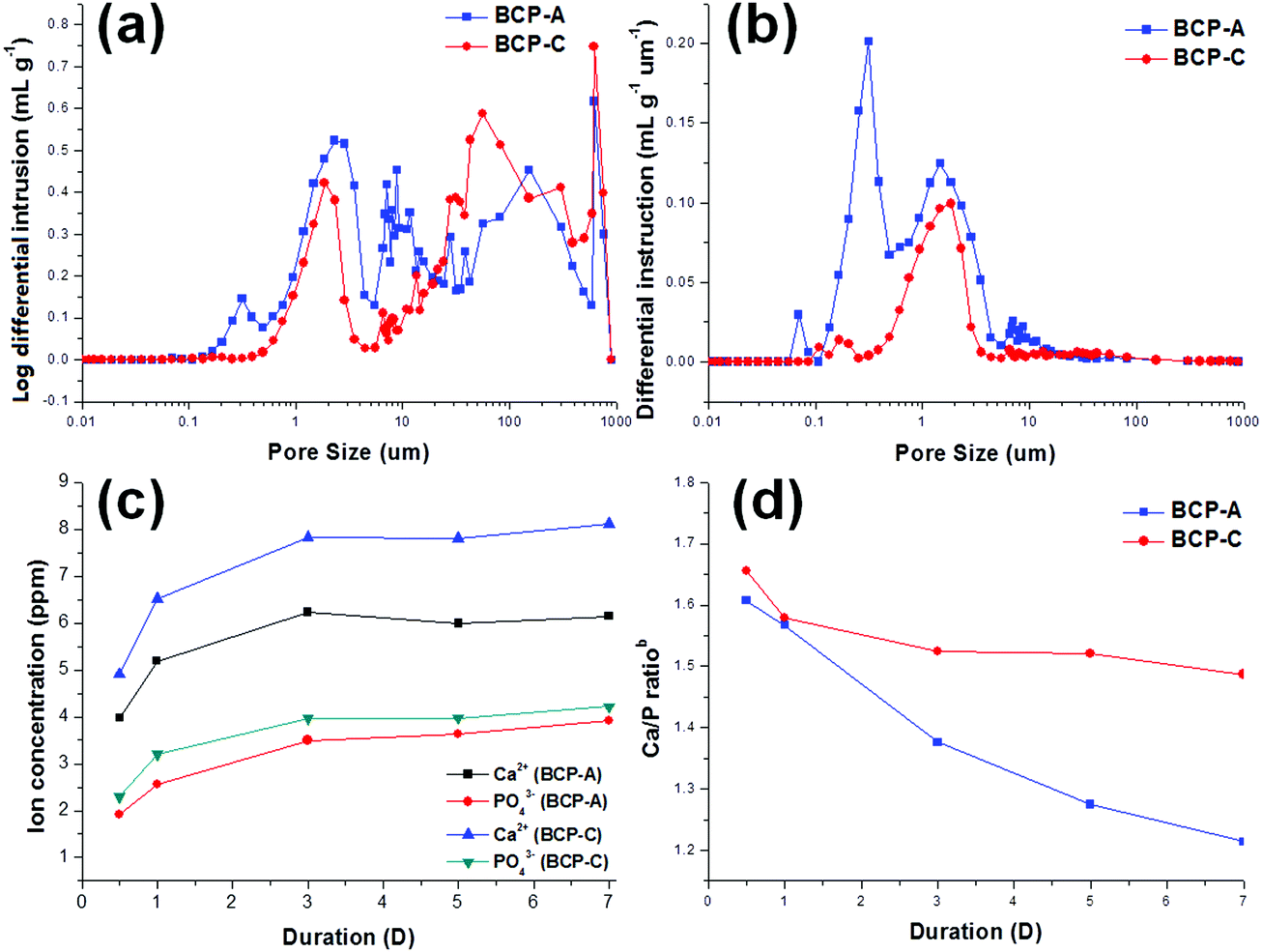 | ||
| Fig. 4 Pore structure and ion releasing abilities (immersed in Tris–HCl solution) of BCP-C and BCP-A: (a) pore size distributions, (b) pore quantity distributions, (c) Ca2+ and PO43− concentrations released from BCP-A and BCP-C, (d) Ca/P ratiosb in the Tris–HCl solution. | ||
3.2 In vitro biological performances
 | ||
| Fig. 5 The surface topographies of BCP-A and BCP-C before and after immersion in SBF for 3 days (before: a and b, BCP-C; c and d, BCP-A; after: e and f, BCP-C; g and h, BCP-A). | ||
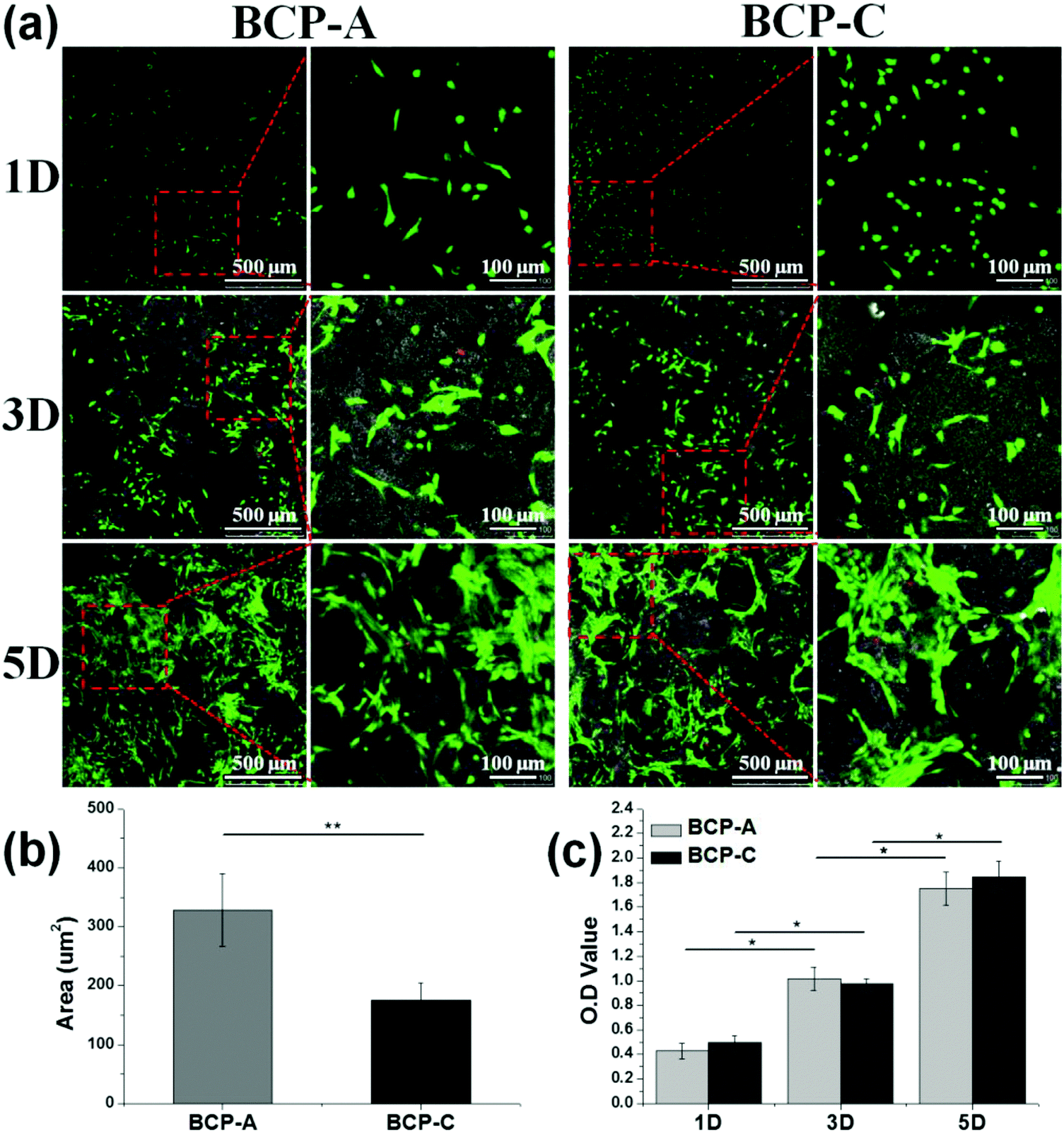 | ||
| Fig. 6 (a) CLSM observations for BMSC growth onto BCP-A and BCP-C at day 1, 3 and 5; (b) quantification of the cell area of BMSCs at day 1; (c) CCK-8 results for the proliferation of BMSCs cultured on BCP-A and BCP-C. Values are expressed as the mean ± SD (n = 3), * refers to p < 0.05, and ** refers to p < 0.01. | ||
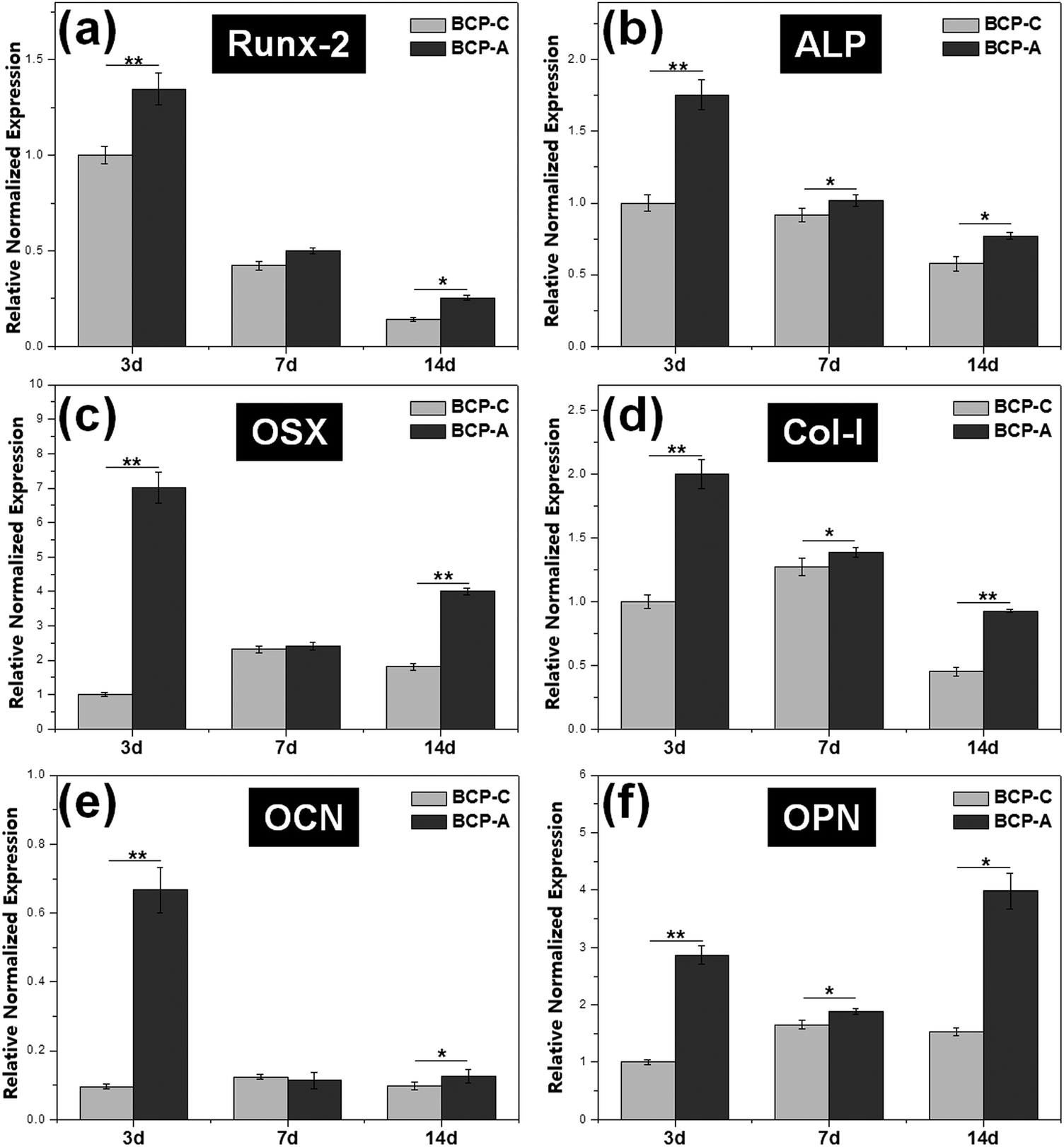 | ||
| Fig. 7 The expression of osteogenic genes (including Runx-2, ALP, OSX, Col-I, OCN and OPN) in BMSCs cultured on BCP-C and BCP-A for 3, 7 and 14 days. Values are expressed as the mean ± SD (n = 3), * refers to p < 0.05, and ** refers to p < 0.01. | ||
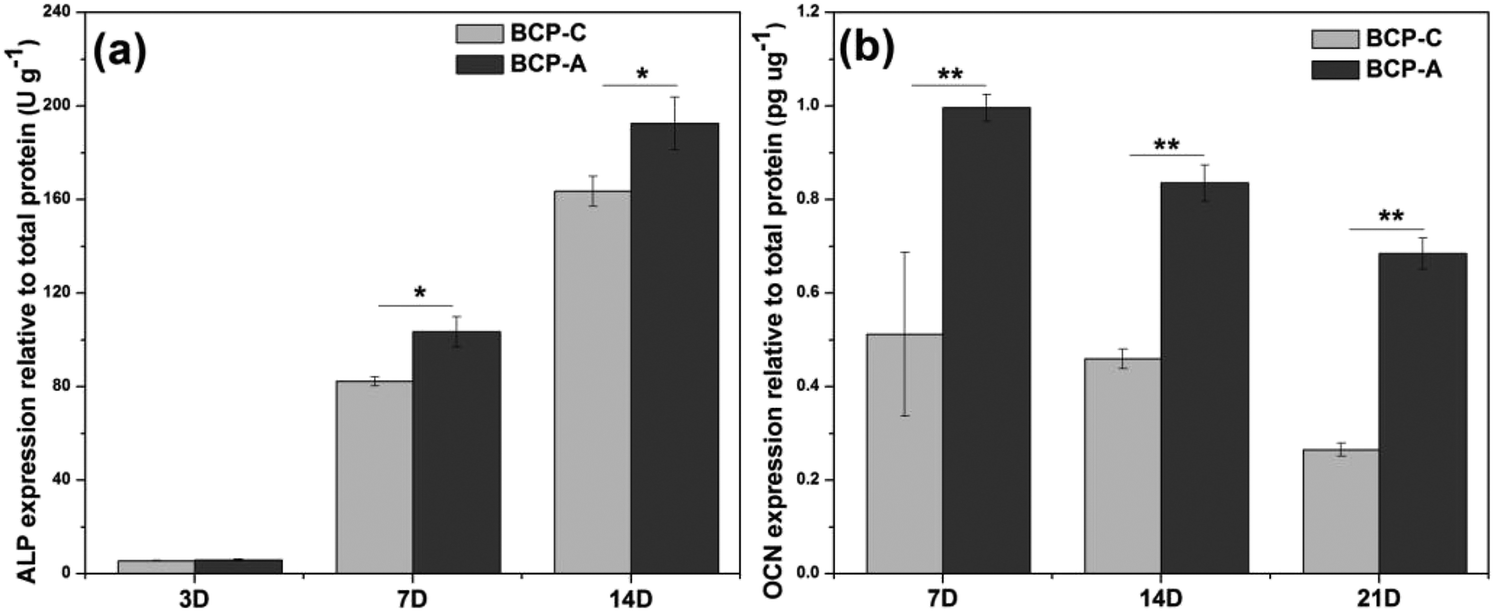 | ||
| Fig. 8 Intracellular ALP activity and OCN production in BMSCs cultured on BCP-C and BCP-A. Values are expressed as the mean ± SD (n = 3), * refers to p < 0.05, and ** refers to p < 0.01. | ||
3.3 In vivo osteoinductivity
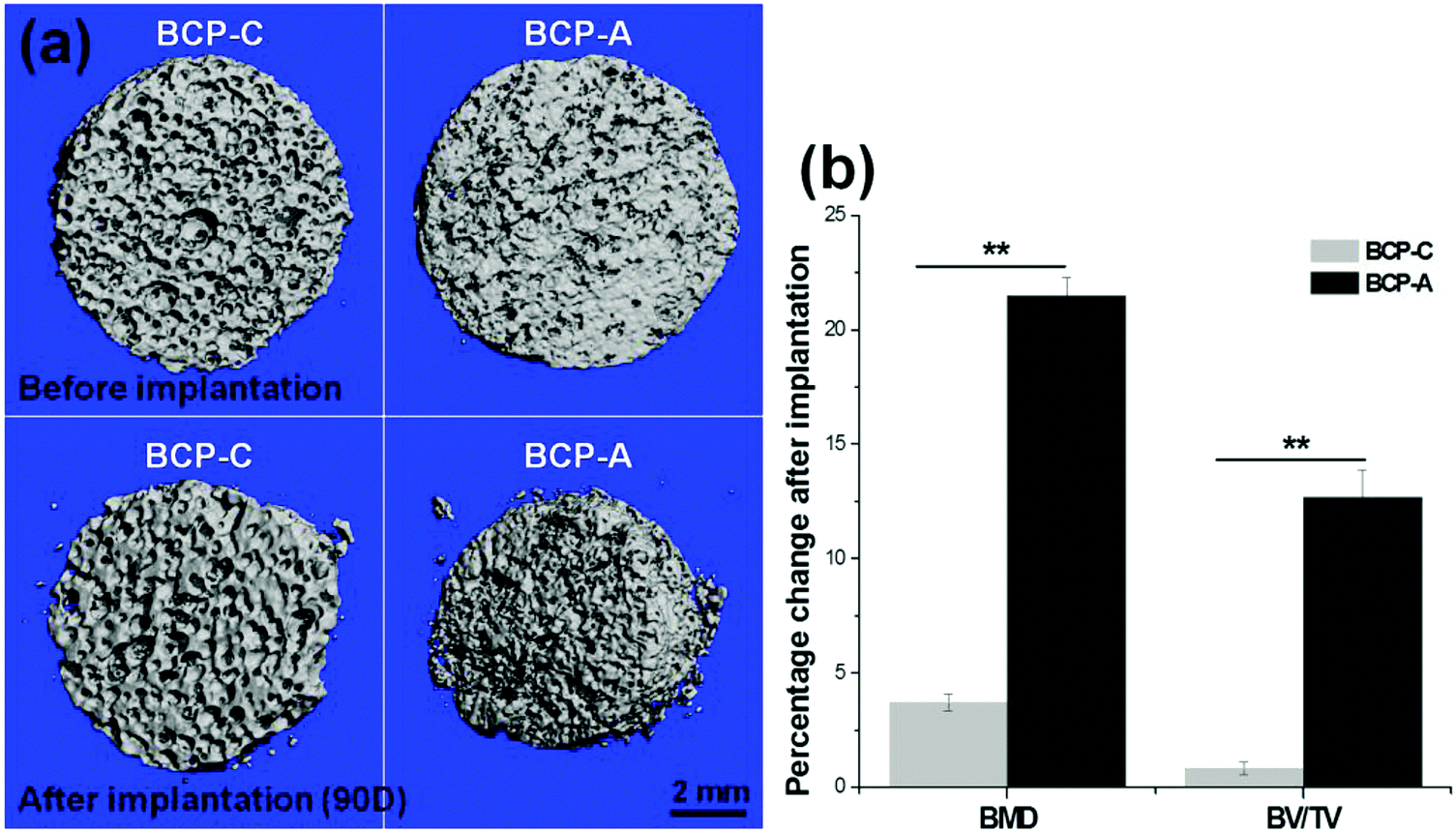 | ||
| Fig. 9 Images and parameters rendered by μ-CT: (a) 3D visualization of BCP-C and BCP-A before and after implantation for 90 days; (b) quantitative comparison of the changes in bone mineral density (BMD) and bone volume/total volume ratio (BV/TV). Values are expressed as the mean ± SD (n = 3), * refers to p < 0.05, and ** refers to p < 0.01. | ||
 | ||
| Fig. 10 Histological analysis of BCP-C and BCP-A implanted intramuscularly in dogs: (a) H&E staining of BCP-C and BCP-A after implantation for 45 and 90 days (M = material, Ft = fibrous tissue, V = new capillary vessel, B = new bone); (b) bone forming incidence; (c) bone area in inner pores. Values are expressed as the mean ± SD (n = 9), * refers to p < 0.05, and ** refers to p < 0.01. | ||
4. Discussions
With the development of regenerative medicine, traditional biomaterials are facing many challenges. A promising frontier is endowing biomaterials with biomimetic properties in order to gain the desirable biological functions and induce tissue regeneration by simulating the composition and structure of natural tissues.1,2,38 The discovery of the osteoinductive capacity of porous Ca–P ceramics has highlighted the potential to regenerate bone tissues by means of the bioactive scaffold itself without the addition of exogenous cells and molecules.5 However, the present Ca–P ceramics still have a lower bioactivity than natural bone. Thus, enhancing the biological properties of Ca–P ceramics seems to be important and urgent. One alternative is biomimicry. It is well known that CDHA is more similar to natural bone mineral in both chemical composition and crystal structure as compared with other synthetic Ca–P materials. Therefore, introducing a high content of CDHA in Ca–P ceramics may hold promise in improving their biological performances.Indeed, the initial precipitation was only composed of the CDHA phase during the wet-chemical synthetic process of BCP powder, and CDHA would decompose into HA and β-TCP phases within 600–700 °C according to reaction (1), which was reflected in the XRD patterns of BCP-C sintered at 600 °C and 700 °C (Fig. 1b). However, the decomposition of CDHA in BCP-A took place above 700 °C (Fig. 1a), demonstrating that the addition of the alginate in BCP-A might increase the decomposing temperature of CDHA. After the decomposition of CDHA, although both BCP-C and BCP-A contained the HA and β-TCP phases, the HA content in BCP-A (74.6–83.7%) was much higher than that in BCP-C (about 20%) as listed in Table 1. These findings indicated that the addition of alginate could increase the content of the HA phase in BCP ceramics.
During the sintering process of BCP-A, some calcium sources (i.e. CaO) could be decomposed from calcium alginate according to reaction (2). But no diffraction peak of CaO or other residues were detected in the XRD patterns of BCP-A (Fig. 1a), which might be because the content was too low to be detected or the residue had reacted with the ceramic body. Since the pH values of the solution immersed by BCP-A were nearly neutral (7.17) as listed in Table 2, it was demonstrated that the CaO residue might have reacted in the sintering process.
| Ca10−x(HPO4)x(PO4)6−x(OH)2−x → (1 − x)Ca10(PO4)6(OH)2 + 3xCa3(PO4)2 + xH2O | (1) |
| 4[(C6H7O6)2Ca]x + 13xO2 → 4xCaO + 24xCO2↑ + 14xH2O | (2) |
Based on the relatively higher content of the HA phase in BCP-A (83.7%) than BCP-C (19.2%), it could be inferred that CaO might react with the β-TCP phase to form the HA phase according to reaction (3) in BCP-A. However, the amount of CaO decomposed from calcium alginate was quite low. Even if the M and G units in sodium alginate all bind to Ca2+, the generated CaO could only cause about 35% increase in the HA content of BCP-A according to the theoretical calculation. The ICP-OES results further confirmed that the amount of CaO that entered into BCP-A was quite small, as the Ca/P ratioa of BCP-A (1.577) was slightly higher than that of BCP-C (1.534) as listed in Table 2. By calculating the lattice parameters of the HA phase in BCP-A (Table 1) from the XRD patterns (Fig. 1a), it could be found that the lattice parameters (i.e., a/b-axis, c-axis and cell volume) of the HA phase in BCP-A were higher than those of the standard HA references (090432), which is similar to those of CDHA.39 Therefore, it was presumed that CaO might react with the β-TCP phase to form CDHA through reaction (4) in BCP-A, and the addition of alginate might stabilize most of the CDHA or inhibit its decomposition (reaction (1)) in the sintering process of BCP-A.
| 3Ca3(PO4)2 + CaO + H2O → Ca10(PO4)6(OH)2 | (3) |
| 3Ca3(PO4)2 + (1 − x)CaO + (1 + x)H2O → Ca10−x(HPO4)x(PO4)6−x(OH)2−x | (4) |
The formation of CDHA may be also confirmed in the FT-IR spectra. Because there was no new functional group, the FT-IR spectrum (Fig. 2) of BCP-A was similar to those of BCP-C. But the bending vibration peak of the OH group at 630 cm−1, and the splitting degree of the bending vibration peaks of the PO43− group (v4, at about 550 and 600 cm−1) in BCP-A were obviously stronger than those in BCP-C (Fig. 3), which further proved the formation of the CDHA phase in BCP-A. The bending vibration peak (v4) shapes of the PO43− group in BCP-A were similar to those of HA ceramics, whereas the peak shapes in BCP-C were similar to those of β-TCP ceramics.40 In addition, the characteristic peak of HPO42− (875 cm−1) was not found in the FT-IR spectra of BCP-A, demonstrating that the CDHA in BCP-A might be mainly in the form of Ca10−x(PO4)6(OH)2−2x. In other words, the addition of alginate might stabilize the existence of CDHA in BCP-A by inhibiting the occurrence of reaction (1), and only the dehydration reaction of CDHA (reaction (5)) occurred in the sintering process.
| Ca10−x(HPO4)x(PO4)6−x(OH)2−x → Ca10−x(PO4)6(OH)2−2x + xH2O | (5) |
It is known that the porous structure plays an important role in determining the biological performances of BCP ceramics, as pores can favor the exchange of nutrient and accommodate the ingrowth of cells.41 The interconnected pore structure and relatively high porosity of both BCP-A and BCP-C were confirmed by the MIP results (Fig. 4a, b and Table 2). Furthermore, more abundant micropores (<10 μm) could be generated from the decomposition of alginate in BCP-A compared with BCP-C (Fig. 4b). The specific surface area (SSA) results showed that the SSA of BCP-A (Table 2) was also higher than that of BCP-C. Besides, the addition of alginate could also decrease the crystal (Table 1) and grain sizes (Fig. 5c and d) of BCP-A, which is consistent with our previous findings.28 The abundant micropores, high SSA and small grain size of BCP-A could contribute to the increase of surface roughness and active sites, which might exert beneficial effects on enhancing its biological performances.
Moreover, bioactive ions (e.g. Ca2+ and PO43−) released from the degradation of Ca–P ceramics also play an important role in the process of osteogenesis, as they generate a local ionic micro-environment to trigger the osteogenic differentiation of osteo-progenitors.7,11 In this study, it was found that the Ca2+ and PO43− ion releasing concentrations and the Ca/P ratiosb (Fig. 4c and d) of the Tris–HCl solution with immersed BCP-A were lower than those of BCP-C. This might be because the degradation of BCP ceramics was a dissolution and re-deposition process, and the re-deposition of apatite took place easier in BCP-A than BCP-C. During the degradation process, Ca2+ and PO43− released from CDHA could further redeposit as apatite, which might dramatically decrease the Ca/P ratiosb in the solution with the immersed BCP-A. In fact, the high content of CDHA in BCP-A might provide abundant crystal nucleation points for the deposition of apatite, which was also confirmed in the SBF immersing experiment.
SBF immersion, protein adsorption and cell culture were carried out to compare the biocompatibility and bioactivity of BCP-C and BCP-A. It is known that the ability of bone-like apatite formation has become a direct criterion to evaluate the bioactivity of implants in vitro.33 After immersion for 3 days (Fig. 5), the surface of BCP-A was totally covered with a layer of net-like crystals, indicating that BCP-A had better bone-like apatite forming abilities than BCP-C, which might be partly because BCP-A had more appropriate ion releasing concentrations and was more favorable for re-depositing apatite as compared with BCP-C. This result suggested that BCP-A might show better osteogenic potential than BCP-C.
Generally, it is believed that protein adsorption is the initial event occurring on the surface of biomaterials after implantation, and determines the following cell behaviors and tissue responses.42,43 Compared with BCP-C, BCP-A could adsorb more protein (listed in Table 2), which might be because BCP-A containing a high content of CDHA had a smaller grain size, more micropores and a higher specific surface area, for providing more active sites for protein adsorption than BCP-C.
Moreover, an excellent bioactive scaffold must be beneficial for cell adhesion and growth. CLSM observations (Fig. 6a) and CCK-8 results (Fig. 6c) demonstrated that obvious BMSC proliferation occurred on BCP-A and BCP-C, and with the prolongation of incubation time, BMSCs began to migrate into the porous structure and proliferated quickly. These findings indicated that both BCP-A and BCP-C showed good biocompatibility. However, the initial attachment of BMSCs onto BCP-A and BCP-C was different, as BMSCs grown on BCP-A had a significantly larger cell area than those on BCP-C (Fig. 6b), indicating that BCP-A might be more favorable for cell adhesion and spreading at an early stage. Moreover, BMSCs cultured on BCP-A also exhibited a more elongated and slender cell shape, which might favor their osteogenic differentiation, as previous findings suggested that cell elongation could increase actomyosin contractility to promote osteogenesis.44,45
Previous studies6,44–47 have confirmed that Ca–P ceramics could up-regulate the mRNA expression of various osteogenic markers in BMSCs, including Runx2, OSX, Col-I, ALP, OCN, OPN and so on. Among them, Runx2, OSX, Col-I and ALP are commonly considered as the early markers for pre-osteoblasts, whereas OCN and OPN are believed to be the late markers for mature osteoblasts.44,48–51 qRT-PCR analyses (Fig. 7) found that BCP-A could promote the expression of almost all osteogenic specific markers at day 3 and day 14 compared with BCP-C. Although the expression of Runx-2, OSX and OCN genes in BCP-C and BCP-A had no significant differences at day 7, BCP-A exhibited a significantly higher expression of ALP, Col-I and OPN than BCP-C. These results demonstrated that BCP-A containing a high content of CDHA could dramatically up-regulate the expression of osteogenic genes. Furthermore, BCP-A could significantly enhance the ALP activity and OCN production of BMSCs as compared with BCP-C (Fig. 8). This might be because that compared with BCP-C, BCP-A is more similar to natural bone mineral, and exhibited a significantly stronger binding capacity for serum protein (Table 2), which might provide a provisional matrix to promote cell differentiation via integrin-related signaling pathways.44 Moreover, a certain concentration of ions (i.e. Ca2+ and PO43−) released from BCP-A in the microenvironment might also induce the osteogenic differentiation of BMSCs via several G-protein coupled receptors (GPCRs) or ion channels in cell membranes.52 Therefore, these integrated factors (such as protein binding capacity and Ca2+/PO43− releasing ability) might partially explain our findings that osteogenic differentiation of BMSCs was enhanced by BCP-A compared with BCP-C.
The intramuscular implantation experiment suggested that both BCP-C and BCP-A could induce ectopic bone formation. The μ-CT quantitative measurement found that BCP-A had higher values of BMD and the BV/TV ratio than BCP-C (Fig. 9b). The histomorphometric analysis also showed that the area of newly formed bone in BCP-A was significantly higher than in BCP-C after 45- and 90-day implantation (Fig. 10c), and all of the BCP-A samples could induce ectopic bone formation at day 90 (Fig. 10b). And the in vivo degradation ability of BCP-A was stronger than that of BCP-C. Furthermore, new bone tissues in the stained sections of BCP-C specimens were relatively independent and showed a “doughnut-like” shape (Fig. 10a); but in those of BCP-A, new bone tissues not only grew along the pore walls, but also interconnected together to integrate into a large area of bone. These findings were also consistent with the results of in vitro studies like SBF immersion, serum protein adsorption, and BMSC osteogenic differentiation, suggesting that the high content of CDHA in BCP-A might be responsible for improving the osteoinductive potential of BCP ceramics. As the high content of CDHA made BCP-A more similar to natural bone mineral, BCP-A might be more easily recognized by the osteogenesis-related proteins and cells in comparison to BCP-C. In addition, the high content of CDHA in BCP-A might also build a suitable local ion micro-environment, which could be beneficial to the growth of cells and tissues into the porous ceramics. Although BCP-A exhibited a relatively low ion releasing ability in the in-vitro Tris–HCl immersing experiment (Fig. 4c and d), its local ion environment might be more favorable for osteogenesis than BCP-C. It has been proved that the appropriate ion microenvironment provided by Ca–P ceramics plays a critical role in osteoinductivity, as too high or too low ion concentrations are not conducive to the osteogenesis process.7,11 Furthermore, BCP-A with a high content of CDHA also had more micropores than BCP-C, and previous studies have also demonstrated that the micropores might correlate to their propensity to stimulate the osteogenic differentiation of stem cells in vitro and induce ectopic bone formation in vivo.4 To sum up, the existence of CDHA in BCP ceramics could enhance their biological performances, suggesting that BCP-A should have better potential as bone grafts for tissue regeneration compared with BCP-C.
The underlying mechanism on the effect of CDHA on the biological performances (esp. osteoinductivity) of BCP ceramics requires further study.
5. Conclusion
The present study introduced an effective approach to stabilize the CDHA in BCP ceramics by adding alginate, as during the sintering process, some residue (i.e. CaO) decomposing from alginate might react with the ceramic body to form abundant CDHA in the obtained BCP ceramics (BCP-A). Further in vitro studies and in vivo intramuscular implantation suggested that compared with the conventional ones (BCP-C), BCP-A with a high content of CDHA exhibited better bioactivity and stronger osteoinductivity. Overall, these findings demonstrate a promising strategy to improve the bioactivity of conventional BCP ceramics by introducing a high content of CDHA to mimic the bone mineral, and these novel BCP ceramics have good prospects for applications in the regenerative medicine of hard tissues.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFB0700804), the Sci & Tech Support Project of Sichuan Province (No. 2016SZ0009), and the National Science Foundation of China (31400819, 31670985).References
- A. Oryan, S. Alidadi, A. Moshiri and N. Maffulli, J. Orthop. Surg. Res., 2014, 9, 18–44 CrossRef PubMed.
- X. Yu, X. Tang, S. V. Gohil and C. T. Laurencin, Adv. Healthcare Mater., 2015, 4, 1268–1285 CrossRef CAS PubMed.
- J. J. Li, D. L. Kaplan and H. Zreiqat, J. Mater. Chem. B, 2014, 2, 7272–7306 RSC.
- H. Yuan, H. Fernandes, P. Habibovic, J. D. Boer, A. M. C. Barradas, A. D. Ruiter, W. R. Walsh, C. A. V. Blitterswijk and J. D. D. Bruijn, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13614–13619 CrossRef CAS PubMed.
- Y. Hong, H. Fan, B. Li, B. Guo, M. Liu and X. Zhang, Mater. Sci. Eng., R, 2010, 70, 225–242 CrossRef.
- X. Li, M. Wang, Y. Deng, X. Chen, Y. Xiao and X. Zhang, ACS Biomater. Sci. Eng., 2017, 3, 1557–1566 CrossRef CAS.
- Z. Tang, Y. Tan, Y. Ni, J. Wang, X. Zhu, Y. Fan, X. Chen, X. Yang and X. Zhang, Mater. Sci. Eng., C, 2017, 70, 1000–1010 CrossRef CAS PubMed.
- Y. Chen, Z. Sun, Y. Li and Y. Hong, J. Mater. Chem. B, 2017, 5, 807–816 RSC.
- J. Bouler, P. Pilet, O. Gauthier and E. Verron, Acta Biomater., 2017, 53, 1–12 CrossRef CAS PubMed.
- M. Ebrahimi, M. G. Botelho and S. V. Dorozhkin, Mater. Sci. Eng., C, 2017, 71, 1293–1312 CrossRef CAS PubMed.
- J. Wang, Y. Chen, X. Zhu, T. Yuan, Y. Tan, Y. Fan and X. Zhang, J. Biomed. Mater. Res., Part A, 2014, 102, 4234–4243 Search PubMed.
- Z. Tang, X. Li, Y. Tan, H. Fan and X. Zhang, Regener. Biomater., 2017, 1–17 Search PubMed.
- J. D. Pasteris, B. Wopenka and E. Valsami-Jones, Elements, 2008, 4, 97–104 CrossRef CAS.
- K. Ishikawa, P. Ducheyne and S. Radin, J. Mater. Sci.: Mater. Med., 1993, 4, 165–168 CrossRef CAS.
- S. V. Dorozhkin, Acta Biomater., 2010, 6, 715–734 CrossRef CAS PubMed.
- R. Biltz and E. Pellegrino, J. Dent. Res., 1983, 62, 1190–1195 CrossRef CAS PubMed.
- N. D. Ravi, R. Balu and T. S. Kumar, J. Am. Ceram. Soc., 2012, 95, 2700–2708 CrossRef CAS.
- S. Radin and P. Ducheyne, J. Biomed. Mater. Res., Part A, 1993, 27, 35–45 CrossRef CAS PubMed.
- P. Kasten, R. Luginbühl, M. Van Griensven, T. Barkhausen, C. Krettek, M. Bohner and U. Bosch, Biomaterials, 2003, 24, 2593–2603 CrossRef CAS PubMed.
- P. Kasten, J. Vogel, F. Geiger, P. Niemeyer, R. Luginbühl and K. Szalay, Biomaterials, 2008, 29, 3983–3992 CrossRef CAS PubMed.
- H. Guo, J. Su, J. Wei, H. Kong and C. Liu, Acta Biomater., 2009, 5, 268–278 CrossRef CAS PubMed.
- S. Raynaud, E. Champion, D. Bernache-Assollant and J. P. Laval, J. Am. Ceram. Soc., 2001, 84, 359–366 CrossRef CAS.
- A. Destainville, E. Champion, D. Bernache-Assollant and E. Laborde, Mater. Chem. Phys., 2003, 80, 269–277 CrossRef CAS.
- A. Y. P. Mateus, C. C. Barrias, C. Ribeiro, M. P. Ferraz and F. J. Monteiro, J. Biomed. Mater. Res., Part A, 2008, 86A, 483–493 CrossRef CAS PubMed.
- S. Kulanthaivel, S. R. Vs, T. Agarwal, S. Pradhan, K. Pal, S. Giri, T. K. Maiti and I. Banerjee, J. Mater. Chem. B, 2017, 5, 4177–4189 RSC.
- J. R. Lakkakula, T. Matshaya and R. W. Krause, Mater. Sci. Eng., C, 2017, 70, 169–177 CrossRef CAS PubMed.
- C. C. Ribeiro, C. C. Barrias and M. A. Barbosa, Biomaterials, 2004, 25, 4363–4373 CrossRef CAS PubMed.
- X. Li, Y. Deng, X. Chen, Y. Xiao, Y. Fan and X. Zhang, Mater. Lett., 2016, 185, 428–431 CrossRef CAS.
- J. Wang, D. Liu, B. Guo, X. Yang, X. Chen, X. Zhu, Y. Fan and X. Zhang, Acta Biomater., 2017, 51, 447–460 CrossRef CAS PubMed.
- X. Ye, C. Zhou, Z. Xiao, Y. Fan, X. Zhu, Y. Sun and X. Zhang, Mater. Lett., 2014, 128, 179–182 CrossRef CAS.
- M. Kohri, K. Miki, D. E. Waite, H. Nakajima and T. Okabe, Biomaterials, 1993, 14, 299–304 CrossRef CAS PubMed.
- B. Yuan, Y. Chen, H. Lin, Y. Song, X. Yang, H. Tang, E. Xie, T. Hsu, X. Yang and X. Zhu, ACS Biomater. Sci. Eng., 2016, 2, 977–986 CrossRef CAS.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- H. Y. Yeung, L. Qin, K. M. Lee, K. S. Leung and J. C. Y. Cheng, Advanced Bioimaging Technologies in Assessment of the Quality of Bone and Scaffold Materials, Springer, 2007, pp. 289–305 Search PubMed.
- S. Raynaud, E. Champion, D. Bernacheassollant and P. Thomas, Biomaterials, 2002, 23, 1065–1072 CrossRef CAS PubMed.
- J. P. Soares, J. E. Santos, G. O. Chierice and E. T. G. Cavalheiro, Eclética Química, 2004, 29, 57–64 CrossRef CAS.
- J. Zhang, Q. Ji, X. Shen, Y. Xia, L. Tan and Q. Kong, Polym. Degrad. Stab., 2011, 96, 936–942 CrossRef CAS.
- S. I. Stupp, Biomaterials and Regenerative Medicine, MRS Bulletin, 2014, 30, 546–553 CrossRef.
- R. Wilson, J. Elliott and S. Dowker, Am. Mineral., 1999, 84, 1406–1414 CrossRef CAS.
- X. Li, G. Bo, Y. Xiao, T. Yuan, Y. Fan and X. Zhang, J. Mater. Sci.: Mater. Med., 2016, 27, 1–10 Search PubMed.
- F. Bobbert and A. Zadpoor, J. Mater. Chem. B, 2017, 5, 6175–6192 RSC.
- H. Chen, C. Wang, X. Zhu, K. Zhang, Y. Fan and X. Zhang, Mater. Sci. Eng., C, 2014, 43, 182–188 CrossRef CAS PubMed.
- X. D. Zhu and H. Y. Fan, Acta Biomater., 2009, 5, 1311–1318 CrossRef CAS PubMed.
- X. Chen, J. Wang, Y. Chen, H. Cai, X. Yang, X. Zhu, Y. Fan and X. Zhang, J. Mater. Chem. B, 2016, 4, 2280–2289 RSC.
- X. Chen, X. Fu, J. G. Shi and H. Wang, Nanotechnol. Biol. Med., 2013, 9, 1283–1292 CrossRef CAS PubMed.
- Z. Tang, Z. Wang, F. Qing, Y. Ni, Y. Fan, Y. Tan and X. Zhang, J. Biomed. Mater. Res., Part A, 2015, 103, 1001–1010 CrossRef PubMed.
- Y. Chen, J. Wang, X. Zhu, Z. Tang, X. Yang, Y. Tan, Y. Fan and X. Zhang, Acta Biomater., 2015, 11, 435–448 CrossRef PubMed.
- T. Komori, Cell Tissue Res., 2010, 339, 189–195 CrossRef CAS PubMed.
- K. Nakashima, X. Zhou, G. Kunkel, Z. Zhang, J. M. Deng, R. R. Behringer and B. de Crombrugghe, Cell, 2002, 108, 17–29 CrossRef CAS PubMed.
- J. M. Oliveira, M. T. Rodrigues, S. S. Silva, P. B. Malafaya, M. E. Gomes, C. A. Viegas, I. R. Dias, J. T. Azevedo, J. F. Mano and R. L. Reis, Biomaterials, 2006, 27, 6123–6137 CrossRef CAS PubMed.
- F. Long, Nat. Rev. Mol. Cell Biol., 2012, 13, 27–38 CrossRef CAS PubMed.
- A. M. Barradas, H. A. Fernandes, N. Groen, Y. C. Chai, J. Schrooten, J. van de Peppel, J. P. van Leeuwen, C. A. van Blitterswijk and J. de Boer, Biomaterials, 2012, 33, 3205–3215 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb02620j |
| This journal is © The Royal Society of Chemistry 2018 |