
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the activity and stability of Ir catalysts for PEM electrolyzer anodes by SnO2:Sb aerogel supports: does V addition play an active role in electrocatalysis?†
Li
Wang
a,
Feihong
Song
a,
Guillaume
Ozouf
b,
Dorin
Geiger
c,
Tobias
Morawietz
d,
Michael
Handl
d,
Pawel
Gazdzicki
a,
Christian
Beauger
b,
Ute
Kaiser
c,
Renate
Hiesgen
d,
Aldo S.
Gago
*a and
K. Andreas
Friedrich
*ae
aInstitute of Engineering Thermodynamics, German Aerospace Center (DLR), Pfaffenwaldring 38-40, 70569 Stuttgart, Germany. E-mail: aldo.gago@dlr.de; andreas.friedrich@dlr.de
bMINES ParisTech, PSL Research University, PERSEE-Centre procédés énergies renouvelables et systems énergétiques, CS 10207, Rue Claude Daunesse, 06904 Sophia-Antipolis Cedex, France
cGroup of Electron Microscopy of Materials Science, Central Facility for Electron Microscopy, University of Ulm, 89081 Ulm, Germany
dUniversity of Applied Sciences Esslingen, Dep. of Basic Science, Kanalstrasse 33, Esslingen, 73728, Germany
eInstitute of Energy Storage, University of Stuttgart, Pfaffenwaldring 31, 70569 Stuttgart, Germany
First published on 26th January 2017
Abstract
Low Ir loading oxygen evolution reaction (OER) catalysts with superior activity and durability for proton exchange membrane (PEM) electrolyzers are an important topic in industry and academia. One possible strategy for addressing this challenge is the use of support materials that are stable under highly corrosive acidic environments at a high working potential (>1.4 V). Moreover, highly porous structure is another key criteria for OER catalyst support to achieve a high electrochemical surface area. Here, we report a novel Ir supported on a SnO2:Sb aerogel OER catalyst (Ir/SnO2:Sb-mod-V), which was prepared under ambient pressure by using vanadium additives. It shows an unrivaled activity and enhanced stability, on which vanadium does not play any active role but demonstrates the influences that changes the porosity of the aerogel support and affects the impurity content of the chlorine. By taking advantage of the high porosity of the aerogel substrate, Ir/SnO2:Sb-mod-V allows a decrease of more than 70 wt% for precious metal usage in the catalyst layer while keeping a similar OER activity compared to its unsupported counterpart.
PEM electrolyzers have attracted great attention as a promising technology to store intermittent electricity from solar or wind energy in the last decade. To date, Ir-based electrocatalysts are still the only technically feasible option as an anode catalyst to promote the oxygen evolution reaction (OER) because they combine both high activity and considerable stability.1 However, a large amount of Ir (3–0.5 mg cm−2) is necessary, compared to less than 0.3 mg cm−2 of Pt on the cathode side, to overcome the sluggish OER reaction kinetics, which lowers the efficiency of the electrolyzer.1,2 Considering the high cost and scarcity of Ir, extensive research has been performed to address this challenge.3–7
Recently, two main strategies were used for reducing the Ir loading: (i) developing highly active amorphous IrOx based catalysts and (ii) using an electro-conductive ceramic as a substrate material.3,4 Regarding the first strategy, Cherevko et al. and Danilovic et al. investigated amorphous IrOx films on Ir; they showed higher activities than annealed IrO2 layers, yet a higher dissolution rate.8,9 Nanoparticles of IrOx–Ir and Ir-black also show much higher activities compared to state-of-the-art thermally treated IrO2.7 Markovic and co-workers reported one IrRuOx electrocatalyst with improved stability due to the segregation of the Ir into Ir nano-domains by annealing the catalyst.10 Seitz et al. developed an IrOx/SrIrO3 catalyst by leaching Sr from the surface of SrIrO3, outperforming even RuOx systems.3
By taking the second approach, our group demonstrated improved Ir utilization compared to Ir-black by depositing metallic Ir nano-particles on Magnéli phase Ti4O7 without further thermal treatment.6 Chen et al. also reported an amelioration for the OER in acidic medium when using Ti4O7 as a support for Ir-based catalysts.11 In recent years, the well-known electro-ceramic tin oxide doped with antimony (SnO2:Sb) has attracted significant attention from the research community due to its considerable electronic conductivity and high electrochemical stability.12–14 Puthiyapura et al. prepared and investigated the rutile phase of IrO2 supported on commercial SnO2:Sb particles, showing improved Ir utilization.15 In addition, Strasser and co-workers developed a highly active catalyst consisting of IrNiOx supported on mesoporous SnO2:Sb and revealed a metal/metal oxide support interaction (MMOSI) between amorphous IrOx and SnO2:Sb, resulting in an enhancement of the intrinsic OER activity.4,16
The precious metal utilization and stability of nanostructured Ir anodes of PEM electrolyzers have yet to be further improved, the aforementioned efforts notwithstanding. In this context, three-dimensional (3D) aerogel structures of carbon17 and SnO2:Sb18 are widely studied as catalyst supports for the oxygen reduction reaction (ORR) due to their ultra-high intrinsic specific surface area. In the present work, we develop an iridium supported on a SnO2:Sb aerogel catalyst (Ir/SnO2:Sb-mod-V) for OER with well-retained highly porous structure by using V additives during synthesis, showing an unprecedented activity concurrent with an excellent stability compared to Ir/SnO2:Sb and unsupported IrOx. The retained porosity of aerogel substrate after synthesis yields an increased electrochemical surface area, which is directly correlated with catalyst performance. Previous works have reported that vanadium can enhance the electrocatalyst performance either by forming M–V (M is precious metal) alloy19–21 or by surface vanadium redox species modification.22,23 However, our results demonstrate that vanadium species are not an active part for OER, rather showing the significant influences on retaining the aerogel structure under atmospheric drying and lowering the impurity content of the chlorine. To the best of our knowledge, we present for the first time the exploitation of the highly porous 3D structure of aerogel support for enhancing OER activity. The synthesized catalysts were characterized by X-ray photoelectron spectroscopy (XPS), scanning electron microscope (SEM), energy-dispersive X-ray spectroscopy (EDS), high-resolution transmission electron microscopy (HRTEM) and atomic force microscopy (AFM). Cyclic voltammetry (CV), chronopotentiometry and copper underpotential deposition (Cu-UPD) were carried out to evaluate the performance and stability of the catalysts.
An aerogel of SnO2:Sb was formed by drying a previously synthesized gel using a sol–gel route from metal alkoxide precursors in supercritical conditions.24 Nanoparticles of Ir were reduced under an Ar atmosphere, according to the previously reported synthesis,6 and deposited on the aerogel substrate. In the case of Ir/SnO2:Sb-mod-V, an ammonium metavanadate (NH4VO3) was introduced after the addition of the surfactant cetyltrimethylammonium bromide (CTAB) to the solvent. Unsupported IrOx was achieved by the same method without adding the SnO2:Sb aerogel. All the prepared catalysts were used without further thermal treatment, avoiding the formation of the IrO2 rutile phase, which shows lower OER activity than the amorphous IrOx produced via electrochemical oxidation.9
Using XPS analysis, shown in Fig. 1, vanadium was clearly identified in the Ir/SnO2:Sb-mod-V sample. According to the elemental composition profile depicted in Fig. 1a, the atomic concentration of vanadium was 3–4 at%, which was approximately 3 times less than the concentration of iridium, corresponding to a wt% Ir![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V ratio of approximately 10:1. It is worth mentioning that 30 wt% of V (vs. IrV) was calculated as the nominal ratio (atomic V:Ir ratio of approximately 1.6:1) for the synthesis, which is much higher than the content detected by XPS, meaning that more than half of V was not reduced and rinsed away afterwards.
:V ratio of approximately 10:1. It is worth mentioning that 30 wt% of V (vs. IrV) was calculated as the nominal ratio (atomic V:Ir ratio of approximately 1.6:1) for the synthesis, which is much higher than the content detected by XPS, meaning that more than half of V was not reduced and rinsed away afterwards.
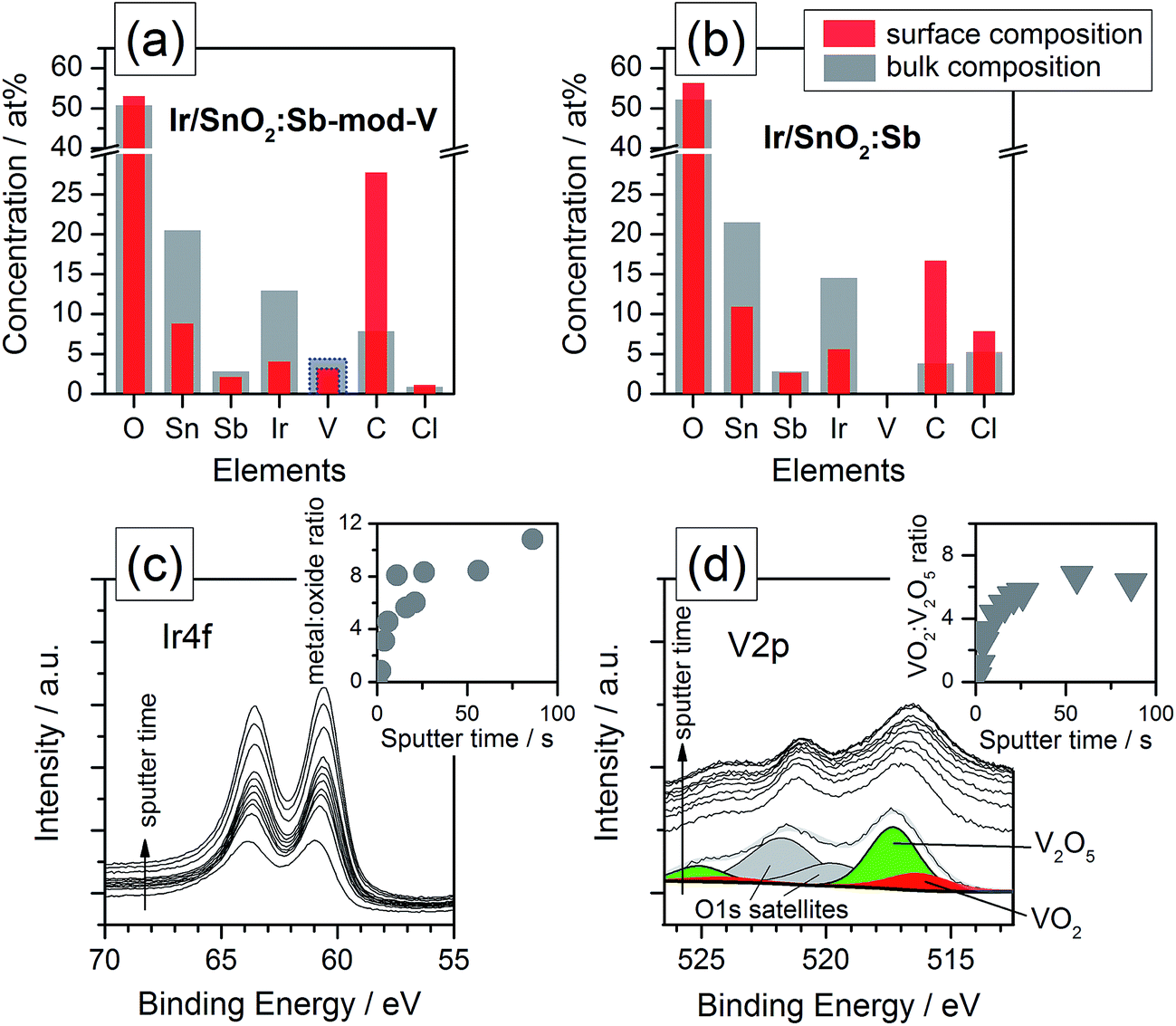 | ||
| Fig. 1 XPS analysis: surface and bulk element composition profiles of (a) Ir/SnO2:Sb-mod-V and (b) Ir/SnO2:Sb. Panels (c) and (d) show detailed Ir4f and V2p spectra, respectively. In the insets of panels (c) and (d), the respective atomic ratios of Ir-metal:Ir-oxide and VO2:V2O5 are plotted. | ||
The elemental composition profile of Ir/SnO2:Sb, depicted in Fig. 1b for comparison, exhibits very similar elemental concentrations, but with no vanadium (see ESI†), a slightly reduced carbon signal that was detected predominantly on the sample surface. In both cases, the samples were dominated by Sn and O. The bulk Sn:O ratio equals 0.4, which was slightly lower than the expected stoichiometric ratio of the SnO2 support material. The additional O could be due to additional oxidized components or possibly contamination. It is worth to mention that the Cl− contaminates show an apparent increase in the sample of Ir/SnO2:Sb, 5.25 at% compared to 0.88 at% of Ir/SnO2:Sb-mod-V, which normally has a strong negative effect on precious metal electrocatalysts.25,26 This change is ascribed to NH4VO3 additions during synthesis, NaBH4 shows a more powerful reducing properties when V species present in the reaction solution, resulting in a different residual Cl− content.
The Ir composition, obtained by peak fitting of the Ir4f detailed spectra shown in Fig. 1c, is depicted in the inset. Apparently, the surface iridium was fully oxidized, while the metal:oxide ratio increased to 8–10 for the bulk (tsputter > 20 s). Vanadium was detected by measuring the V2p orbital spectra. To discriminate between the V2p peaks and overlapping O1s satellite peaks, the fitted signals before sputtering were added in Fig. 1d (see also ESI†). According to the fitted V2p3/2 levels at peak positions at 516.4 and 517.3 eV, the corresponding vanadium species has been identified as VO2 and V2O5; no metallic vanadium was detected, which was expected between 512–513 eV.27,28 The ratio profile of the vanadium-oxide species, provided as an inset, shows that V2O5 clearly dominates at the sample surface, but mainly VO2 was found to occur in the bulk.
EDS combined with SEM was used for the elemental distribution analysis to analyze the presence of vanadium and chlorine impurity further. The results (Table S2†) show the wt% ratio of Ir:V equals approx. 10:1 in the sample of Ir/SnO2:Sb-mod-V, in agreement with the XPS measurements described above. Ir/SnO2:Sb was also analyzed under the same conditions for comparison purposes (Table S3†), and the absence of the V signals indicates that the V observed on Ir/SnO2:Sb-mod-V was indeed derived from the precursor NH4VO3. Chlorine impurity changes shows the same trend as observed from XPS, ca. 5 times higher in the case of Ir/SnO2:Sb.
The morphology of the Ir/SnO2:Sb-mod-V was analyzed with SEM by using the secondary electron as shown in Fig. 2a (also Fig. S2(b), see ESI†). By comparison with the original SnO2:Sb aerogel (Fig. S2(a), see ESI†), the highly porous structure of aerogel was retained after Ir deposition when the V compounds were present on its surface. Clearly, the highly porous structure of Ir/SnO2:Sb was damaged in the samples without V, as apparent from the dense (Fig. S2(c), see ESI†), flat (Fig. S3(d),† low resolution) morphologic surface. One plausible explanation is that the vanadium compounds, either VO2 or V2O5, reduce the surface tension of the ethanol solution, thereby also reducing the capillary pressure force during the atmospheric drying of the catalyst. It has been reported that replacing a high surface tension solvent with a low surface tension solvent leads to the retention of the 3D cross-linked structure of aerogels during atmospheric drying.29 The corresponding back scattered electron SEM images (Fig. S3(a) and (c), see ESI†) show a uniform Ir dispersion on both Ir/SnO2:Sb-mod-V and Ir/SnO2:Sb on a larger scale, in which Ir particles are present as bright tiny spots.
 | ||
| Fig. 2 (a) SEM image of Ir/SnO2:Sb-mod-V; (b, c) 300 kV aberration-corrected HRTEM images of Ir/SnO2:Sb-mod-V at different magnifications, SnO2:Sb particles are lined with yellow, while Ir-containing areas are marked in red; (d) 300 kV aberration-corrected HRTEM image of Ir/SnO2:Sb. | ||
Fig. 2b shows the HRTEM image of Ir/SnO2:Sb-mod-V. Particles, such as those surrounded by yellow dashed lines, with a size of ca. 10 nm were identified as a single particle of SnO2:Sb aerogel. The small dark particles, indicated by red circles and arrows, were attributed to Ir nano-particles with a smaller particle size of ca. 2–3 nm. As we can see, some of the Ir particles were situated on the substrate as single particles and were well-dispersed on the aerogel structure, while some show agglomeration as illustrated in the red circle of Fig. 2b. One selected area of the Ir/SnO2:Sb-mod-V is shown in Fig. 2c at higher magnification, where a single Ir particle is next to SnO2:Sb particles. Crystallographic analysis enabled the (111) lattice plane of the Ir nano-particles and the (110) lattice plane of the SnO2 particles to be discerned. Interestingly, no evidence of V was found at the nano-scale, as shown in Fig. 2b and c, which implies that Ir and V did not form a solid solution and that the V was distributed heterogeneously in the sample of Ir/SnO2:Sb-mod-V. For comparison, a HRTEM image of Ir/SnO2:Sb is shown in Fig. 2d. As expected, Ir and SnO2:Sb were recognized based on the Ir (002) lattice plane and the SnO2 (101) lattice, with characteristic distances of 1.92 Å and of 2.67 Å, respectively. A similar structure of Ir/SnO2:Sb-mod-V and Ir/SnO2:Sb at the nano-scale indicates that the presence of V had no significant influence on the microstructure of the Ir catalysts supported on SnO2:Sb.
Electronic conductivity, which normally influences performance, is an important parameter for evaluating electrocatalysts. An AFM conductivity measurement was carried out for this purpose. Two SnO2:Sb aerogel supported Ir catalysts were deposited on a Si wafer and electrically connected to the metal substrate with conductive silver paste. Fig. 3a and b show the current mapping of Ir/SnO2:Sb-mod-V and Ir/SnO2:Sb, respectively. The conductive area was (95 ± 4)% for Ir/SnO2:Sb-mod-V and (91 ± 3)% for Ir/SnO2:Sb. The magnitude of the conductive area percentage with a varied threshold current is shown in Fig. 3c. No significant difference in conductivity between Ir/SnO2:Sb-mod-V and Ir/SnO2:Sb was observed. Also a similar mean size of the conductive agglomerates was determined at ∼28 nm for Ir/SnO2:Sb-mod-V and ∼27 nm for Ir/SnO2:Sb.
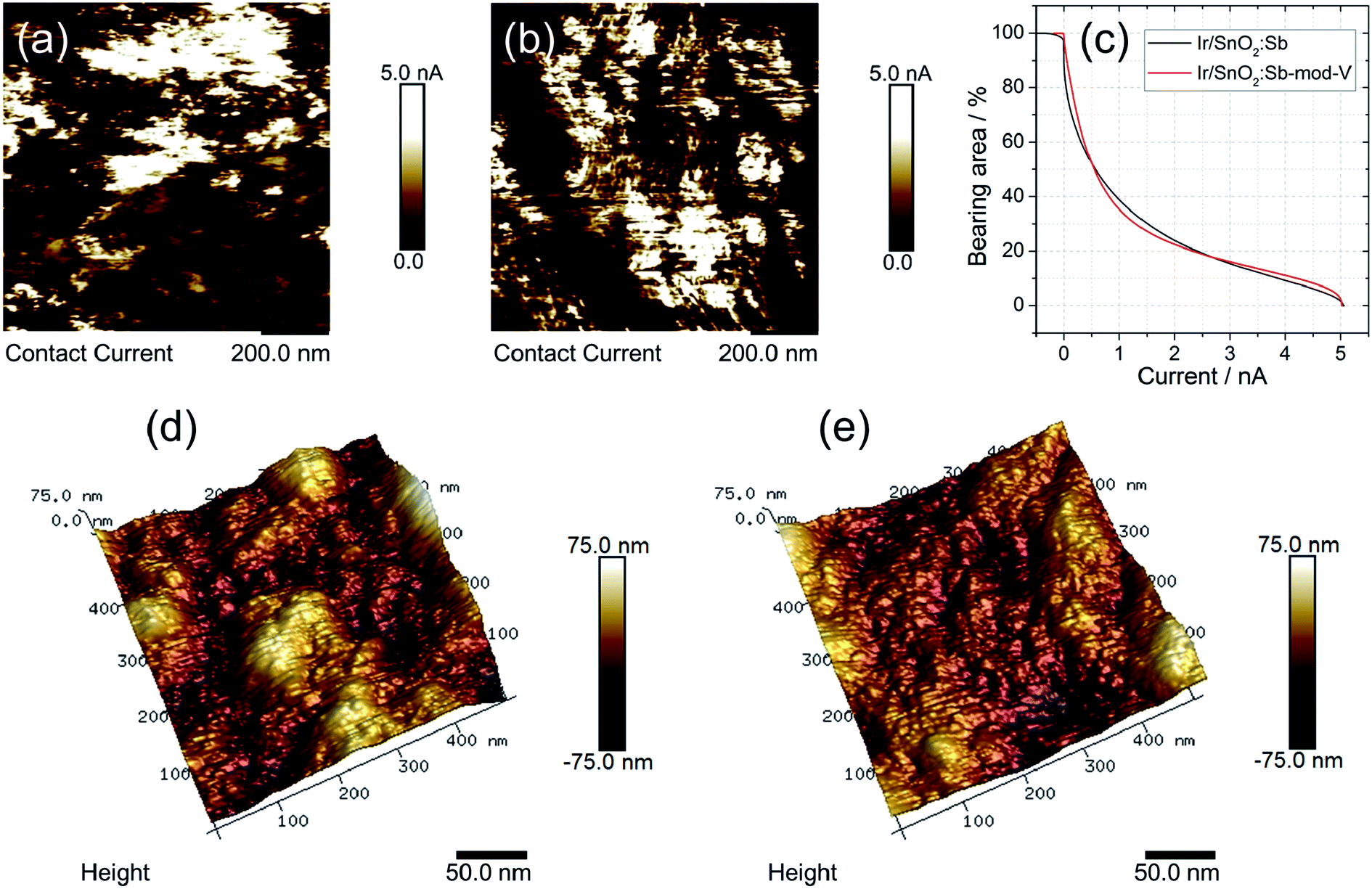 | ||
| Fig. 3 AFM measurements: contact current mapping of (a) Ir/SnO2:Sb-mod-V and (b) Ir/SnO2:Sb; panel (c) shows the bearing area vs. current for the two supported catalysts, evaluated from AFM current mappings; 3D topography of (d) Ir/SnO2:Sb-mod-V and (e) Ir/SnO2:Sb. | ||
Surface roughness can be an indicator for the porosity of powder samples. The porosity is another key parameter for evaluating electrocatalysts, especially OER catalysts, because the porosity influences water transport and the detachment of oxygen. For Ir/SnO2:Sb-mod-V and Ir/SnO2:Sb, the roughness (Ra) determined from three different spots on the samples at the 3 μm scale was (100 ± 20) nm for Ir/SnO2:Sb-mod-V and (80 ± 20) nm for Ir/SnO2:Sb. However, one should be aware of the large error bars, ±20 nm in both cases, and the importance of inter-agglomerate roughness. The mean error is a result of the heterogeneities of the samples at this scale. Note that the determination of (Ra) on a smaller area taking care that only the surface of one agglomerate is measured (evaluating 300 × 300 nm2 areas) leads to a reduced roughness (16 ± 1 nm for Ir/SnO2:Sb-mod-V and 12 nm ± 1 nm for Ir/SnO2:Sb). It was, however, larger for the V-modified sample. In Fig. 3d and e, topography images of Ir/SnO2:Sb-mod-V and Ir/SnO2:Sb are given. Their difference in surface roughness implies that the 3D porous structure of Ir/SnO2:Sb-mod-V was retained after synthesis, while the porous structure partially collapsed in the case of Ir/SnO2:Sb. This was in agreement with the SEM morphology analysis.
Three catalysts were electrochemically oxidized by 10 cycles of CV between 0 V and 1.6 V vs. a reversible hydrogen electrode (RHE) (Fig. S7, see ESI†). Afterwards, 3 cycles of CV were carried out from 1.0 V to 1.6 V to examine OER activity. The first CV cycle after capacitance correction is shown in Fig. 4a. Both SnO2:Sb aerogel supported Ir catalysts displayed significantly enhanced OER activity normalized by Ir loading compared to the unsupported IrOx catalyst that we previously published.7 Furthermore, Ir/SnO2:Sb-mod-V exhibited slightly higher OER mass activity compared with Ir/SnO2:Sb. Ir mass activity was compared at an overpotential of 280 mV (Fig. 4b), where mass transport effects were negligible. At this potential, unsupported IrOx, Ir/SnO2:Sb and Ir/SnO2:Sb-mod-V reached 33.2 A g−1, 94.6 A g−1 and 121.5 A g−1, respectively. For comparison, IrOx/com.-ATO and IrNiOx/meso-ATO reported by Nong et al.,4 reached ca. 32 A g−1 and 89 A g−1, respectively. It is worthwhile to note that any comparison of mass activities is difficult because the measured values depend greatly on the electrode preparation method, the amount of ionomer in the catalyst layer, the electrolyte concentration, etc. Even so, the most significant result is the fact that the three catalysts showed similar geometric OER activity, while the supported samples contained only ca. 28 wt% Ir on the electrodes (Fig. S8, see ESI†), showing that the SnO2:Sb aerogel support had successfully improved Ir utilization. In more practical terms, one can foresee that it is possible to retain high efficiencies with less than 70 wt% Ir loading in the membrane electrode assembly (MEA). In this context, the use of aerogel supports paves a way to develop ultra-low loading Ir anodes for PEM electrolyzers.
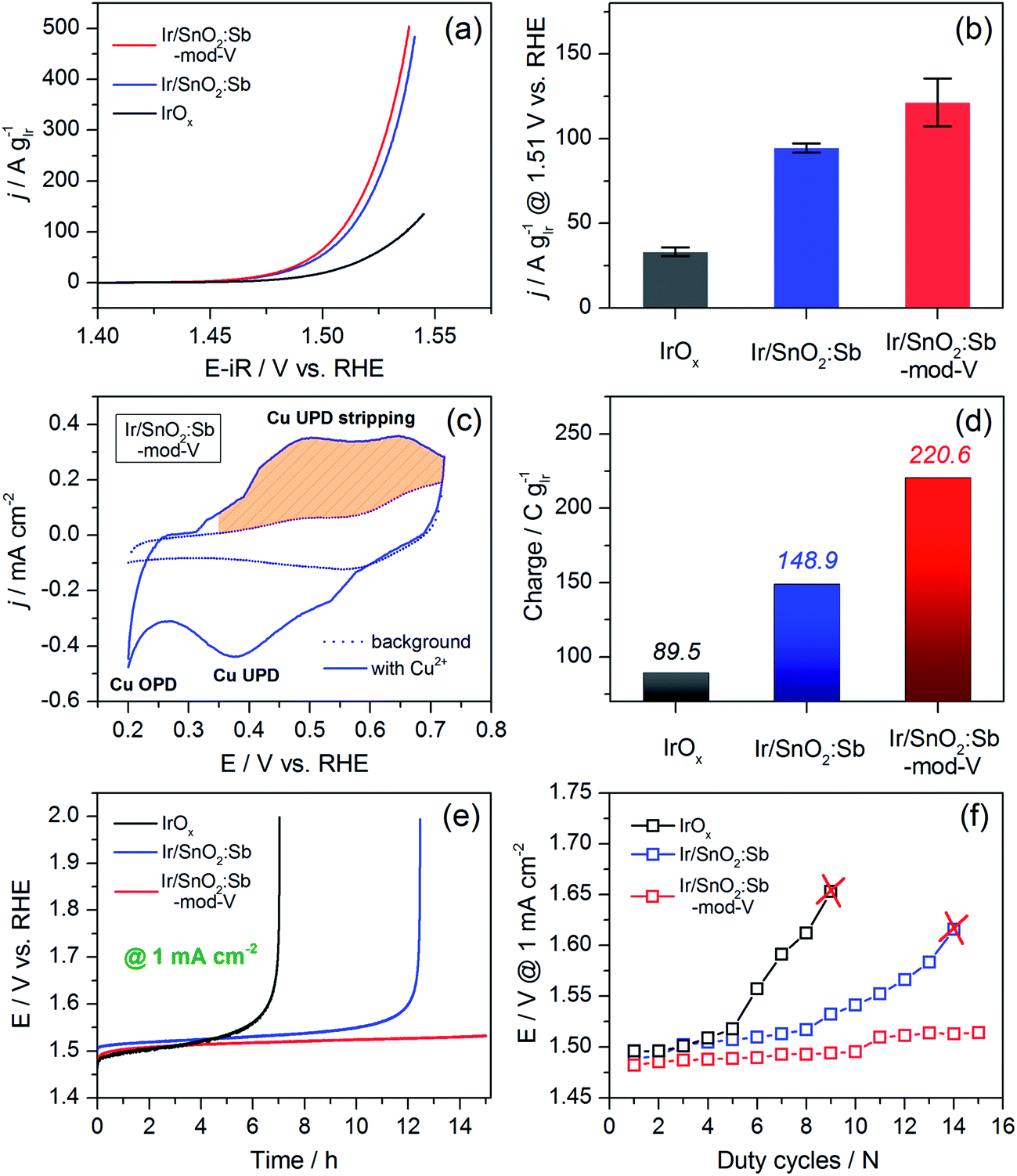 | ||
| Fig. 4 (a) OER activity performance of Ir/SnO2:Sb-mod-V, Ir/SnO2:Sb and unsupported IrOx, 25 °C, N2-saturated 0.05 M H2SO4, 5 mV s−1, 1600 rpm; (b) mass activity comparison under an overpotential of 280 mV; (c) Cu-UPD profile of Ir/SnO2:Sb-mod-V; (d) charge comparison based on Cu-UPD stripping peaks among three catalysts; (e) stability tests: chronopotentiometry with a current density of 1 mA cm−2 for 15 hours, 25 °C, N2-saturated 0.05 M H2SO4; (f) duty cycle measurements: the corresponding potential under 1 mA cm−2 after each duty cycle was recorded. Ir loading on electrodes: 60 μg cm−2 for IrOx, 17 μg cm−2 for Ir/SnO2:Sb-mod-V and Ir/SnO2:Sb. | ||
The Tafel slope of three catalysts are given in Table 1, 43.7 mV dec−1, 41.8 mV dec−1 and 40.9 mV dec−1 for IrOx, Ir/SnO2:Sb and Ir/SnO2:Sb-mod-V, respectively. The slight difference between supported catalysts and unsupported catalyst can be attributed to the influence from MMOSI.16 Similar Tafel slope implies that three catalysts have the same active center, namely Ir, and V does not contribute to the OER catalysis in the case of Ir/SnO2:Sb-mod-V. This is confirmed by XPS analysis on electrode surface (Fig. S10, ESI†) after electrochemical oxidation (seq. 1 and seq. 2 in Table S1†), a ca. 95 at% decrease of vanadium content strongly indicates that V is only a transient before catalyst electrochemical activation. It does not play any active role in OER catalysis, rather influencing the porosity of aerogel support and residual chlorine content.
| Catalyst | IrOx | Ir/SnO2:Sb | Ir/SnO2:Sb-mod-V |
|---|---|---|---|
| Tafel slope (mV dec−1) | 43.7 | 41.8 | 40.9 |
The catalyst particles of Ir/SnO2:Sb-mod-V after 10 CV cycles of electrochemical oxidation (EC) were collected and analyzed by HRTEM. The image (Fig. S4, see ESI†) showed that Ir and SnO2:Sb retained a similar size and structure, ca. 2 nm and 10 nm, respectively, compared to the fresh sample. This indicates that no significant changes, either for Ir particles or SnO2:Sb particles, were observed after the EC protocol.
To further elucidate the OER activity difference among the three catalysts, a Cu-UPD measurement was performed to determine the charge during Cu-UPD stripping, which is related to the electrochemical surface area. Copper is a suitable choice for UPD on iridium because both elements have similar atomic radii: Cu (0.128 nm) and Ir (0.136 nm).30 The integration of the Cu-UPD stripping peak area analysis enables the calculation of the electrochemical surface area of Ir, supposing one copper atom adsorbed onto one surface iridium atom and that 2 electrons are transferred.31Fig. 4c shows the Cu-UPD profile of Ir/SnO2:Sb-mod-V and its background CV curve measured in the same electrolyte without Cu2+. The Cu-UPD peak appeared in the cathodic scan at ca. 0.38 V vs. RHE. This was followed by copper overpotential deposition (Cu-OPD) from ca. 0.25 V.32 The anodic peak from 0.35 V to 0.72 V vs. RHE was attributed to the Cu-UPD stripping and used for evaluating the UPD charge; the values are provided in Fig. 4d.
Based on their Cu-UPD stripping peaks (Fig. S11, ESI†), a charge of 220.6 C g−1 relative to the Ir loading was observed for Ir/SnO2:Sb-mod-V, whereas the charge for Ir/SnO2:Sb and unsupported IrOx was 148.9 C g−1 and 89.5 C g−1, respectively. This result was in agreement with the Ir mass normalized OER activity under an overpotential of 280 mV, implying that the Cu-UPD stripping charge can be used as an indicator of the electrochemical surface area for metallic Ir-based OER catalysts.
Chronopotentiometry was used for evaluating the stability, presented in Fig. 4e. As we can see, the potential of unsupported IrOx and Ir/SnO2:Sb, under a constant current density of 1 mA cm−2, increased sharply to 2.0 V vs. RHE after ca. 7 and 12 h, respectively. On the contrary, no significant potential increase was observed for Ir/SnO2:Sb-mod-V during the 15 h test. The result demonstrates that the stability of the electrodes with supported catalysts was clearly promoted by using SnO2:Sb aerogel substrate, and it was further enhanced in the case of Ir/SnO2:Sb-mod-V due to its higher support porosity and less Cl− contaminates, resulting in a higher accessible electrochemical surface area.
The stability of the catalysts were further confirmed by performing a “duty-cycle” protocol (details see ESI†), which was proposed by Strasser and co-workers to simulate PEM electrolyzer working conditions,4 to provide a dynamic operation environment. The results are shown in Fig. 4f. Unsupported IrOx and Ir/SnO2:Sb completely lost their activities after 9 and 14 duty-cycles, respectively, while the overpotential of Ir/SnO2:Sb-mod-V at 1 mA cm−2 increased only slightly after 15 duty-cycles, in agreement with the stability trend shown in Fig. 4e.
Based on the above investigation, the boosted OER activity and superior stability of Ir/SnO2:Sb-mod-V can be explained by the following: (1) Ir utilization was significantly increased by using an aerogel structure as a support because of its 3D cross-linked porous structure, confirmed by Cu-UPD measurements; (2) possibly, the MMOSI between Ir and SnO2:Sb additionally enhanced the OER;16 (3) V compounds (VO2 or V2O5), which present as a transient, help to retain the highly porous 3D structure of the aerogel and lower the chlorine impurity content, leading to a high electrochemical surface area.
In summary, conductive ceramic aerogel, SnO2:Sb, has been introduced for the first time as OER catalyst supports for PEM electrolyzers, showing promising applications. The highly porous structure of SnO2:Sb aerogel was successfully retained by using V additives under atmospheric drying. Ir/SnO2:Sb-mod-V demonstrates an unprecedentedly improved Ir utilization and enhanced stability, which is attributed to its high porosity of aerogel substrate and low chlorine impurity content, resulting in a high accessible electrochemical surface area. V addition importantly influences the porosity of catalyst support and Cl− contaminates content, but does not play any active role in OER catalysis. Chlorine-contained precursor should be avoided in future work to get rid of Cl− contaminations. Further understanding of surface and electronic interactions between the SnO2:Sb aerogel substrate and Ir active sites requires fine structure spectroscopies and in operando characterization techniques. These studies are part of our ongoing work. In addition, atomic layer deposition (ALD) method, which is a novel approach for preparing advanced Pt-based catalysts for PEM fuel cells,33 should be used and compared as a reference in the future work in order to compete with the cutting-edge technique.
Acknowledgements
The authors appreciate Ina Plock (DLR, Stuttgart) and Suzanne Jacomet (Armines, France) for the SEM and EDS analysis, also acknowledge Anke Lützner (DLR, Stuttgart) for the XPS measurements and Pierre Ilbizian (Armines, France) for the supercritical drying. The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007–2013) for Fuel Cell and Hydrogen Joint Technology Initiative under Grant No. 621237 (INSIDE) and Grant No. 325239 (NanoCAT).References
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- C. Rozain, E. Mayousse, N. Guillet and P. Millet, Appl. Catal., B, 2016, 182, 153–160 CrossRef CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- H. N. Nong, H. S. Oh, T. Reier, E. Willinger, M. G. Willinger, V. Petkov, D. Teschner and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 2975–2979 CrossRef CAS PubMed.
- E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, C. Schlaup, M. Malizia, T. W. Hansen, S. Horch, I. E. L. Stephens and I. Chorkendorff, Chem. Sci., 2015, 6, 190–196 RSC.
- L. Wang, P. Lettenmeier, U. Golla-Schindler, P. Gazdzicki, N. A. Cañas, T. Morawietz, R. Hiesgen, S. S. Hosseiny, A. S. Gago and K. A. Friedrich, Phys. Chem. Chem. Phys., 2016, 18, 4487–4495 RSC.
- P. Lettenmeier, L. Wang, U. Golla-Schindler, P. Gazdzicki, N. A. Cañas, M. Handl, R. Hiesgen, S. S. Hosseiny, A. S. Gago and K. A. Friedrich, Angew. Chem., Int. Ed., 2016, 128, 752–756 CrossRef.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J. Grote, A. Savan, B. Ratna, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. Lett., 2014, 5, 2474–2478 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2014, 53, 14016–14021 CrossRef CAS PubMed.
- G. Chen, S. R. Bare and T. E. Mallouk, J. Electrochem. Soc., 2002, 149, A1092–A1099 CrossRef CAS.
- R. Kötz, S. Stucki and B. Carcer, J. Appl. Electrochem., 1991, 21, 14–20 CrossRef.
- H. S. Oh, H. N. Nong and P. Strasser, Adv. Funct. Mater., 2015, 25, 1074–1081 CrossRef CAS.
- F. Vicent, E. Morallón, C. Quijada, J. L. Vázquez, A. Aldaz and F. Cases, J. Appl. Electrochem., 1998, 28, 607–612 CrossRef CAS.
- V. K. Puthiyapura, M. Mamlouk, S. Pasupathi, B. G. Pollet and K. Scott, J. Power Sources, 2014, 269, 451–460 CrossRef CAS.
- H. S. Oh, H. N. Nong, D. Teschner, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schloegl and P. Strasser, J. Am. Chem. Soc., 2016, 138, 12552–12563 CrossRef CAS PubMed.
- H. Du, L. Gan, B. Li, P. Wu, Y. Qiu, F. Kang, R. Fu and Y. Zeng, J. Phys. Chem. C, 2007, 111, 2040–2043 CAS.
- G. Ozouf, G. Cognard, F. Maillard, L. Guétaz, M. Heitzmann and C. Beauger, ECS Trans., 2015, 69, 1207–1220 CrossRef.
- L. G. R. A. Santos, K. S. Freitas and E. A. Ticianelli, Electrochim. Acta, 2009, 54, 5246–5251 CrossRef CAS.
- H. Yano, M. Kataoka, H. Yamashita, H. Uchida and M. Watanabe, Langmuir, 2007, 23, 6438–6445 CrossRef CAS PubMed.
- B. Li, D. C. Higgins, D. Yang, H. Lv, Z. Yu and J. Ma, Int. J. Hydrogen Energy, 2013, 38, 5813–5822 CrossRef CAS.
- C. Gutsche, C. J. Moeller, M. Knipper, H. Borchert, J. Parisi and T. Plaggenborg, Electrocatalysis, 2015, 6, 455–464 CrossRef CAS.
- Y. Li, H. Xu, H. Zhao, L. Lu and X. Sun, J. Appl. Electrochem., 2016, 46, 183–189 CrossRef CAS.
- G. Ozouf and C. Beauger, J. Mater. Sci., 2016, 51, 5305–5320 CrossRef CAS.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 508, 41–47 CrossRef CAS.
- I. Katsounaros, J. C. Meier and K. J. J. Mayrhofer, Electrochim. Acta, 2013, 110, 790–795 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- Handbook of X-ray Photoelectron Spectroscopy, ed. J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Physical Electronics, Inc., 1995 Search PubMed.
- J. H. Harreld, W. Dong and B. Dunn, Mater. Res. Bull., 1998, 33, 561–567 CrossRef CAS.
- J. C. Slater, J. Chem. Phys., 1964, 41, 3199 CrossRef CAS.
- C. L. Green and A. Kucernak, J. Phys. Chem., 2002, 2, 11446–11456 CrossRef.
- S. C. F. Araujo, Master degree thesis, University of North Texas, August 2016.
- N. Cheng, Y. Shao, J. Liu and X. Sun, Nano Energy, 2016, 29, 220–242 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, energy-dispersive X-ray spectroscopy (EDS) analysis, additional XPS, SEM, HRTEM and electrochemical measurements results. See DOI: 10.1039/c7ta00679a |
| This journal is © The Royal Society of Chemistry 2017 |