
Development of demineralized bone matrix-based implantable and biomimetic microcarrier for stem cell expansion and single-step tissue-engineered bone graft construction†
Zhenxing
Wang‡
ab,
Dingyu
Wu‡
ab,
Jiwei
Zou‡
c,
Quan
Zhou
d,
Wei
Liu
ab,
Wenjie
Zhang
ab,
Guangdong
Zhou
ab,
Xiansong
Wang
ab,
Guoxian
Pei
*c,
Yilin
Cao
*ab and
Zhi-Yong
Zhang
*abef
aDepartment of Plastic and Reconstructive Surgery, Shanghai 9th People's Hospital, Shanghai Key Laboratory of Tissue Engineering, School of Medicine, Shanghai Jiao Tong University, Shanghai 200011, China. E-mail: mr.zhiyong@gmail.com; Fax: +86-021-34292305; Tel: +86-21-34291002
bNational Tissue Engineering Center of China, Shanghai 200241, China. E-mail: yilincao@yahoo.com; Fax: +86-021-34292305; Tel: +86-21-34291002
cDepartment of Orthopedics, Xijing Hospital, The Fourth Military Medical University, Xi’an 710032, China. E-mail: nfperry@163.com; Fax: +86-29-84773524; Tel: +86-29-84773524
dHunan Cancer Hospital, Hunan 410000, China
eChina Orthopedic Regenerative Medicine Group (CORMed), Hangzhou, Zhejiang 310058, China
fTranslational Research Center for Regenerative Medicine and 3D Printing Technologies, The Third Affiliated Hospital of Guangzhou Medical University, Guangzhou, 510150, China
First published on 28th October 2016
Abstract
Tissue engineered bone grafts (TEBG) using mesenchymal stem cells (MSCs) demonstrate great potential for bone defect treatment. However, current MSC expansion techniques and multiple-step TEBG construction strategy have problems such as repeated trypsinization, limiting further clinical application. Microcarriers present promising solutions, but conventional microcarriers are either non-implantable or have insufficient biomimetic potential to maintain effective cellular function. Here, we developed a biomimetic and implantable microcarrier using demineralized bone matrix (DBM-MC), which preserves the essential biochemical composition, architecture and surface topography of natural bone tissue. Furthermore, based on this DBM-MC, we established a single-step micro-sized TEBG (μTEBG) construction strategy integrating multiple procedures of cell seeding, expansion, and differentiation. Benchmarked with Cytodex 3, a widely used microcarrier, DBM-MC shared similar physical properties, and supported efficient cell adhesion and proliferation with MSC characteristics being well maintained. However, when implanted ectopically, the MSC/DBM-MC constructs achieved more neo-bone formation with better vascularization than MSC/Cytodex 3. Moreover, μTEBG generated via our single-step strategy can successfully heal a critical-sized cranial defect with two-fold more bone regeneration. This new DBM-MC and single-step μTEBG construction strategy can provide an enclosed, large-scale, reduced-trypsinization, and semi-automatic fabrication process to generate μTEBGs with outstanding osteogenic and angiogenic capacity, demonstrating great potential for clinical application.
Introduction
Large bone defects and fractures are major clinical burdens, representing a strong clinical need for effective bone grafts. However, the clinical use of traditional bone grafts such as autografts, allografts, and synthetic grafts is generally limited by their inherent disadvantages, including limited availability, compromised healing efficacy, and incapability for remodeling.1 Tissue engineering strategies have been considered as some of the most promising alternative approaches to address this ever-increasing clinical need by developing tissue engineered bone grafts (TEBGs), which can promote bone regeneration and remodeling and be available unlimitedly.2Cells play an essential role in constructing effective TEBGs by promoting bone tissue regeneration through the paracrine release of osteoinductive factors and/or the direct contributions to bone tissue formation via cellular differentiation into osteogenic cells.3 Of the currently available cell sources, mesenchymal stem cells (MSCs) are frequently used for bone tissue engineering (BTE) applications, due to their favorable characteristics such as in vitro expansion capacity, low or no immunogenicity, robust osteogenic capacity, and lack of tumorigenicity.4–6 However, some of the major hurdles limiting the clinical application of MSC-based BTE strategy are that the current MSC cellular expansion technique and the multiple-step TEBG construction strategy are rather complicated, labor-intensive and less cost-effective and may adversely affect the osteogenic potential of TEBG.7 Firstly, the clinical use of MSCs typically requires a very large cell number; and the conventional petri dish-based cell expansion method involves repeated trypsinization during cellular subpassaging, which continuously destroys the secreted extracellular matrix (ECM), leading to rapid cellular aging and declining osteogenesis.8 Secondly, the current TEBG construction strategy includes multiple operation steps such as cell expansion, cell/scaffold seeding, osteogenic priming; these procedures are generally separated from each other and are rather complicated and labor-intensive; thus they are prone to operators’ variation and risk of contamination.8
Recently, microcarrier culture systems, especially commercial microcarriers (such as Cytodex 3), have attracted increasing interest for their potential application to increase the expansion efficiency of MSCs.8–10 Microcarriers can greatly enlarge the surface area for cell proliferation and harness the advantages of the suspension culture, facilitating an easy scale-up and more cost-effective procedure with less trypsinization intervention, as compared to the petri dish culture methods.11 Nevertheless, the currently available commercial microcarriers were not originally designed for tissue engineering application and are usually made of non-degradable materials with poor osteoconductivity and biocompatibility, thus being unsuitable for in vivo implantation.12 As a result, the use of these non-implantable microcarriers requires a prolonged trypsinization procedure to retrieve MSCs from the microcarriers before in vivo application, and this may compromise the yield, viability and functionality of MSCs for BTE application, as demonstrated in our previous studies.8 Recently, studies have been reported of the successful development of biodegradable microcarriers for cell expansion, using materials such as PLA,13 gelatin,14 PLGA,15 chitosan,16 PLLA;17 however, these biodegradable microcarriers are generally made of single-component material and could not ideally provide the favorable biomimetic matrix microenvironment for effective cell growth and function, due to the lack of the essential biochemical composition, architecture, and surface topography of natural bone tissue. Furthermore, most of those studies only focused on the use of these microcarriers for stem cell expansion, whereas the potential use of the microcarriers as tissue engineering scaffolds to construct tissue engineered micro-grafts was rarely investigated.13–15,17,18
To overcome these drawbacks, in this study, we successfully developed a biomimetic and implantable microcarrier, which is made of demineralized bone matrix (DBM), especially for BTE application. DBM material (derived from natural bone tissue) was purposefully selected, because it can preserve the essential biochemical composition, architecture and surface topography of natural bone tissue.19,20 In addition, DBM has been widely used in clinics for different orthopedic defect treatments, due to its favorable characteristics in terms of biocompatibility, osteoconductivity and biodegradability, which can facilitate the regulatory approval and clinical translation of the DBM microcarrier (DBM-MC).21,22 Moreover, based on this DBM-MC, we further established an integrated construction technique of TEBG to simplify the current complicated, multiple-step fabrication procedures (Fig. 1). In particular, MSCs were seeded and expanded dynamically onto the DBM-MCs in a stirring bioreactor, followed by osteogenic priming via media change from growth media to differentiation media (Fig. 1). With this integrated and continuously dynamic culture procedure, MSCs can rapidly expand, robustly differentiate and deposit ECM onto the DBM-MCs, formulating micro-sized TEBGs (μTEBGs) with DBM-MC cores as scaffolds (Animation 1, ESI†).
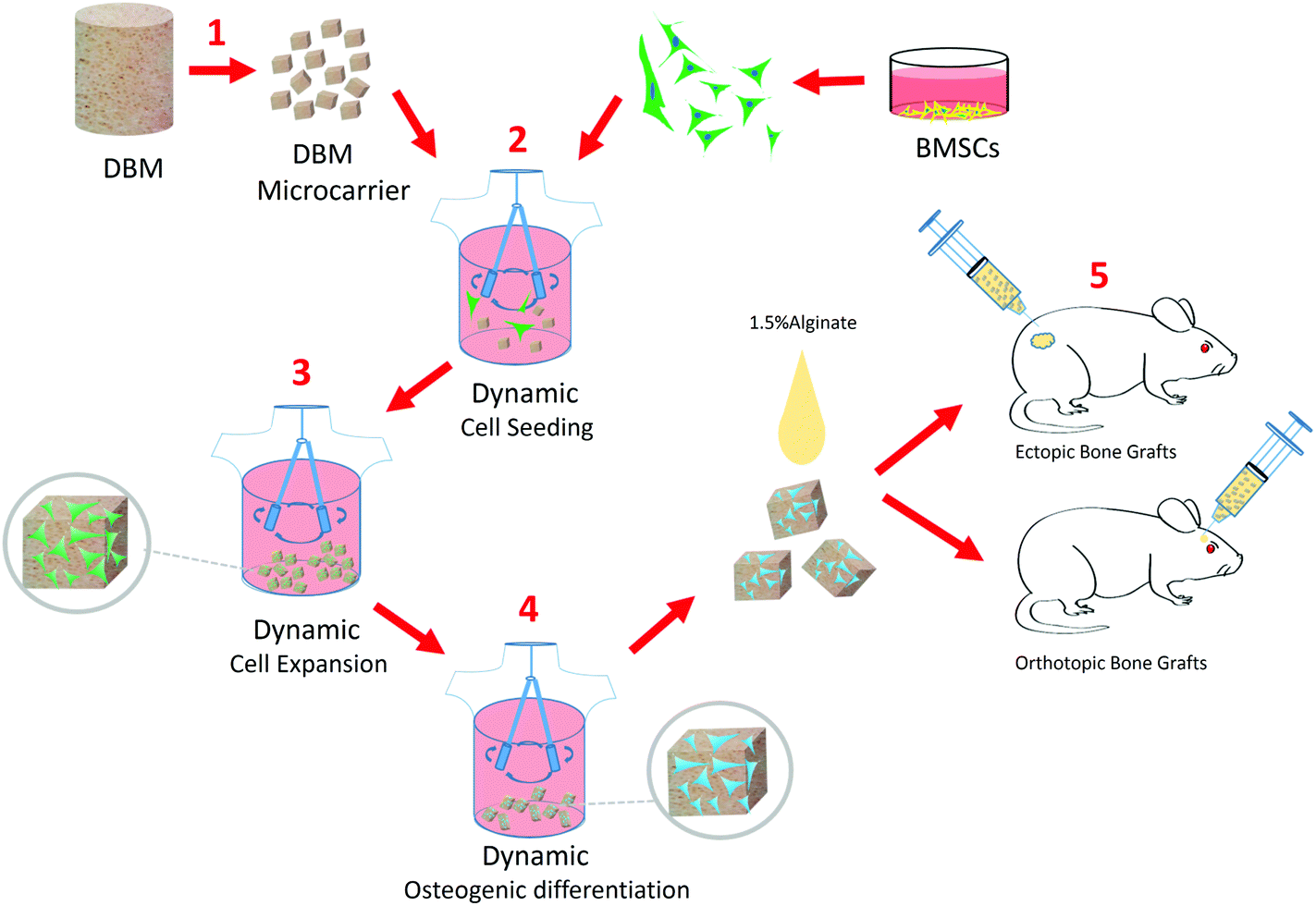 | ||
| Fig. 1 The experimental design showing the DBM-MC integrated dynamic μTEBG fabrication system for injectable bone tissue engineering applications. (1) The DBM-MCs were fabricated from large blocks of DBM via pulverization, sieving, and lyophilization. (2) MSCs were dynamically seeded onto the DBM-MCs and (3) rapidly expanded on the DBM-MC constructs with growth media under continuous stirring. (4) The MSC/DBM-MC constructs were primed into μTEBGs using osteogenic differentiation media. (5) The μTEBGs were harvested, suspended in alginate hydrogel, and then subcutaneously injected to evaluate ectopic bone formation or injected directly into cranial defects to study orthotopic bone formation in a rat model. | ||
In the present study, firstly, we developed and characterized the DBM-MC, benchmarking with Cytodex 3, a widely used commercial microcarrier for MSC expansion.8 Then, we systematically compared the DBM-MC with Cytodex 3 in terms of MSC expansion (MSC immunophenotype, proliferation and differentiation potential) as well as for μTEBG construction (in vivo ectopic bone formation capacity). Finally, the proof-of-concept testing of this integrated μTEBG construction system for large bone defect treatment was conducted in a critical-sized cranial defect model.
Results
Physical characterization of the DBM-MCs
The fabricated DBM-MCs and Cytodex 3 are shown in 10 cm culture dishes in Fig. 2B and E. Under phase contrast microscopy, the DBM-MCs appeared irregularly shaped, while the Cytodex 3 had a spherical appearance (Fig. 2C and F). The two types of carriers shared similar density (DBM: 1.06 ± 0.05 g mL−1; Cytodex 3: 1.04 ± 0.02 g mL−1; Fig. 2H). However, Cytodex 3 has a much higher approximate number per gram (dry weight) than DBM-MC (DBM: 1.45 ± 0.55 × 106 g−1; Cytodex 3: 2.51 ± 0.18 × 106 g−1; Fig. 2I).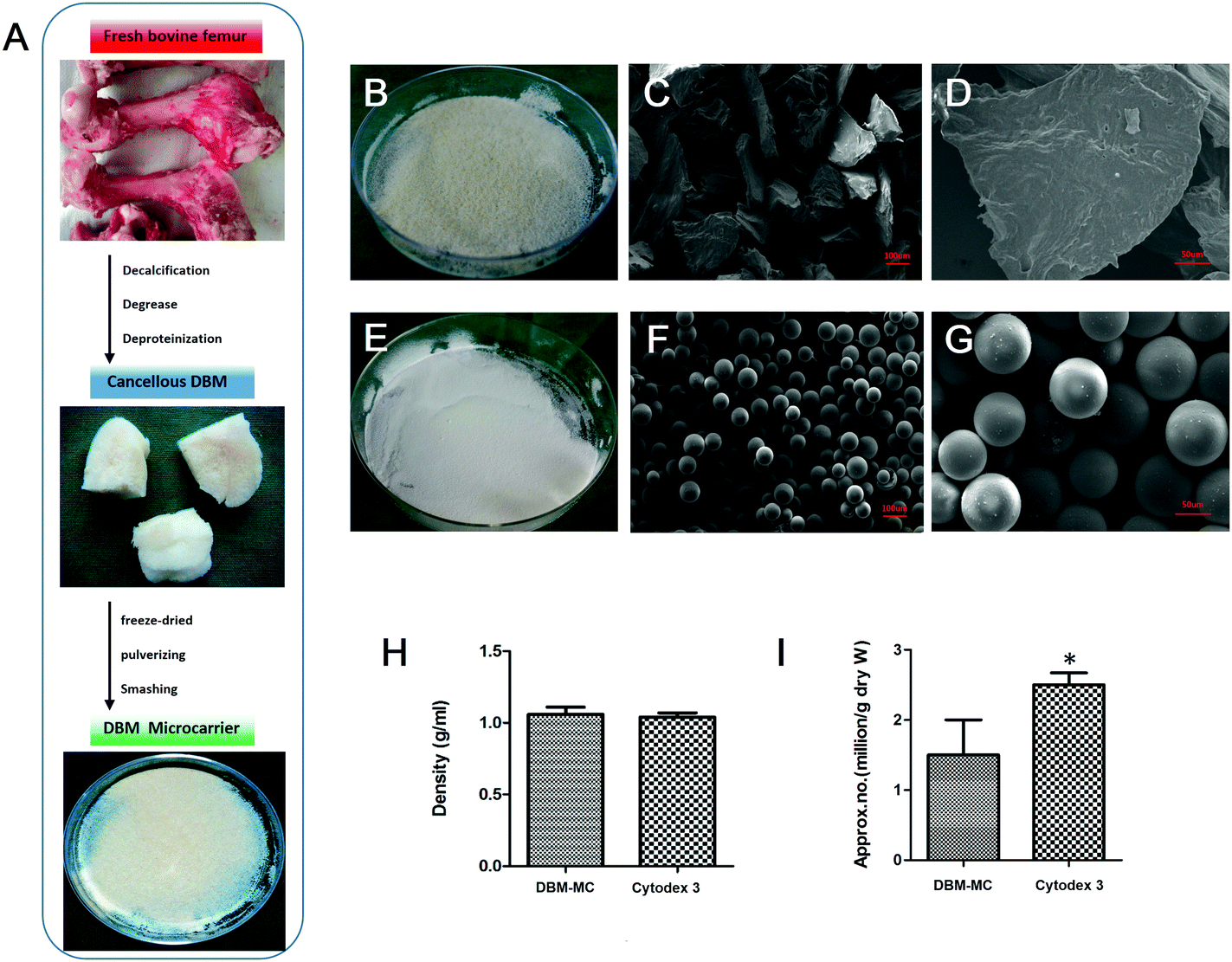 | ||
| Fig. 2 Fabrication and characterization of the DBM-MCs. (A) In the fabrication process, fresh bovine femurs were harvested, lyophilized, pulverized, and sieved to obtain DBM-MCs. Macroscopic views of (B) the DBM-MCs and (E) Cytodex 3. Cytodex 3 appeared spherical (F and G), while the DBM-MCs appeared irregularly shaped (C and D) by SEM. The densities per gram (H) of the DBM-MCs and Cytodex 3 were similar, while Cytodex 3 showed higher approximate particle number per gram than DBM-MC (I). (Scale bars in C, F: 100 μm; D, G: 50 μm; *p < 0.05.) | ||
Dynamic seeding and expansion efficiency of MSCs
The MSCs were dynamically seeded and expanded upon DBM-MCs or Cytodex 3 in two different stirring bioreactors (Fig. 3A). The efficiency of the dynamic MSC seeding was assessed at 24 hours by counting the number of unattached cells in the culture medium. The dynamic seeding efficiencies of the two carriers were similar (DBM: 73 ± 8%; Cytodex 3: 78 ± 5%; Fig. 3B). Quantification of dsDNA was used to evaluate the proliferation and expansion efficiency of MSCs on the microcarriers. The dsDNA content of the Cytodex 3 specimens began to increase quickly at day 3, while the dsDNA content of the DBM-MCs began to increase at day 5. At day 11, the DNA content of the Cytodex 3 sample was 6-fold larger than had been seeded, while the content of the DBM-MCs was 4-fold larger (Fig. 3C). Scanning electron microscopy (SEM) images showed that the acellular DBM-MCs had a rough surface, while the Cytodex 3 had a smoother surface and more uniform size distribution (Fig. 3D and G). The MSCs adhered well on both microcarriers, secreting abundant ECM upon the carriers (white arrow, Fig. 3E, F, H and I).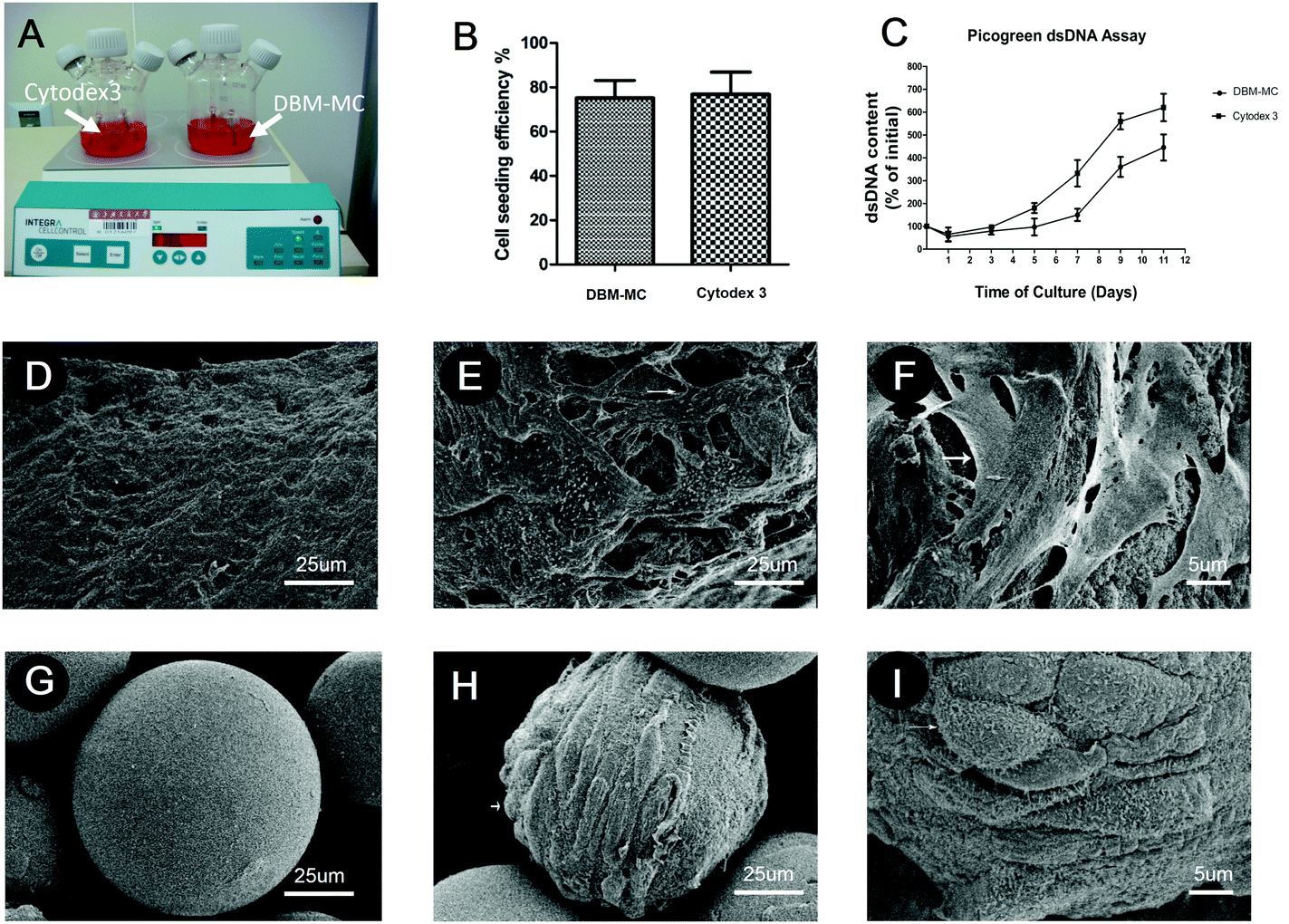 | ||
| Fig. 3 The MSCs adhered to and expanded on the microcarriers in the dynamic stirring bioreactor system. (A) The MSCs were dynamically seeded onto the DBM-MCs and Cytodex 3, and then expanded for 14 days in the stirring bioreactor. (B) The seeding efficiencies of the DBM-MCs and Cytodex 3 were comparable. (C) Despite a relatively slower rate of expansion compared with Cytodex 3, the MSCs expanded 4-fold on the DBM-MCs, as demonstrated by dsDNA content. A robust cellular adhesion and extracellular matrix deposition after 14 days of dynamic expansion were observed in both (D–F) the DBM-MCs and (G–I) the Cytodex 3, demonstrating successful cell seeding and proliferation. (*p < 0.05.) | ||
Characteristics of the MSCs before and after expansion on the microcarriers
MSCs harvested from the DBM-MCs and Cytodex 3 were cultured in 2D monolayers and assessed for their CFU, growth curve, morphology, and surface markers after expansion. No significant difference was observed in MSCs before and after extensive expansion on both microcarriers. The CFU of the MSCs from the DBM carriers (43 ± 10%) and from Cytodex 3 (50 ± 4%) were similar (Fig. 4E–G and H). In addition, the MSCs from the DBM-MCs appeared spindle-like (Fig. 4A–C) and showed a similar growth curve to those from the Cytodex 3 (Fig. 4D).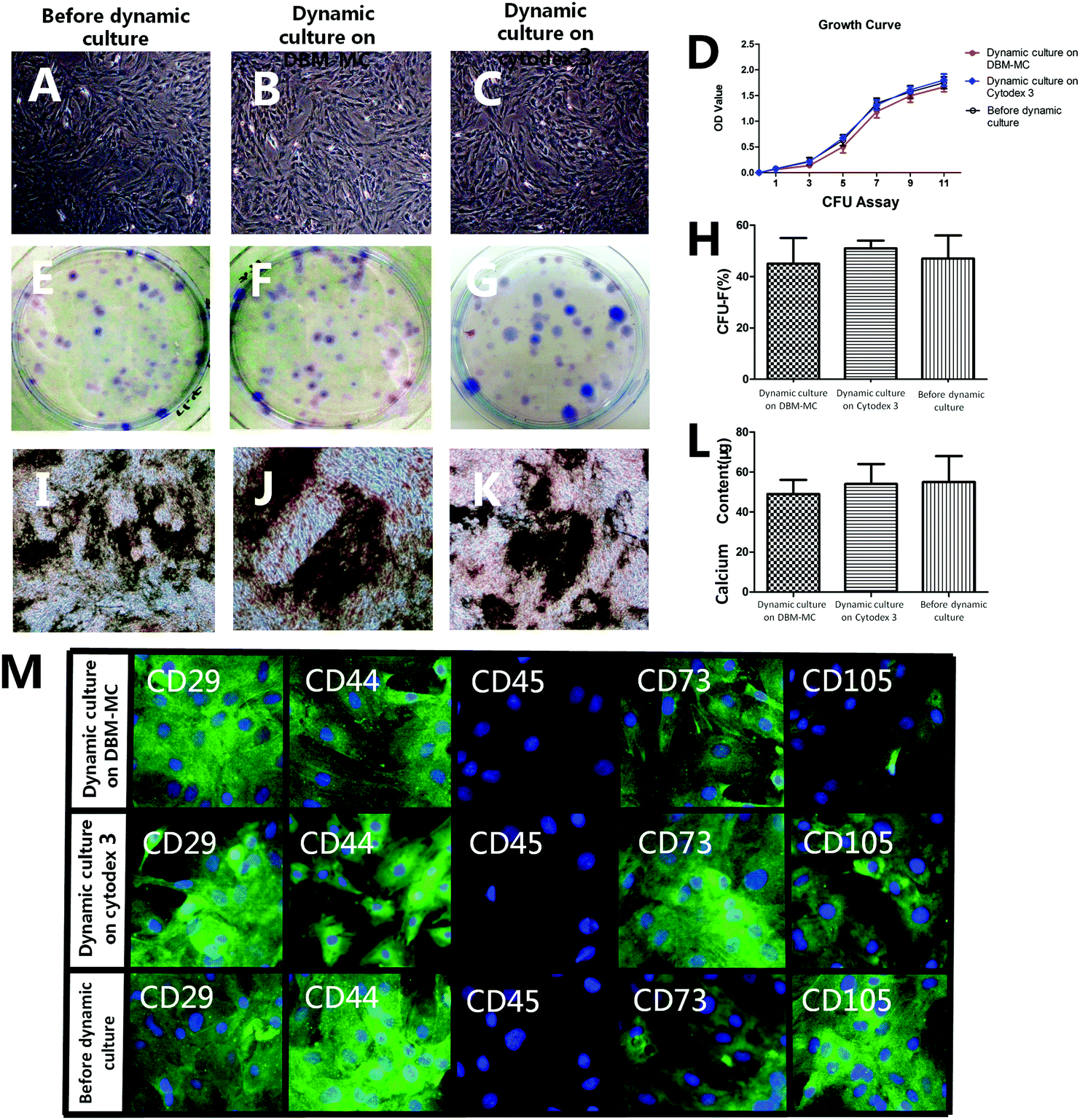 | ||
| Fig. 4 Characterization of the MSCs before and after microcarrier dynamic expansion. No significant differences were found among the MSCs before expansion and those isolated from the DMB microcarrier and Cytodex 3 after 14 days of expansion in terms of cell morphology (A–C), proliferation rate (D), self-renewal capacity (E–H), or osteogenic potential via von Kossa staining (I–K) and calcium deposition calculation (L). (M) Immunocytochemistry demonstrated similar immunophenotypes of the MSCs before and after dynamic expansion: positive for MSC markers (CD73, CD105) and adhesion molecule markers (CD29, CD44), but negative for hematopoietic marker (CD45). (Scale bars in D, E, G, H: 25 μm; F, I: 5 μm.) | ||
When exposed to osteogenic medium for 14 days, distinct calcium deposits were found by von Kossa staining in both groups (Fig. 4I–K). Furthermore, similar levels of calcium were quantified in the media (DBM: 41 ± 7 μg; Cytodex 3: 50 ± 9 μg; Fig. 4L). Immunocytochemistry staining showed that both groups were negative for hematopoietic marker (CD45), but positive for MSC markers (CD105, CD73) and cell adhesion molecule markers (CD29, CD44) (Fig. 4M).
Ectopic bone formation of MSC-mediated microcarrier constructs
Both types of microcarriers were compared head-to-head for their efficacy in generating μTEBGs. After dynamic expansion and osteogenic priming, MSC-mediated microcarrier constructs (μTEBGs) were implanted subcutaneously with alginate hydrogel in rat to evaluate their in vivo bone formation capacity. Macroscopically, the ectopically formed bone tissue appeared to be larger in the MSC-mediated DBM-MC group than in the Cytodex 3 group at 1 and 3 months (Fig. 5A–D). Micro-CT images confirmed that more calcification tissue was formed in the MSC/DBM-MC group than in the MSC/Cytodex 3 group (Fig. 5E–H) with significantly nearly two-fold higher bone volume and three-fold greater BV/TV (bone volume/tissue volume) ratio in the DBM group than in the Cytodex 3 group (Fig. 5I and J; *p < 0.05).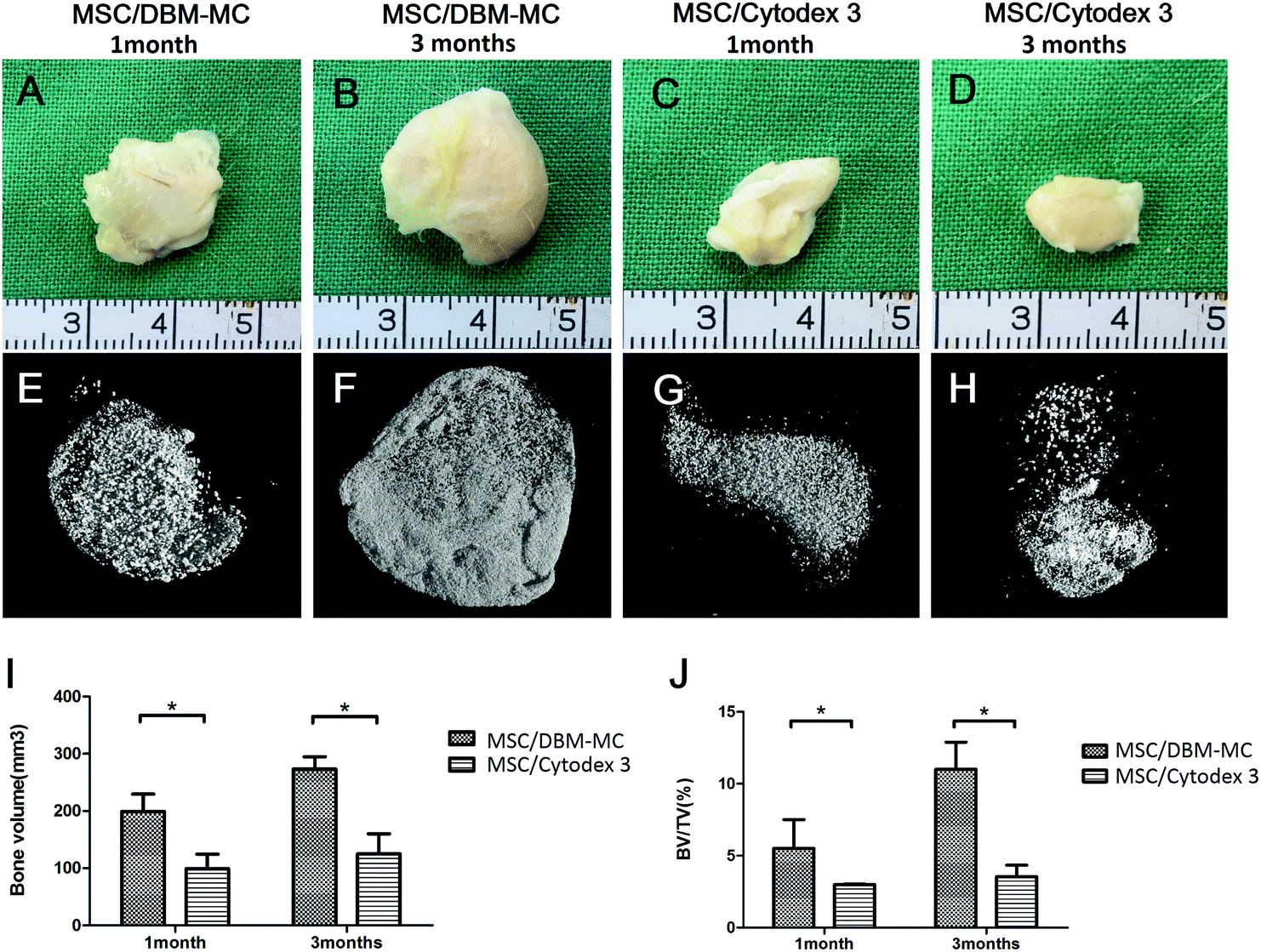 | ||
| Fig. 5 The evaluation of in vivo osteogenic potential of MSC-microcarrier constructs by an ectopic bone formation model. Macroscopic views of the MSC-microcarrier constructs subcutaneously implanted in rats showed that more ectopic bone formation occurred in the MSC/DBM-MC groups at (A) 1 and (B) 3 months than in the MSC/Cytodex 3 groups at (C) 1 and (D) 3 months. 3D reconstructions of the micro-CT data supported the finding that more mineralization occurred in the MSC/DBM-MC group at (E) 1 and (F) 3 months than in the MSC/Cytodex 3 group at (G) 1 and (H) 3 months. Quantitative analysis of the micro-CT data showed more bone volume (I) and a larger bone volume/tissue volume ratio (J) in the MSC/DBM-MC group than in the Cytodex 3 group at both 1 and 3 months. (*p < 0.05.) | ||
Histological analyses of hematoxylin and eosin (H&E) staining and Masson's trichrome staining further corroborate the macroscopic and micro-CT findings. At 3 months, a large amount of regenerated bone tissue was found in MSC-mediated DBM-MCs, in which dense bone tissue was homogenously distributed among the DBM-MCs throughout the whole of the implants (Fig. 6A, B, E and F) and good neo-vascularization was observed surrounding the DBM-MCs (Fig. 6B, yellow arrows), indicating the great angiogenic potential of MSC/DBM constructs. On the contrary, empty cavities were observed in the MSC-mediated Cytodex 3 constructs, with scarce new bone formation and a poor vascular network, implying an occurrence of core necrosis after implantation (Fig. 6C, D, G and H).
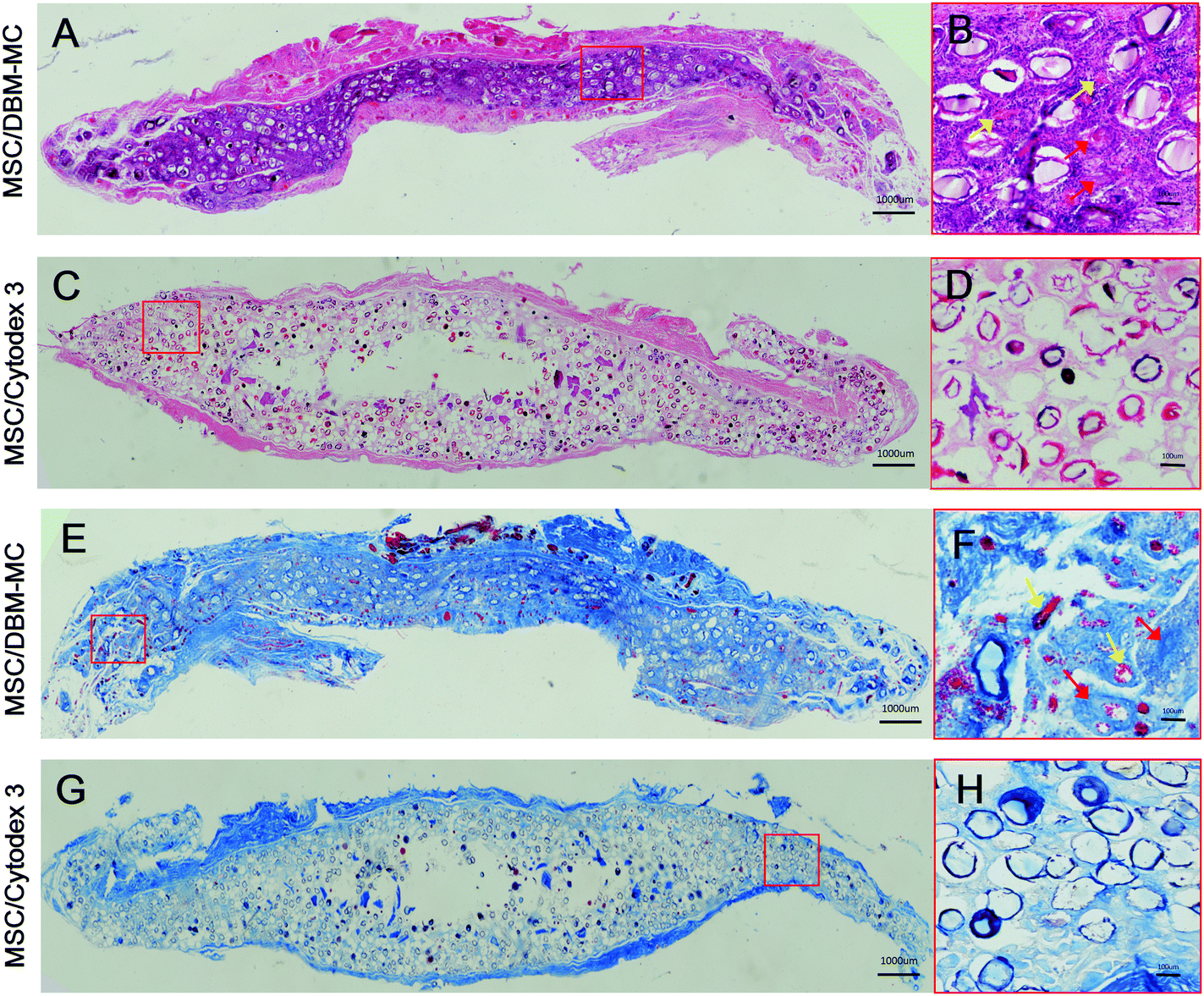 | ||
| Fig. 6 Histological analysis of the ectopic bone formation in the MSC-microcarrier constructs after 3 months’ implantation. H&E staining of the (A and B) DBM-MC and (C and D) Cytodex 3 groups. Masson's Trichrome staining of the (E and F) DBM-MC and (G and H) Cytodex 3 groups. In the MSC/DBM-MC group, much more and denser bone tissue (red arrow) was formed and homogenously distributed through the whole implants (A, B, E and F), whereas the MSC/Cytodex 3 group experienced very scarce new bone formation with empty cavities in the core region (C, D, G and H). Furthermore, good neo-vascularization was observed surrounding the DBM-MCs (B, H, yellow arrow), while poor vascular network was found in MSC/Cytodex 3 group, implying core necrosis occurring after implantation. Yellow arrow: neo-vessels. (Scale bars in A, C, E, G: 1000 μm; B, D, F, H: 100 μm.) | ||
Use of the integrated TEBG construction system for critical-sized bone defect treatment
μTEBGs were formulated using the DBM-MC-based integrated construction system and injected with alginate hydrogel into 5 mm critical-sized cranial defects (Fig. 7A) to investigate their therapeutic efficacy for large bone defect treatment. The macroscopic view of the defects after 3 months’ treatment showed that the defects in the μTEBGs group were fully covered by dense new bone tissue (Fig. 7B, indicated by the red arrow), whereas for the other three groups defects were partially filled by fibrous tissue with a semi-transparent appearance (Fig. 7B, blue arrow). Micro-CT analysis supported the macroscopic observation result that there was a much larger volume of bone tissue in the μTEBGs group at 1 and 3 months (Fig. 7C), with more than two-fold higher BV and three-fold greater BV/TV ratio than in the other groups (p < 0.05) (Fig. 7D and E) and achieving almost complete healing of the large defects at 3 months (Fig. 7C). H&E staining of the cranial sections further demonstrated that the defects in the μTEBGs group achieved full bridging and became filled up with dense bone tissue, whereas the untreated group (blank), alginate gel group (implantation of gel alone) and DBM-MC group (implantation with DBM-MC and gel alone) all had a compromised effect in defect healing, with defects partially or completely occupied by fibrous tissue (Fig. 8). Some remaining DBM carriers were found in DBM-MC groups (Fig. 8, red arrow)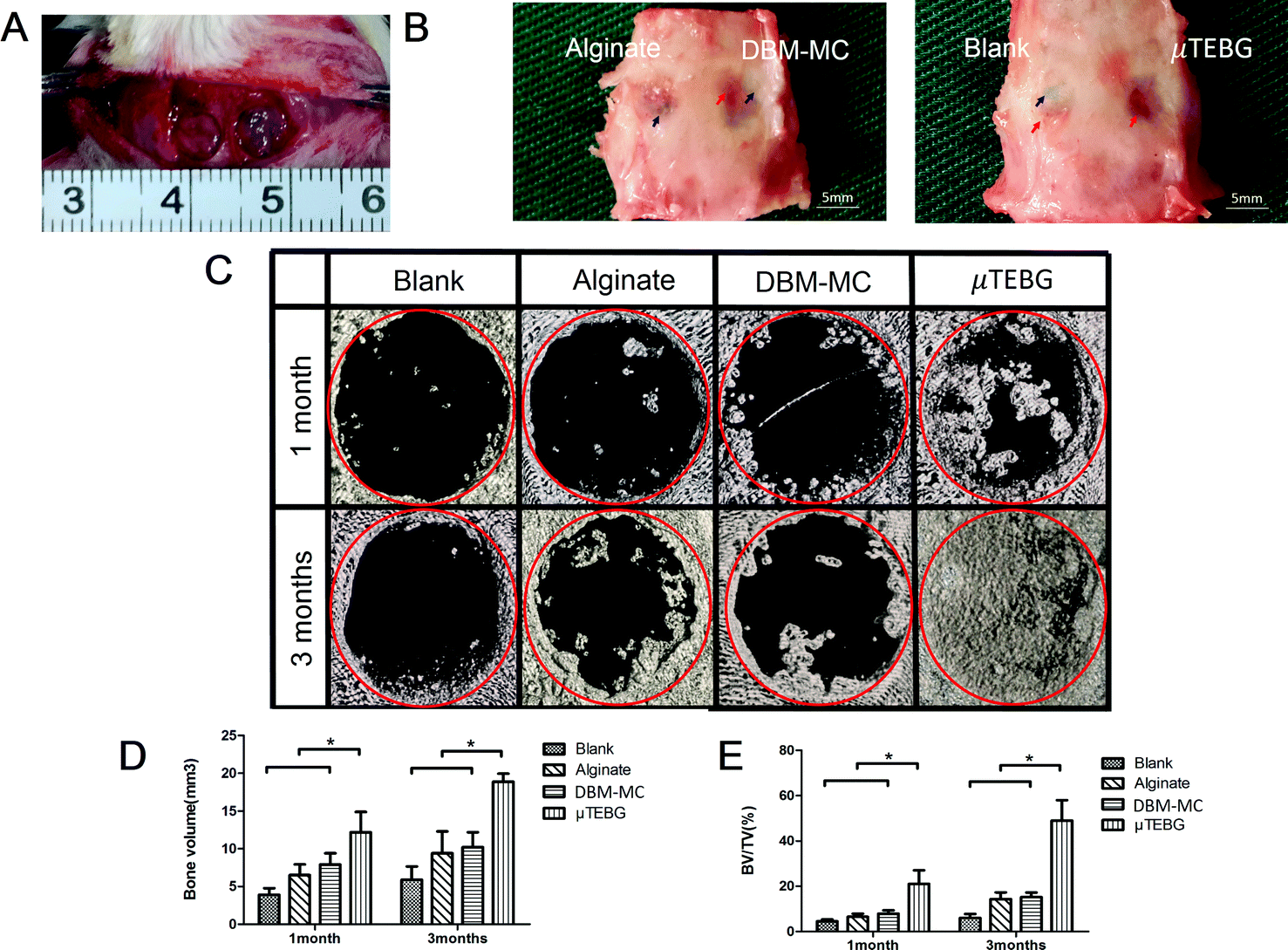 | ||
| Fig. 7 Functional testing of the integrated DBM-MC-based μTEBG construction system for large defect treatment in a rat model. (A) The surgical procedure included creation of two 5 mm critical-sized cranial defects in each rat before injection of μTEBGs (MSC/DBM-MC in alginate), acellular DBM-MC in alginate, or alginate alone. (B) Macroscopic views of the rat cranial defects 3 months after treatment: the defects in the μTEBG group were fully covered by dense new bone tissue (red arrow), whereas in the other three groups defects were partially filled by fibrous tissue with semi-transparent appearance (blue arrow). Red circle: defect area. Blue arrows: areas of non-repair. Red arrows: new bone formation. (C) Micro-CT 3D reconstruction analysis of the defect areas showed an almost complete healing of the large defects at 3 months in μTEBG group; however, the other groups experienced very limited healing. Red circle: surgical defect. Quantitative analyses of (D) the BV and (E) BV/TV ratio demonstrated more than two-fold higher BV and three-fold greater BV/TV ratio in μTEBG group than in the other groups (p < 0.05). (Scale bars in B: 1 mm; *p < 0.05.) | ||
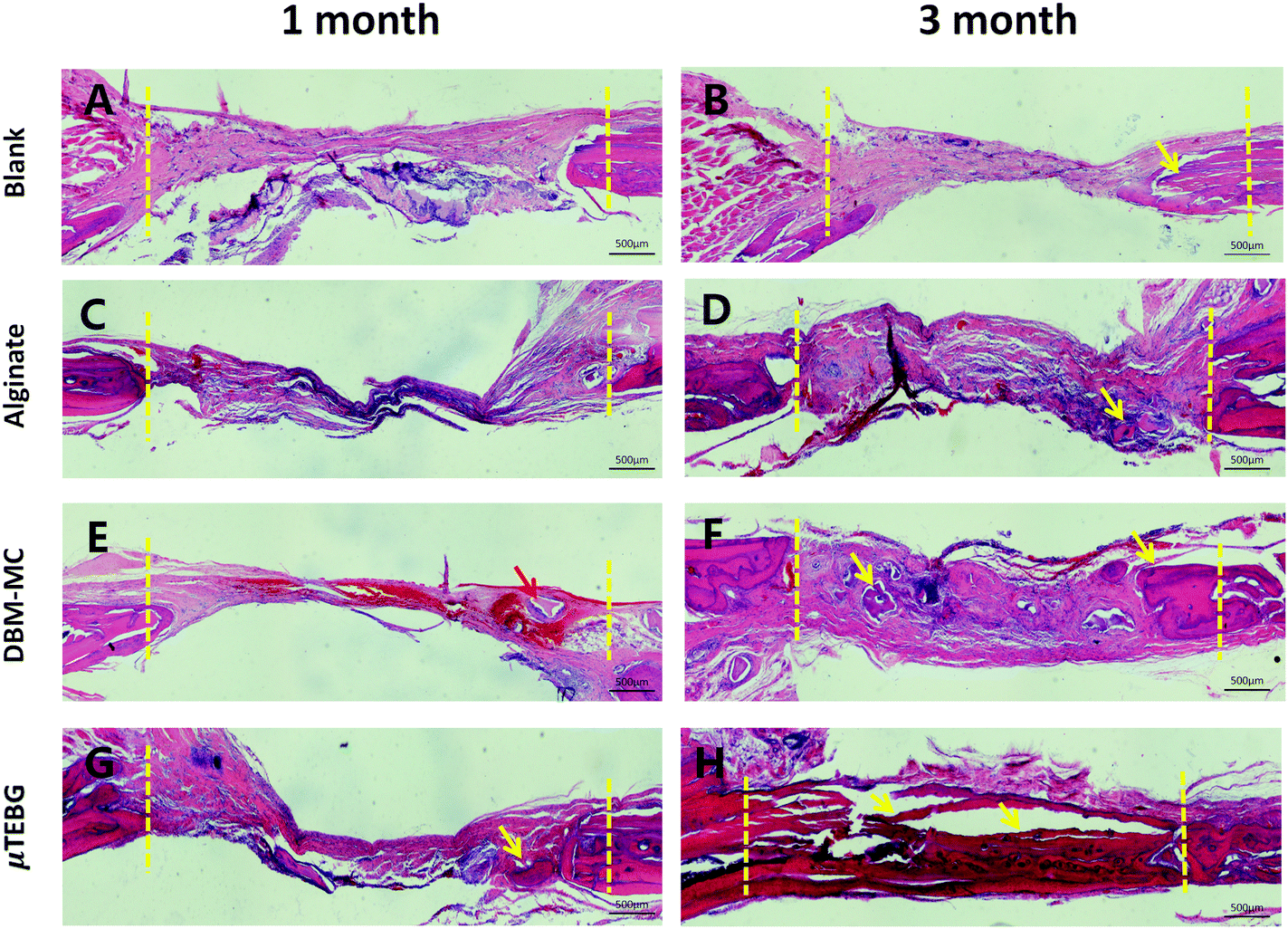 | ||
| Fig. 8 Histology of the repair of the critical-sized cranial defects. H&E staining of the (A and B) untreated group (blank), (C and D) alginate gel group (implantation of gel alone), (E and F) DBM-MC group (implantation with DBM-MC and gel alone), and (G and H) μTEBG group at (A, C, E and G) 1 and (B, D, F and H) 3 months. The defects in the μTEBG group have achieved full bridging and filled with dense bone tissue, whereas the other three groups have resulted in compromised defect healings, with defects partially or completely occupied by fibrous tissue. Red arrows: remaining DBM carriers. Yellow arrows: new bone formation. Dotted yellow line: initial defect edge. (Scale bars: 500 μm.) | ||
Discussion
Bone tissue engineering has demonstrated promising potential in generating highly osteogenic TEBGs for repairing large defects; however, the current cell expansion and TEBG construction strategies are rather complicated, labor-intensive and inefficient. Therefore, we developed a unique implantable DBM-MC for efficient MSC expansion and, furthermore, established an integrated TEBG fabrication strategy (Animation 1, ESI†). Through this study, we have demonstrated the capability of this system to support an enclosed, large-scale and semi-automatic cell and tissue construction process, which can seamlessly integrate the conventional multiple operation steps, including cell seeding, expansion, and osteogenic priming, under a continuous dynamic rotation condition. When compared to the conventional MSC expansion technique and TEBG construction strategy, the integrated μTEBG fabrication system possesses the following advantages: (1) it can greatly simplify the operation procedures and increase the fabrication efficiency, which will make the fabricated TEBG more cost-effective for clinical application; (2) it can significantly reduce the use of trypsinization and the cyclic destruction of ECM, with the potential to further improve the viability and functionality of TEBG; (3) it can provide an enclosed, large-scale, semi-automatic fabrication process from MSC seeding, to expansion and through to TEBG construction, which should considerably decrease the probability of contamination and quality variation. The μTEBGs generated via this system showed great potential as injectable TEBGs for large bone defect treatment, with outstanding osteogenic and angiogenic efficacy.It is well known that during stem cell expansion and differentiation, ECM plays a critical role by providing effective biochemical and biomechanical cues to modulate the cellular behavior of stem cells.23 Natural ECM consists of proteoglycans and fibrous proteins such as collagen, elastin, fibronectin, and laminins, which can not only provide essential physical scaffolding for the cellular constituents but also supply surface topography structure and biochemical components which are required for cellular morphogenesis, differentiation, and homeostasis.24 Microcarriers were firstly introduced by Van Wezel and have been proven to be an effective large-scale cell expansion system for immortalized cell line expansion to produce vaccines, antibodies, and so on.25 Despite their increasing interest and great potential for large-scale stem cell expansion, the current microcarriers are not biomimetically designed for the production of high-quality stem cells for clinical application. On the one hand, many commercial microcarriers such as Cytodex, Cultisphere-G, and SphereCol have been investigated for MSC expansion;26,27 however, these commercial microcarriers are not purposely designed for in vivo application and are usually made of non-biodegradable materials (e.g. Cytodex 3 is made of dextran), and thus are not suitable for use as scaffolds for tissue engineering application. On the other hand, research efforts have been spent to develop biodegradable microcarriers using PLGA, gelatin, PLLA and other materials; however, these biodegradable microcarriers generally lack the osteoinductivity and osteoconductivity required to serve as favorable scaffold matrices. In general, both the commercial microcarriers and the current biodegradable microcarriers are made of single-component material and fail to mimic and provide the basic structure, biochemical components, and 3D surface topographic cues of natural bone ECM needed to promote optimal stem cell expansion and in vivo bone tissue regeneration.
In order to develop a biomimetic microcarrier to provide effective biochemical, structural and topographic cues for stem cell expansion and differentiation, DBM was purposefully selected as the raw material to fabricate an implantable microcarrier and also as a tissue engineering scaffold for generating effective TEBGs. DBM derived from natural bone tissue was first explored as an allogenic bone graft material in 1974.28 It can preserve the composite biochemical components (such as collagen, bone morphogenic proteins (BMP), transforming growth factor-β (TGF-β) etc.) of natural bone. Matrix proteins, such as various collagens, provide an osteoconductive matrix, while BMP and TGF-β are potent osteoinductive glycoproteins and growth factors; therefore DBM can present a favorable biochemical substrate to promote better cell differentiation.29 Secondly, it has been demonstrated that the 3D structure and topography (porous structure and surface roughness) of a material have profound influences on cell morphology and biological activity and DBM can preserve the trabecular porous structure, surface roughness and hydrophilic properties of native bone tissue, all of which are required for cell adhesion and proliferation.30 For example, researchers showed that DBM exhibited a higher seeding efficiency and proliferation rate of BMSCs when compared to hydroxyapatite or β-tricalcium phosphate.31 Last but not least, when compared to many biodegradable polymers such as PLGA and PLLA, which may cause a drop in pH and lead to inflammation during rapid degradation, DBM degraded at a modest speed with mild by-products that are not harmful to cells and tissues.31 In the present study, MSCs were seeded with high efficiency and expanded well on the DBM-MC, despite the lower proliferation efficiency than Cytodex 3 microcarriers (Fig. 3C). This could possibly be explained by the differences in the swelling coefficient between DBM and Cytodex 3 microcarriers. Cytodex 3 can swell dramatically after immersion in water, leading to a larger surface area than the DBM-MC and higher cellular expansion efficiency. Nevertheless, on the other hand, the ectopic bone formation assay demonstrated that the DBM-MCs still can generate sufficient cellular expansion and work with MSCs to generate highly osteogenic and angiogenic TEBGs, leading to the formation of dense and well-vascularized bone tissue. In contrast, the Cytodex 3 microcarrier expanded and produced TEBGs that could achieve only a very limited amount of ectopic bone formation, with a large necrotic cavity being found in the center of the implant (Fig. 6C).
The commonly used cellular expansion technique to obtain sufficient cells for tissue engineering application is mainly based on the use of petri dishes or culture flasks.32 The typical cell expansion process involves plating cells on a 2D petri dish, allowing for cellular proliferation to confluence with time, and then carrying out enzymatic digestion and the uniform re-plating of cells in a new petri dish. However, this process has several problems. Firstly, the current procedures of digestion, centrifugation, cell seeding and culture medium change are rather labor-intensive; the quality of cultured cells relies heavily on the operator's experience and may easily be subject to operator-dependent variation. In addition, during the whole culture process, contact inhibition may occur frequently, which can reduce the multiple differentiation potential of MSCs when they grow to over-confluence.33 Secondly, the current petri dish- or culture flask-based cell expansion technique has a very low utilization efficiency of culture reagent and space, making the BTE therapeutic treatment very costly. Thirdly, the present cell culture operation is generally an open and manual process, with higher risk of contamination during the repeated culture media change and sub-passaging. On the contrary, the integrated TEBG construction system involves the use of a bioreactor for generating the suspension cultures of MSCs loaded on to the DBM microcarrier; because the parameters can be well standardized and the process can be easily automated, operator-dependent error and variation can be greatly minimized. Additionally, it can easily be scaled up to make full use of the culture media and space, achieving much more cost-effectiveness in clinical application. Last but not least, this system has seamlessly integrated the conventional multiple operation steps including cell seeding, expansion, and osteogenic priming within an enclosed bioreactor chamber, which can dramatically reduce the risk of contamination as compared to the traditional manual multiple-step process.
Better cranial defect repair with significantly more new bone formation was found after treatment with the fabricated μTEBGs compared with the other treatment groups (Fig. 7). This may be attributed to the better preservation of ECM, which was deposited by MSCs, during the fabrication of TEBG using our integrative construction system. ECM is well known to play an important role by providing an adhesive substrate and microenvironment for cells during migration, morphogenesis, differentiation and homeostasis, through the signaling of biochemical and biomechanical cues to cells. Glycosaminoglycans, collagen, adhesive molecules such as fibronectin, growth factors, and hormones are all found in ECM.34 It has been implied by some studies that during TEBG construction, the preservation of ECM will be very helpful in promoting bone regeneration. For example, it is reported that microfibers may be formed using alginate and ECM-derived components and it has been shown that these microfibers can promote the osteogenic differentiation and mineralization of cells that are cultured on them.35 In addition, researchers reported that the incorporation of in vivo-deposited ECM into ceramic scaffolds can potentially improve bone tissue regeneration.24 Traditional multiple-step TEBG construction would destroy and discard the ECM, due to the enzymatic treatment during cell passages, leading to cell senescence and aging, as well as to compromised bone regeneration potential.36 On the contrary, the present integrated TEBG construction system did not require the use of trypsinization to sub-passage and expand the cells, thus allowing the preservation of ECM as much as possible, which could possibly explain the robust bone regeneration capacity of μTEBGs (Fig. 7).37
In the current study, the μTEBG fabricated via this integrated system was essentially microtissue. The generation of microtissues instead of large-block tissue is a relatively new tissue engineering strategy that has recently attracted increasing interest.38 Previous studies have reported that cell-laden microcarriers could be used to form bone grafts in bottom-up bone tissue engineering.39 Many approaches have been developed to fabricate microtissues. Micro hanging-drop plates, microfluidic chips, and arrayed microwells are typical examples.40 Microwell-mesh platform has been used to provide an elegant mechanism to retain microtissues in microwells.41 Hydrogel has been used to encapsulate cells into microspheres to engineer 3D microtissues with diverse shapes.40 Here, we developed a new way to generate microtissue (μTEBGs) using implantable microcarriers, within which MSCs attached, proliferated, and became osteogenically differentiated. In contrast to current approaches without the use of a core material, our μTEBGs have a DBM core as scaffold, and thus possess better mechanical properties for bone tissue engineering application. In addition, the provision of a dynamic culture environment with appropriate flow shear stress stimulation via the use of a stirring bioreactor can nurture the μTEBGs to possess better osteogenic and angiogenic capacity (Fig. 6F),1 in line with previous reports that collagen–fibrin composite microtissues emulated improved vasculogenesis.42 Apart from the mechanical stimulation imparted by the dynamic system, the better angiogenesis results found for the μTEBG group might be attributed to the loose structure of μTEBGs and the possibility of release of angiogenesis growth factors by the seed cells on the microcarriers.
Conclusion
In the current study, we developed a biomimetic and implantable DBM microcarrier for BTE application and showed its favorable properties for cell adhesion and proliferation. Moreover, on the basis of this microcarrier, we further established an integrated TEBG fabrication system to seamlessly integrate the conventional multiple operation procedures. This integrated system allows the easy generation of μTEBG constructs in an enclosed, large-scale, semi-automatic and single-step fabrication process. When implanted ectopically or orthotopically in vivo, the μTEBG can promote robust in vivo bone tissue regeneration with good vascularization and can successfully heal a critical-sized bone defect. Therefore, this DBM microcarrier and the integrated TEBG construction system based on it demonstrate promising potential as an advanced technique to facilitate the clinical translation of BTE technology.Methods
All procedures in the present study were undertaken strictly in accordance with state regulations and laws and in accordance with the Standing Committee on Ethics in China (State Scientific and Technological Commission of China).42 The animal experiments were approved by Shanghai Jiao Tong University Committee on Animal Care and were conducted at the Key Laboratory of Tissue Engineering at Shanghai Ninth People's Hospital, affiliated with Shanghai Jiao Tong University. All experiments involving animals were performed in accordance with the approved guidelines.Preparation of DBM-MCs
Freshly harvested limbs from different bovines (more than 3 donors) were decalcified, degreased, and deproteinized before use as DBM, as previously described.28 The DBM was frozen at −80 °C for 48 hours before use. Next, the DBM was thoroughly pulverized while being maintained at temperatures below 0 °C. Then, the particles were strained through 200–500 μm sieving meshes. The 200–500 μm DBM particles were collected, strained again through 200–500 μm meshes, freeze-dried, and sterilized with ethylene oxide. The resulting DBM-MCs were preserved at 4 °C (Fig. 2A).Physical characteristics of DBM-MCs
The physical characteristics of the DBM-MCs were compared with those of Cytodex 3, including macroscopic appearance and size using optical microscopy (Nikon, Tokyo, Japan) and a microscopic image analyzer (JX-2000, Nikon, Tokyo, Japan). The density was calculated as ρmicrocarrier = (m/(m + m1 − m2)) × ρwater, where m1 is the mass of a bottle full of distilled water, m2 is the mass of the same bottle filled with distilled water and a mass m of the carrier, and ρwater is the density of the distilled water at 25 °C. The approximate number of the two kinds of microcarriers per gram (dry weight) was estimated by counting average numbers of microcarriers in 1 microgram under optical microscopy (*p < 0.05). All of the experiments were performed at 25 °C and in triplicate.Rat MSC isolation and in vitro culture
Newborn Sprague Dawley rats (3–5 days old) were euthanized by cervical dislocation. After being immersed in 75% alcohol for 5 minutes to disinfect the skin, the femurs and tibias were separated from the attached muscles and soft tissues. The long bone marrow cavities were repeatedly aspirated with syringes until the cavities appeared white, and the fresh bone marrow tissue was seeded onto 10 cm culture dishes with 7 mL of low-glucose DMEM (Hyclone, Logan, UT, USA) supplemented with 10% FBS (Hyclone), and 1% penicillin and streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). The culture dishes were incubated in a humidified environment (5% CO2, 37 °C) and the culture medium was changed every 3 days. When the attached cells became confluent, the MSCs were digested with 0.25% trypsin/1 mM EDTA (Thermo Fisher Scientific) and passaged.Dynamic seeding, expansion, and osteogenic differentiation of MSCs on DBM-MCs and Cytodex 3
Comparing MSCs expanded on DBM-MCs and Cytodex 3
The colony-forming unit (CFU) capacity of the collected MSCs was assessed by plating 200 cells per dish in DMEM for 21 days, followed by staining with 2% crystal violet after 4% paraformaldehyde fixation for 30 minutes at room temperature. Each colony greater than or equal to 1 mm in diameter was counted.
The growth kinetics of the MSCs were assessed by seeding 1000 cells per well in 96-well plates and culturing for 12 days. Cell counting was performed with a kit (CCK-8, CK04-500, Dojindo, Kumamoto, Japan) on five different wells every 3 days, and the optical density values were assessed with a microplate reader (Thermo Fisher Scientific).
MSC phenotype
MSCs isolated from the DBM carriers and Cytodex 3 after dynamic expansion were seeded onto glass coverslips at 0.1 × 106 mL−1. Once the cells had covered the entire glass surface, the coverslips were processed for immunocytochemistry as previously described.6 The presence of CD29, CD44, CD45, CD73, and CD105 (Abcam, Cambridge, UK) was detected using fluorescence microscopy.Scanning electron microscopy
After the dynamic cell expansion period, 0.5 mL of the DBM carriers and Cytodex 3 suspensions were aspirated from the stirring bottles. Microcarriers were separated and fixed in glutaraldehyde, dehydrated through a graded ethanol series, air dried, and sputtered with gold. The specimens were then assessed using SEM (S3400N, Hitachi, Tokyo, Japan) at 5 kV under high vacuum.Cell expansion efficiency
Samples of 1 mL from each of the stirring bottles were aspirated on days 1, 3, 5, 7, 9, and 11. The total numbers of cells on the DBM-MCs and Cytodex 3 were assessed using a PicoGreen dsDNA quantification assay kit (Molecular Probes Inc., Eugene, OR), following the manufacturer's instructions.In vivo subcutaneous ectopic bone formation in rats
After dynamic osteogenic differentiation, 20 mg of DBM-MCs were aspirated, separated from the medium, mixed with 1 mL of 1.5% alginate solution (Sigma), and allowed to crosslink with the addition of 100 mM CaCl2 for 2 minutes, forming a DBM-MC-alginate gel (20 mg mL−1). The same mass of Cytodex 3 was treated in the same way to obtain a Cytodex 3-alginate gel. Twenty male adult Sprague Dawley rats (age: 3 months; weight: 200–250 g) supplied by the National Tissue Engineering Center were randomly divided into two groups (n = 10 rats per group). All animals were intraperitoneally injected with chloral hydrate (10% mass fraction, 50 mg/100 g) for anesthesia. A tiny incision was then made in the dorsal side of the skin and a muscle pouch was created using blunt tissue forceps. The same amount of DBM-MCs or Cytodex 3 was mixed with 1 mL alginate and aspirated with a 1 mL syringe and then injected into muscle pouches so as to remain in situ.In vivo cranial defect orthotopic bone formation in rats
Another 20 male adult Sprague Dawley rats (age: 3 months; weight: 200–250 g) supplied by the National Tissue Engineering Center were randomly divided into two groups (n = 10 rats per group). Two parallel 5.0 mm critical cranial defects were drilled into each rat under anesthesia with an intraperitoneal injection of 10% chloral hydrate. In Group 1 (DBM-MC/alginate), the left-hand defects were implanted with 40 μL of acellular DBM carriers-alginate gel (0.8 mg of μTEBGs), and the right-hand defects were implanted with the same volume of alginate gel. In Group 2 (TEBG/untreated), the left-hand defects were implanted with the same volume of TEBG-alginate gel, and the right-hand defects were left untreated.Micro-computed tomography
At 4 and 12 weeks after defect creation and treatment, the skulls from groups 1 (acellular DBM/alginate) and 2 (TEBG DBM/untreated), as well as the ectopic injection sites, were harvested (n = 5 per group per time point). All of the specimens were evaluated using micro-CT (μ-80, Scanco Medical, Zurich, Switzerland). Regions of interest were defined as a cylinder (diameter: 30 mm; height: 5 mm) that included both bilateral defects. The scan data were reconstructed in VG studio (Volume Graphics GmbH, Heidelberg, Germany) to establish 3D models (mean threshold value = 226). Quantitative morphometric analyses were performed to determine the bone volume (BV) and bone volume/total volume (BV/TV), using micro-CT assistant software (Scanco Medical, Zurich, Switzerland).Histology
After the micro-CT evaluations, all of the specimens were fixed in 4% paraformaldehyde for 1 week, and then decalcified in 10% EDTA for 30 days. After being dehydrated through a graded ethanol series, the specimens were embedded in paraffin, sectioned to 5 μm, and stained with hematoxylin and eosin (Sigma) and Masson's Trichrome (Sigma) to show the tissue morphology and new bone formation.Statistical analyses
Parametric data were assessed using Student's t-tests. Multi-group comparisons of the means were carried out by one-way analysis of variance (ANOVA) tests with post hoc contrasts by Student–Newman–Keuls test. P-Values less than 0.05 were considered statistically significant.Author contributions
Zhenxing Wang, Dingyu Wu, and Jiwei Zou performed experiments; Zhenxing Wang and Zhi-Yong Zhang analyzed the data and wrote the manuscript; Zhi-Yong Zhang, Guoxian Pei, and Yilin Cao designed the study; Quan Zhou performed data acquisition; Junjun Fan and Dan Li contributed reagents and analytical tools; Wei Liu, Wenjie Zhang, Guangdong Zhou, and Xiansong Wang contributed scientific advice. All authors reviewed the manuscript.Competing financial interests
The authors declare no competing financial interests.Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (no. 81572137, 81101353 and 81371964), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (no. 1220000187), the National Young Thousand-Talent Scheme, the Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant (20152522), and the Shanghai Jiao Tong University Medicine-Engineering Integrated Research Grant (YG2012MS44) to Zhang Zhi-Yong.References
- Z. Zhang, S. Teoh, M. S. K. Chong, E. S. M. Lee, L. Tan, C. N. Mattar, N. M. Fisk, M. Choolani and J. Chan, Biomaterials, 2010, 31, 608–620 CrossRef CAS PubMed.
- P. S. Lienemann, S. Metzger, A. S. Kivelio, A. Blanc, P. Papageorgiou, A. Astolfo, B. R. Pinzer, P. Cinelli, F. E. Weber, R. Schibli, M. Behe and M. Ehrbar, Sci. Rep., 2015, 5, 10238 CrossRef CAS PubMed.
- P. Gao, H. Zhang, Y. Liu, B. Fan, X. Li, X. Xiao, P. Lan, M. Li, L. Geng, D. Liu, Y. Yuan, Q. Lian, J. Lu, Z. Guo and Z. Wang, Sci. Rep., 2016, 6, 23367 CrossRef CAS PubMed.
- Z. Y. Zhang, S. H. Teoh, J. H. Hui, N. M. Fisk, M. Choolani and J. K. Chan, Biomaterials, 2012, 33, 2656–2672 CrossRef CAS PubMed.
- M. Krampera, S. Glennie, J. Dyson, D. Scott, R. Laylor, E. Simpson and F. Dazzi, Blood, 2003, 101, 3722–3729 CrossRef CAS PubMed.
- Z. Y. Zhang, S. H. Teoh, M. S. Chong, J. T. Schantz, N. M. Fisk, M. A. Choolani and J. Chan, Stem Cells, 2009, 27, 126–137 CrossRef CAS PubMed.
- A. I. Caplan and D. Correa, Cell Stem Cell, 2011, 9, 11–15 CrossRef CAS PubMed.
- T. K. Goh, Z. Zhang, A. K. Chen, S. Reuveny, M. Choolani, J. K. Y. Chan and S. K. Oh, BioRes. Open Access, 2013, 2, 84–97 CrossRef CAS PubMed.
- D. Schop, F. W. Janssen, E. Borgart, J. D. de Bruijn and R. van Dijkhuizen-Radersma, J. Tissue Eng. Regener. Med., 2008, 2, 126–135 CrossRef CAS PubMed.
- K. Kumagai, A. Vasanji, J. A. Drazba, R. S. Butler and G. F. Muschler, J. Orthop. Res., 2008, 26, 165–175 CrossRef PubMed.
- P. C. Tseng, T. H. Young, T. M. Wang, H. W. Peng, S. M. Hou and M. L. Yen, Biomaterials, 2012, 33, 556–564 CrossRef CAS PubMed.
- D. Schop, R. van Dijkhuizen-Radersma, E. Borgart, F. W. Janssen, H. Rozemuller, H. J. Prins and J. D. de Bruijn, J. Tissue Eng. Regener. Med., 2010, 4, 131–140 CrossRef CAS PubMed.
- P. Kumar, M. Z. Hosain, J. H. Kang, M. Takeo, A. Kishimura, T. Mori and Y. Katayama, J. Biomater. Sci., Polym. Ed., 2015, 26, 1465–1474 CrossRef CAS PubMed.
- S. Pettersson, J. Wettero, P. Tengvall and G. Kratz, J. Tissue Eng. Regener. Med., 2009, 3, 450–460 CrossRef CAS PubMed.
- H. Thissen, K. Y. Chang, T. A. Tebb, W. B. Tsai, V. Glattauer, J. A. Ramshaw and J. A. Werkmeister, J. Biomed. Mater. Res., Part A, 2006, 77, 590–598 CrossRef CAS PubMed.
- X. B. Wu, C. H. Peng, F. Huang, J. Kuang, S. L. Yu, Y. D. Dong and B. S. Han, Hepatobiliary Pancreatic Dis. Int., 2011, 10, 509–515 CrossRef CAS.
- W. Wang, M. Dang, Z. Zhang, J. Hu, T. W. Eyster, L. Ni and P. X. Ma, Acta Biomater., 2016, 36, 63–72 CrossRef CAS PubMed.
- S. J. Florczyk, M. Leung, Z. Li, J. I. Huang, R. A. Hopper and M. Zhang, J. Biomed. Mater. Res., Part A, 2013, 101, 2974–2983 CrossRef PubMed.
- J. R. Mauney, S. Sjostorm, J. Blumberg, R. Horan, J. P. O'Leary, G. Vunjak-Novakovic, V. Volloch and D. L. Kaplan, Calcif. Tissue Int., 2004, 74, 458–468 CrossRef CAS PubMed.
- J. R. Mauney, J. Blumberg, M. Pirun, V. Volloch, G. Vunjak-Novakovic and D. L. Kaplan, Tissue Eng., 2004, 10, 81–92 CrossRef CAS PubMed.
- G. Liu, Y. Zhang, B. Liu, J. Sun, W. Li and L. Cui, Biomaterials, 2013, 34, 2655–2664 CrossRef CAS PubMed.
- L. Cui, B. Liu, G. Liu, W. Zhang, L. Cen, J. Sun, S. Yin, W. Liu and Y. Cao, Biomaterials, 2007, 28, 5477–5486 CrossRef CAS PubMed.
- H. Kim, K. Lee, D. Kim and D. Hwang, Biomaterials, 2013, 34, 1041–1050 CrossRef CAS PubMed.
- R. D. Ventura, A. R. Padalhin, Y. K. Min and B. T. Lee, Tissue Eng., Part A, 2015, 21, 2649–2661 CrossRef CAS PubMed.
- A. L. van Wezel, Dev. Biol. Stand., 1976, 37, 143–147 CAS.
- Q. A. Rafiq, K. Coopman, A. W. Nienow and C. J. Hewitt, Biotechnol. J., 2016, 11, 473–486 CrossRef CAS PubMed.
- M. Chen, X. Wang, Z. Ye, Y. Zhang, Y. Zhou and W. S. Tan, Biomaterials, 2011, 32, 7532–7542 CrossRef CAS PubMed.
- M. R. Urist and B. S. Strates, Clin. Orthop. Relat. Res., 1970, 71, 271–278 CAS.
- R. D. Ventura, A. R. Padalhin, Y. Min and B. Lee, Tissue Eng., Part A, 2015, 21, 2649–2661 CrossRef CAS PubMed.
- X. Wang, Y. Li, R. Han, C. He, G. Wang, J. Wang, J. Zheng, M. Pei and L. Wei, PLoS One, 2014, 9, e116061 Search PubMed.
- P. Kasten, R. Luginbuhl, M. van Griensven, T. Barkhausen, C. Krettek, M. Bohner and U. Bosch, Biomaterials, 2003, 24, 2593–2603 CrossRef CAS PubMed.
- S. Redaelli, A. Bentivegna, D. Foudah, M. Miloso, J. Redondo, G. Riva, S. Baronchelli, L. Dalpra and G. Tredici, Stem Cell Res. Ther., 2012, 3, 47 CrossRef CAS PubMed.
- W. Wagner, P. Horn, M. Castoldi, A. Diehlmann, S. Bork, R. Saffrich, V. Benes, J. Blake, S. Pfister, V. Eckstein and A. D. Ho, PLoS One, 2008, 3, e2213 Search PubMed.
- S. Ravindran and A. George, Exp. Cell Res., 2014, 325, 148–154 CrossRef CAS PubMed.
- M. Angelozzi, M. Miotto, L. Penolazzi, S. Mazzitelli, T. Keane, S. F. Badylak, R. Piva and C. Nastruzzi, Mater. Sci. Eng., C, 2015, 56, 141–153 CrossRef CAS PubMed.
- W. Wagner, P. Horn, M. Castoldi, A. Diehlmann, S. Bork, R. Saffrich, V. Benes, J. Blake, S. Pfister, V. Eckstein and A. D. Ho, PLoS One, 2008, 3, e2213 Search PubMed.
- M. C. Moore, V. Pandolfi and P. S. McFetridge, Biomaterials, 2015, 49, 37–46 CrossRef CAS PubMed.
- C. Millan, E. Cavalli, T. Groth, K. Maniura-Weber and M. Zenobi-Wong, Adv. Healthcare Mater., 2015, 4, 1348–1358 CrossRef CAS PubMed.
- H. A. Declercq, T. De Caluwe, O. Krysko, C. Bachert and M. J. Cornelissen, Biomaterials, 2013, 34, 1004–1017 CrossRef CAS PubMed.
- G. H. Lee, J. S. Lee, X. Wang and L. S. Hoon, Adv. Healthcare Mater., 2015, 5, 56–74 CrossRef PubMed.
- K. Futrega, J. S. Palmer, M. Kinney, W. B. Lott, M. D. Ungrin, P. W. Zandstra and M. R. Doran, Biomaterials, 2015, 62, 1–12 CrossRef CAS PubMed.
- A. W. Peterson, D. J. Caldwell, A. Y. Rioja, R. R. Rao, A. J. Putnam and J. P. Stegemann, Biomater. Sci., 2014, 2, 1497–1508 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb02414a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |