
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyano substituted benzotriazole based polymers for use in organic solar cells†
Abby
Casey
a,
Joshua P.
Green
a,
Pabitra
Shakya Tuladhar
a,
Mindaugas
Kirkus
a,
Yang
Han
ab,
Thomas D.
Anthopoulos
 b and
Martin
Heeney
*a
b and
Martin
Heeney
*a
aDept. Chemistry and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK. E-mail: m.heeney@imperial.ac.uk
bDept. Physics and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK
First published on 6th March 2017
Abstract
A new synthetic route to the electron accepting di-cyano substituted benzo[d][1,2,3]triazole (BTz) monomer 2-(2-butyloctyl)-4,7-di(thiophen-2-yl)-2H-benzotriazole-5,6-dicarbonitrile (dTdCNBTz) is reported. The cyano substituents can be easily introduced to the BTz unit in one step via the nucleophilic aromatic substitution of the fluorine substituents of the fluorinated precursor 2-(2-butyloctyl)-4,7-di(thiophen-2-yl)-2H-benzotriazole-5,6-difluoro (dTdFBTz). Co-polymers were prepared with distannylated benzo[1,2-b:4,5-b′]dithiophene (BDT) monomers containing either 2-ethylhexylthienyl (T-EH) side chains or 2-butyloctylthienyl (T-BO) side chains via Stille coupling to yield the novel medium band gap polymers P1 and P2 respectively. Whilst the organic photovoltaic (OPV) performance of P1 was limited by a lack of solubility, the improved solubility of P2 resulted in promising device efficiencies of up to 6.9% in blends with PC61BM, with high open circuit voltages of 0.95 V.
The incorporation of both electron donating and accepting building blocks into semiconducting polymers has been a highly successful approach when designing new donor polymers for bulk heterojunction (BHJ) organic solar cells.1–3 When co-polymerised, the orbitals of the donor and acceptor monomers hybridise resulting in a reduced band gap through the simultaneous raising of the highest occupied molecular orbital (HOMO) and lowering of the lowest unoccupied molecular orbital (LUMO). However, whilst this reduction in band gap can lead to a desirable increase in the short-circuit photocurrent (JSC) through enhanced light absorption, raising the HOMO level can result in an undesirable reduction in open circuit voltage (VOC).4–6 It is therefore generally preferable to reduce the polymer band gap through selective lowering of the LUMO level whilst maintaining a deep HOMO level. This can be achieved by increasing the strength of the electron accepting monomer.7–13 We have previously shown that the strength of the common electron accepting monomer 4,7-di(thiophen-2-yl)-2,1,3-benzothiadiazole (dTBT) can be increased by substituting one or two cyano groups onto the central 2,1,3-benzothiadiazole (BT) ring.14,15 When these cyano substituted BT acceptor units (see Fig. 1A) were incorporated into donor–acceptor polymers with dithienogermole (DTG), the polymer LUMO level was systematically lowered to reduce the band gap without raising the HOMO level. We found that optimising the position of the polymer LUMO level led to large increases in OPV device efficiency through improved photocurrent without sacrificing VOC.14 Cyano substituents have also been shown to induce strong dipole moments,16 promote balanced ambipolar charge transport and thermal stability in semiconducting polymers,17 as well as allow fine tuning of donor polymer energy levels.18
 | ||
| Fig. 1 (A) Di- and mono-cyano substituted BT derivatives can be synthesised from their fluorinated precursors14,15 (B) di-cyano substituted BTz can be synthesised from its fluorinated precursor. | ||
Further exploring the use of cyano substituents to increase the strength of common electron accepting monomers, we here report the synthesis of the di-cyano substituted benzo[d][1,2,3]triazole (BTz) monomer, 2-(2-butyloctyl)-4,7-di(thiophen-2-yl)-2H-benzotriazole-5,6-dicarbonitrile (dTdCNBTz – see Fig. 1B). Unlike BT, the benzo[d][1,2,3]triazole (BTz) ring can be alkylated in the N-2 position. Alkylating at the N-2 position instead of other sites (such as the 5 and 6 BTz positions or flanking thiophene rings) avoids unfavourable steric interactions along the polymer backbone. This allows higher backbone planarity to be achieved, encouraging π–π stacking between chains which can help to improve charge carrier mobility. However, the BTz unit is significantly less electron withdrawing than the BT unit, as the sulfur atom in the thiadiazole ring is replaced with a more electron donating nitrogen atom in the triazole ring. When un-substituted or fluorinated BTz-based monomers are co-polymerised with benzo[1,2-b:4,5-b′]dithiophene (BDT), the resulting polymers (dTBTz-BDT and dTdFBTz-BDT, Fig. 2) have a relatively wide bandgap (∼2 eV) due to the weak electron accepting ability of the BTz monomer.10,19 Whilst a wide band gap can be useful to obtain high VOC, the JSC can be limited due to reduced light absorption. Despite the large band gaps of these materials, the un-substituted and fluorinated BTz-BDT polymers give promising power conversion efficiencies of over 4% and 7% respectively in OPV devices.19 By adding cyano substituents to the BTz unit and therefore increasing the electron accepting ability, we aimed to reduce the polymer band gap in order to increase the JSC whilst either maintaining or improving the VOC.
 | ||
| Fig. 2 Chemical structures of dTBTz-BDT,19dTdFBTz-BDT,10,19dTdCNBTz-BDT10 and the novel polymers P1 and P2. | ||
Previously we have shown that the mono and di-cyano substituted BT derivatives can be easily synthesised from their fluorinated precursors in one step through nucleophilic aromatic substitution of the fluorine substituents with cyanide (see Fig. 1A). Herein we report the analogous route to the di-cyano-substituted BTz derivative. Whilst the di-cyano substituted BTz monomer has recently been published by You and co-workers,10 we offer an alternative, shorter synthetic route from the common fluorinated analogue. You and co-workers10 co-polymerised the brominated dTdCNBTz monomer with distannylated BDT, dialkylated with 3-butyloctyl chains to form the polymer dTdCNBTz-BDT (see Fig. 2). In this work, we co-polymerise brominated dTdCNBTz with distannylated BDT dialkylated with 2-ethylhexylthienyl and 2-butyloctylthienyl groups to yield the novel two-dimensional conjugated polymers P1 and P2 respectively (see Fig. 2). Alkylthienyl groups have previously been shown to help increase VOC through stabilisation of the HOMO level, as well as increase JSC by red shifting the absorption spectra in comparison to conventional alkoxy chains.20–22 The inclusion of alkylthienyl groups can also help to improve hole mobility.23–25 Unfortunately, P1 was highly insoluble and proved difficult to process. The alkyl chain length was therefore increased to 2-butyloctyl leading to the more soluble P2.
You and co-workers10 found that the cyanated polymer dTdCNBTz-BDT had a reduced band gap in comparison to fluorinated analogue dTdFBTz-BDT due to a stronger stabilisation of the LUMO in comparison to the HOMO level. This is in agreement with work we have carried out previously comparing fluorinated and cyanated BT based donor–acceptor polymers.15 Here we report that the inclusion of thienyl units in the BDT side chains results in P1 and P2 having slightly reduced band gaps in comparison to dTdCNBTz-BDT (see Fig. 2). Promisingly P2 exhibited significantly improved device performance over dTdCNBTz-BDT, with a device power conversion efficiencies up to 6.9% in blends with PC61BM using PEDOT:PSS as the hole transporting layer (HTL).
Synthesis of monomer and polymer
The synthetic route to the dTdCNBTz monomer reported by You and co-workers10 is quite different to the synthesis utilised in this work. You and co-workers reported an elegant eight step synthesis in which a thiophene-flanked triazole-fused 1,4-diketone intermediate undergoes a nucleophilic condensation reaction with succinonitrile to form dTdCNBTz, which is then brominated under the same conditions as reported here. A related system has also recently been reported by the direct arylation of 2-octyl-5,6-dicyano-2H-benzo[d][1,2,3]triazole with a mono-protected thiophene,12 although the synthesis of the starting dicyanated benzotriazole is rather low yielding.26 In this work, we report the initial synthesis of the thiophene-flanked fluorinated precursor (2) via Negishi coupling of commercially available 1 with 2-thienylzinc bromide (Scheme 1). The fluoride substituents of 2 were readily displaced when treated with excess potassium cyanide in the presence of 18-crown-6 to yield the dicyano-substituted product dTdCNBTz (57%). We have previously found that excess molecular bromine with heating was required to brominate the dicyano-substituted BT derivative (see Fig. 1) due to the reduced reactivity of the α1 and α2 thiophene positions of the electron-poor monomer.15 Despite BTz being a weaker electron acceptor than BT, N-bromosuccinimide (NBS) was still found to be ineffective as a brominating agent, and reaction with excess molecular bromine over 30 hours was required to form the final brominated monomer bis-Br(dTdCNBTz).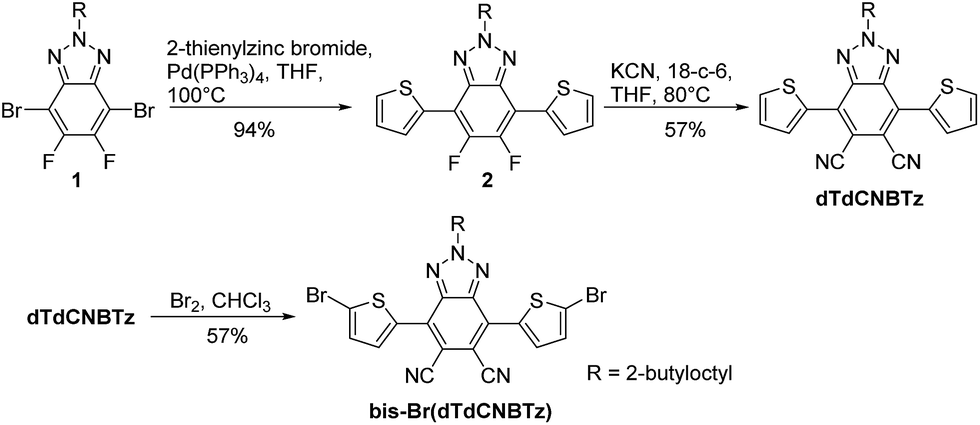 | ||
| Scheme 1 Synthesis of di-cyano substituted benzotriazole monomer bis-Br(dTdCNBTz). | ||
Monomer bis-Br(dTdCNBTz) was reacted with the distannylated BDT monomers BDT(T-EH) (2-ethylhexylthienyl chain) and BDT(T-BO) (2-butyloctylthienyl chain) via Stille coupling under microwave irradiation to yield polymers P1 and P2 respectively (see Scheme 2). After 40 min of microwave heating from 100 °C to 200 °C, the polymers were precipitated into methanol before Soxhlet extraction (methanol, acetone, hexane, chloroform, chlorobenzene). Polymer P1 was highly insoluble, with the hot chloroform and chlorobenzene fractions containing only ∼10 mg (3–4% yield) of polymer each. The remaining undissolved polymer (P1) was heated in 1,2,4-trichlorobenzene (TCB) and precipitated into methanol. However, much of P1 remained undissolved even in hot TCB. The longer branched chains of P2 increased the solubility of the resulting polymer. However, hot chlorobenzene was still required to fully dissolve the higher molecular weight material. Due to the insolubility of polymer P1, the molecular weight could not be determined by gel permeation chromatography (GPC). The molecular weights of the chloroform and chlorobenzene fractions of P2 (P2-CF and P2-CB respectively) were determined using GPC in hot chlorobenzene (80 °C) and are summarised in Table 1. P2-CF (76 mg, 42% yield) had a number-average molecular weight (Mn) of 26.0 kDa with a dispersity index (Đ) of 3.37, whilst P2-CB (88 mg, 49% yield) had a much higher Mn of 78.2 kDa and a lower Đ of 1.46. The more soluble P2-CF was used for subsequent analysis (UV/Vis, cyclic voltammetry, 1H NMR). Neither polymer exhibited any obvious thermal transitions by differential scanning calorimetry up to 300 °C (Fig. S1†).
 | ||
| Scheme 2 Stille polymerisations to yield P1 and P2. | ||
| Polymer | M n (kDa) | M w (kDa) | Đ |
|---|---|---|---|
| P2-CF | 26.0 | 87.5 | 3.37 |
| P2-CB | 78.2 | 114.3 | 1.46 |
Optical properties
The optical properties of P1 and P2 were investigated using UV-Vis absorption spectroscopy. The absorption spectra of both polymers in room temperature and heated (80 °C) 1,2,4-trichlorobenzene (TCB) solutions are shown in Fig. 3A, whilst the absorption spectra of the polymers as thin films are shown in Fig. 3B. The optical properties of the polymers are summarised in Table 2. The room temperature TCB solution and thin film absorption spectra of P1 are almost identical, both having a λmax at 641 nm. This suggests that P1 was aggregated in room temperature TCB solution. Indeed upon heating to 80 °C, the solution λmax blue shifts to 622 nm as the aggregated polymer becomes more solvated (see Fig. 3A). The room temperature solution spectrum of P2 is significantly blue shifted in comparison to that of P1, having a λmax at 627 nm and a high energy shoulder at ∼596 nm. Upon heating (80 °C) the high energy shoulder of the solution absorption spectrum of P1 blue shifts and increases in intensity giving a λmax at 584 nm with a low energy shoulder at ∼618 nm (see Fig. 3A), again indicative of reduced aggregation upon heating. This suggests that both polymers are at least partly aggregated in room temperature TCB. The absorption spectrum of P2 undergoes a small red shift of 9 nm from solution to thin film, and appears almost identical to the solid state spectra of P1.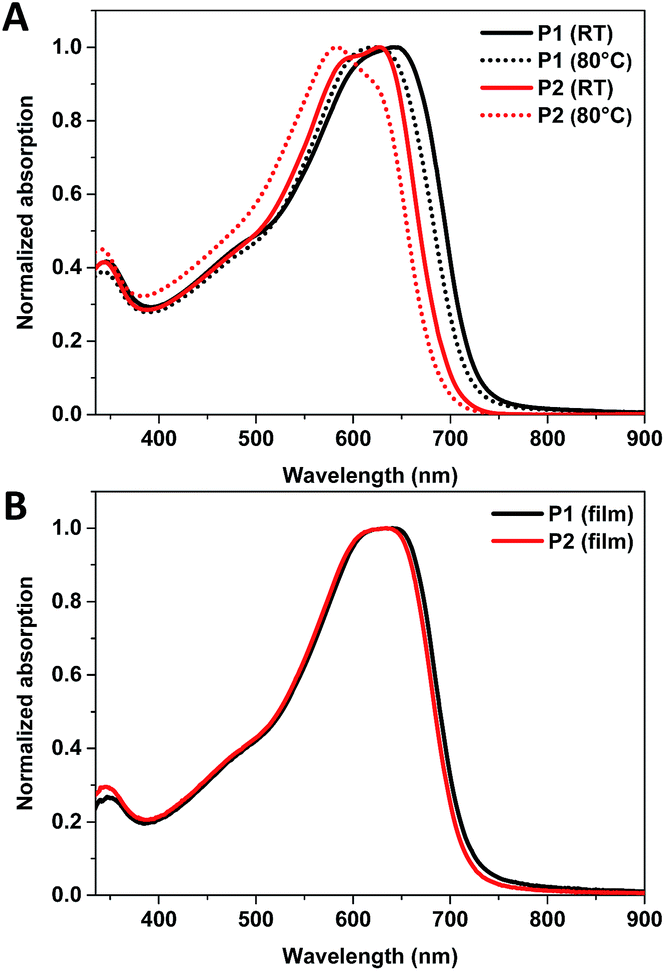 | ||
| Fig. 3 (A) Absorption spectra of P1 and P2 in TCB solution and (B) P1 and P2 in thin film. | ||
The optical band gaps of P1 and P2 are slightly smaller (1.73–1.74 eV) than the reported optical band gap of dTdCNBTz-BDT (1.77 eV). The inclusion of thienyl groups onto the BDT core therefore has the effect of slightly broadening the absorption spectra of P1 and P2.
Electronic properties
The solid state electronic properties of polymers P1 and P2 were investigated using cyclic voltammetry (CV) of films spun onto fluorine-doped tin oxide (FTO). Measurements were performed in anhydrous, degassed solutions of acetonitrile with tetrabutylammonium hexafluorophosphate (0.1 M) electrolyte using an Ag/Ag+ reference electrode. Both polymers gave almost identical oxidation and reduction traces (Fig. S2†), with rather poorly defined oxidation onsets around 1.2 V and reduction onsets around −0.67 V. Assuming that the ferrocene/ferrocenium reference redox couple is 4.8 eV below vacuum level, this corresponds to relatively deep HOMO levels of −5.6 eV and a LUMO level around −3.73 eV. As the polymers only differ by the length of the branched alkyl chain it is not surprising that they show very similar electronic properties. The HOMO levels of P1 and P2 are slightly shallower than those of the reported10 polymer dTdCNBTz-BDT (see Fig. 2), which according to literature has a HOMO level of −5.73 eV. This may be due to the inclusion of the thienyl unit in the side chain, which has previously been shown to influence the polymer HOMO and LUMO levels,21 although we note that comparison with reported oxidation potential is difficult due to the inherent error in the measurement and the different experimental conditions used.To further investigate the role of the thienyl groups, density functional theory calculations (DFT) were performed with a B3LYP level of theory and a basis set of 6-31G(d). The lowest energy conformation of the monomer dTdCNBTz was initially investigated by running a potential energy scan in which the thiophene–benzotriazole angle was systematically changed and the rest of the molecule was allowed to relax to its energy minimum (Fig. S3†). The lowest energy conformer was then used to model trimers of this unit with carbazole (Fig. S4†). These gas phase calculations show that the HOMO was delocalised over the polymer backbone, whereas the LUMO was more localised on the triazole unit. The alkylated thienyl sidegroups were twisted approximately 60° with respect to the BDT unit, limiting their involvement in the frontier molecular orbitals.
OPV performance
The OPV device performances of P1 and P2 were tested in a device configuration of glass/ITO/(PEDOT:PSS or CuSCN)/polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC61BM/Ca/Al. A blend weight ratio of 1:2 polymer:PC61BM was used with a concentration of ∼20 mg mL−1 in TCB for blends using P1 and ∼24 mg mL−1 in TCB for blends using P2. TCB was utilised since the polymers were not sufficiently soluble in CB or CF at the required concentrations. Both P2-CF and P2-CB were tested, whilst the re-precipitated TCB fraction of P1 was tested. Previously You and co-workers reported the device performance of dTdCNBTz-BDT was significantly affected by the choice of hole transport layer (HTL), with overall efficiencies of ∼5.5–6% using CuSCN as the HTL and ∼4% using PEDOT:PSS as HTL.10 The lower efficiency of the devices made using PEDOT:PSS was attributed to a mismatch in energy levels between the deep HOMO of dTdCNBTz-BDT (−5.73 eV) and the work function of PEDOT:PSS (−5.0 eV) compared to CuSCN (−5.5 eV). As such devices with P1 or P2 were made using either PEDOT:PSS or CuSCN as the hole transporting layer (HTL) and then compared. The OPV device characteristics are summarised in Table 3, whilst the J–V curves of average type devices are shown in Fig. 4.
:PC61BM/Ca/Al. A blend weight ratio of 1:2 polymer:PC61BM was used with a concentration of ∼20 mg mL−1 in TCB for blends using P1 and ∼24 mg mL−1 in TCB for blends using P2. TCB was utilised since the polymers were not sufficiently soluble in CB or CF at the required concentrations. Both P2-CF and P2-CB were tested, whilst the re-precipitated TCB fraction of P1 was tested. Previously You and co-workers reported the device performance of dTdCNBTz-BDT was significantly affected by the choice of hole transport layer (HTL), with overall efficiencies of ∼5.5–6% using CuSCN as the HTL and ∼4% using PEDOT:PSS as HTL.10 The lower efficiency of the devices made using PEDOT:PSS was attributed to a mismatch in energy levels between the deep HOMO of dTdCNBTz-BDT (−5.73 eV) and the work function of PEDOT:PSS (−5.0 eV) compared to CuSCN (−5.5 eV). As such devices with P1 or P2 were made using either PEDOT:PSS or CuSCN as the hole transporting layer (HTL) and then compared. The OPV device characteristics are summarised in Table 3, whilst the J–V curves of average type devices are shown in Fig. 4.
| Material | HTL | J SC (mA cm−2) | V OC (V) | FF | PCE (PCEmax) |
|---|---|---|---|---|---|
| a CB = chlorobenzene fraction, CF = chloroform fraction. | |||||
| P1 | PEDOT:PSS | 5.73 ± 0.1 | 0.93 ± 0.01 | 0.30 ± 0.03 | 1.58 ± 0.12 (1.68) |
| P2-CB | PEDOT:PSS | 12.98 ± 0.71 | 0.96 ± 0.03 | 0.42 ± 0.02 | 5.21 ± 0.32 (5.49) |
| P2-CB | CuSCN | 13.21 ± 1.21 | 0.91 ± 0.02 | 0.38 ± 0.02 | 4.48 ± 0.41 (5.00) |
| P2-CF | PEDOT:PSS | 11.25 ± 0.61 | 0.95 ± 0.03 | 0.57 ± 0.03 | 6.12 ± 0.76 (6.93) |
| P2-CF | CuSCN | 11.37 ± 1.46 | 0.88 ± 0.01 | 0.50 ± 0.01 | 5.02 ± 0.60 (5.76) |
 | ||
| Fig. 4 (A) J–V curves for either chlorobenzene (CB) or chloroform (CF) fractions of P2: PC61BM using either PEDOT:PSS or CuSCN as the hole transporting layers, (B) EQE of device made using P2 (chloroform fraction) with PEDOT:PSS as the hole transporting layer. | ||
Solutions containing P1 were difficult to dissolve fully, even when heated for 6 h at 135 °C in TCB, resulting in poor device yields (ca. 50%). Nevertheless devices made from P1 with PEDOT:PSS as the HTL did work, despite the low apparent energy offset between the LUMO of P1 and PCBM. The overall performance was poor, with low JSC (∼5–6 mA cm−2) and FF (∼0.3) resulting in low power conversion efficiencies of ∼1.5% (see Fig. S5†). We ascribe the poor performance to the bad solubility and high aggregation tendency of P1 which likely results in poor mixing between the polymer and PC61BM, limiting the photocurrent. Due to the difficult processing of P1, no further optimisation was attempted.
Both P2-CF and P2-CB were tested in devices using either PEDOT:PSS or CuSCN as the HTL. Despite having higher molecular weight and narrower dispersity (Đ), P2-CB exhibited lower efficiencies in OPV devices (using both CuSCN and PEDOT:PSS) in comparison to the CF fraction. Higher molecular weight polymers generally tend to give improved performance due to improved active layer morphologies and charge transport properties.27,28 Whilst the JSC was improved (∼13 mA cm−2) for P2-CB in comparison to devices made from the lower weight CF fraction (∼11–12 mA cm−2), the fill factor was reduced in both the devices made using PEDOT:PSS (FF = 0.42) and CuSCN (FF = 0.38). Similarly to devices made from P1, this is likely to be due to the reduced solubility of P2-CB causing non-optimal active layer morphology. Films made from P2-CF appeared more visibly homogeneous than those made from P2-CB. Surprisingly, the use of CuSCN as the HTL reduced the efficiency of devices made from both P2-CF and P2-CB in our hands, mainly due to reduction in VOC and FF in both cases. The use of CuSCN did result in an increase in JSC, which we relate to the wide band gap of CuSCN (>3.5 eV) resulting in improved transparency between 400–1100 nm compared to PEDOT:PSS. This has previously been shown to improve light absorption in the active layer of BHJ devices, resulting in increased JSC.29–31
Atomic force microscopy (AFM) was used to investigate the effect of the CuSCN and PEDOT:PSS HTL's on the active layer morphologies of P2-CB and P2-CF. The thin film morphology of just the CuSCN and PEDOT:PSS HTL layers on ITO coated glass are shown in Fig. S6.† In agreement with literature, the film of CuSCN appeared to be nanocrystalline and significantly rougher (root mean square (RMS) roughness of 5.13 nm) than the PEDOT:PSS layer (RMS 0.77 nm).29 AFM topography and phase images of P2-CF and P2-CB coated onto either CuSCN and PEDOT:PSS are shown in Fig. S7 and S8† respectively. Both P2-CF films (RMS 0.71–0.75 nm) are slightly smoother than the P2-CB (RMS 0.89–1.36 nm) on each HTL, in agreement with the improved solubility and processability of P2-CF. The P2-CF and P2-CB films coated onto the CuSCN layer were marginally less smooth than those coated onto the PEDOT:PSS layers but, in general, their surface morphologies appear to be similar. The reduced fill factor and VOC of the P2-CB and P2-CF devices utilising CuSCN may therefore be related to poor contact of the polymer with the rough CuSCN surface. Previous studies using ZnO interlayers in an inverted solar cell device have noted a reduction in both FF and voltage as surface roughness increased.32
Devices made from P2-CF with PEDOT:PSS as the HTL displayed the best performance with overall efficiencies up to 6.93% in combination with a high Voc of 0.95 V. Examination of the external quantum efficiency (EQE) curves (Fig. 4B) show that the blends generates current over the spectral range of the blend, with substantial photocurrent generated by the polymer. The performance is encouraging for a mid-gap and further demonstrates that dicyanobenzotriazole is a promising acceptor for the further development of high ionisation potential polymers.
Conclusions
We report a new route for the synthesis of 5,6-dicyano benzo[d][1,2,3]triazole based monomers via with nucleophilic substitution of a fluorinated precursor with cyanide. We highlight the utility of this route by synthesising the previously reported monomer 2-(2-butyloctyl)-4,7-di(thiophen-2-yl)-2H-benzotriazole-5,6-dicarbonitrile (dTdCNBTz) in one step from the fluorinated precursor dTdFBTz. The resulting monomer was co-polymerised with two distannylated BDT monomers containing either 2-ethylhexylthienyl or 2-butyloctylthienyl side chains via Stille coupling to yield the novel medium band gap polymers P1 and P2 respectively. Polymer P1 was found to be poorly soluble even in hot TCB, whereas the 2-butyloctyl side chains of P2 improved solubility and processability. Both polymers had similar solid state optical and electronic properties, with notably high oxidation potentials around 5.6 eV.Solar cell devices were prepared from both polymers as blends with PC61BM. The performance of P1 was rather poor, mainly due to processability problems but P2 afforded promising device performance with an overall efficiency up to 6.9% and a high open voltage circuit (0.95 V). The performance of P2 was found to be molecular weight dependent, with the lower molecular weight fraction (Mn = 26.0, Đ = 3.37) performing significantly better than the higher molecular weight (Mn = 78.2, Đ = 1.46), which we ascribe to the better solubility of the lower weight fraction. The fact that the lower molecular weight fraction of P2 gave the highest performance shows that this polymer system is highly sensitive to solubility. If the solubility can be improved further, for example by using longer branched alkyl chains, higher molecular weight polymer with a narrower dispersity (Đ) may lead to further improvements in device performance. These results further demonstrate the utility of the 5,6-dicyano benzo[d][1,2,3]triazole building block in developing high ionization potential polymers which can give desirably high voltage solar cells.
Acknowledgements
We thank the UK's Engineering and Physical Sciences Research Council (EPSRC) for financial support via the Doctoral Training Centre in Plastic Electronics EP/G037515/1 and the British Council (Grant Number 173601536).Notes and references
- Z. Zhang and J. Wang, J. Mater. Chem., 2012, 22, 4178 RSC.
- R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 743 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- S. D. Dimitrov and J. R. Durrant, Chem. Mater., 2014, 26, 616 CrossRef CAS.
- M. Shahid, R. S. Ashraf, Z. Huang, A. J. Kronemeijer, T. McCarthy-Ward, I. McCulloch, J. R. Durrant, H. Sirringhaus and M. Heeney, J. Mater. Chem., 2012, 22, 12817 RSC.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- T. C. Parker, D. G. (Dan) Patel, K. Moudgil, S. Barlow, C. Risko, J.-L. Brédas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2014, 2, 22 RSC.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS PubMed.
- H. Zhou, L. Yang, S. C. Price, K. J. Knight and W. You, Angew. Chem., Int. Ed., 2010, 49, 7992 CrossRef CAS PubMed.
- W. Li, L. Yan, H. Zhou and W. You, Chem. Mater., 2015, 27, 6470 CrossRef CAS.
- C. P. Yau, Z. Fei, R. S. Ashraf, M. Shahid, S. E. Watkins, P. Pattanasattayavong, T. D. Anthopoulos, V. G. Gregoriou, C. L. Chochos and M. Heeney, Adv. Funct. Mater., 2014, 24, 678 CrossRef CAS.
- J. Zhang, T. C. Parker, W. Chen, L. Williams, V. N. Khrustalev, E. V. Jucov, S. Barlow, T. V. Timofeeva and S. R. Marder, J. Org. Chem., 2016, 81, 360 CrossRef CAS PubMed.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200 RSC.
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110 CrossRef CAS.
- A. Casey, Y. Han, Z. Fei, A. J. P. White, T. D. Anthopoulos and M. Heeney, J. Mater. Chem. C, 2015, 3, 265 RSC.
- J. Wudarczyk, G. Papamokos, V. Margaritis, D. Schollmeyer, F. Hinkel, M. Baumgarten, G. Floudas and K. Müllen, Angew. Chem., Int. Ed., 2016, 55, 3220 CrossRef CAS PubMed.
- J.-M. Park, S. K. Park, W. S. Yoon, J. H. Kim, D. W. Kim, T. Choi and S. Y. Park, Macromolecules, 2016, 49, 2985 CrossRef CAS.
- H. G. Kim, M. Kim, J. A. Clement, J. Lee, J. Shin, H. Hwang, D. H. Sin and K. Cho, Chem. Mater., 2015, 27, 6858 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625 CrossRef CAS PubMed.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef CAS.
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697 CrossRef CAS PubMed.
- L. Ye, S. Q. Zhang, L. J. Huo, M. J. Zhang and J. H. Hou, Acc. Chem. Res., 2014, 47, 1595 CrossRef CAS PubMed.
- S. Zhang, L. Ye, Q. Wang, Z. Li, X. Guo, L. Huo, H. Fan and J. Hou, J. Phys. Chem. C, 2013, 117, 9550 CAS.
- R. Duan, L. Ye, X. Guo, Y. Huang, P. Wang, S. Zhang, J. Zhang, L. Huo and J. Hou, Macromolecules, 2012, 45, 3032 CrossRef CAS.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397 CrossRef CAS PubMed.
- S. Vagin, A. Frickenschmidt, B. Kammerer and M. Hanack, Eur. J. Org. Chem., 2005, 2005, 3271 CrossRef.
- Z. Xiao, K. Sun, J. Subbiah, T. Qin, S. Lu, B. Purushotharman, D. J. Jones, A. B. Holmes and W. W. H. Wong, Polym. Chem., 2015, 6, 2312 RSC.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456 CrossRef CAS PubMed.
- N. Yaacobi-Gross, N. D. Treat, P. Pattanasattayavong, H. Faber, A. K. Perumal, N. Stingelin, D. D. C. Bradley, P. N. Stavrinou, M. Heeney and T. D. Anthopoulos, Adv. Energy Mater., 2015, 5, 1401529 CrossRef.
- N. D. Treat, N. Yaacobi-Gross, H. Faber, A. K. Perumal, D. D. C. Bradley, N. Stingelin and T. D. Anthopoulos, Appl. Phys. Lett., 2015, 107, 01330 CrossRef.
- A. Patra, N. Chaudhary, R. Chaudhary, J. P. Kesari and S. Chand, J. Mater. Chem. C, 2015, 3, 11886 RSC.
- Z. Mai, Z. Tang, E. Wang, M. R. Andersson, O. Inganas and F. Zhang, J. Phys. Chem. C, 2012, 116, 24462 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5. See DOI: 10.1039/c7ta00835j |
| This journal is © The Royal Society of Chemistry 2017 |