
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stoichiometric water splitting using a p-type Fe2O3 based photocathode with the aid of a multi-heterojunction†
Keita
Sekizawa
 *,
Keiichiro
Oh-ishi
,
Keita
Kataoka
,
Takeo
Arai
,
Tomiko M.
Suzuki
and
Takeshi
Morikawa
*,
Keiichiro
Oh-ishi
,
Keita
Kataoka
,
Takeo
Arai
,
Tomiko M.
Suzuki
and
Takeshi
Morikawa
Toyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute, Aichi 480-1192, Japan. E-mail: e1655@mosk.tytlabs.co.jp
First published on 28th February 2017
Abstract
Fe2O3-based photocathodes are one of the least expensive options for hydrogen generation by water splitting. Although p-type N,Zn-doped Fe2O3 (N,Zn–Fe2O3) has been reported to possess a negative conduction band minimum position sufficient for photocathodic hydrogen generation, the efficiency and stability of the resulting H2 production is low and the reaction is sacrificial. In the present work, analysis by hard X-ray photoelectron spectroscopy (HAXPES) showed that these negative characteristics result from the self-redox reaction of p-type Fe2O3. Based on this result, a TiO2 layer was introduced onto the surface of p-type N,Zn–Fe2O3 to passivate surface defects. In addition, to ensure efficient electron transfer, a thin Cr2O3 layer was also inserted between N,Zn–Fe2O3 and a bottom side conductive oxide layer to generate a favorable band alignment for hole transfer. The resulting Pt/TiO2/N,Zn–Fe2O3/Cr2O3 electrode exhibits a highly stable, significantly enhanced cathodic photocurrent during H2 production under AM 1.5 irradiation. The mechanism providing this improvement was investigated by combining electrochemical impedance spectroscopy, open-circuit voltage decay analysis and scanning tunneling electron microscopy-energy dispersive X-ray spectroscopy. Stoichiometric water splitting without an external electrical bias was also demonstrated by connecting the Fe2O3-based photocathode to an n-type SrTiO3−x photoanode, representing the first-ever example of stoichiometric overall water splitting using an Fe-based photocathode.
Introduction
The conversion of solar energy to storable chemical fuel is an attractive and sustainable means of meeting increasing global energy requirements. Photoelectrochemical (PEC) splitting of water into H2 and O2 is a promising method to directly convert solar energy into fuel. The dual bandgap PEC cell, which connects a p-type photocathode for H2 evolution and an n-type photoanode for O2 evolution, can drive overall water splitting while utilizing a wide wavelength region of solar light and without application of an external voltage. Although efficient water splitting with such PEC cells has been previously reported, most such systems involve the use of expensive p-type semiconductors, such as InP and GaP.1 To expand the practical use of PEC systems for the collection of solar light from a widespread area will require construction from cheap semiconductors composed of abundant elements. Recently, dual PEC cells that utilize low-cost p-type photocathodes such as CaFe2O4 (ref. 2) and surface-modified Cu2O3,4 have been reported. However, the overall water splitting reaction is unstable when using these materials, possibly due to a self-redox reaction that occurs, and the amounts of H2 and O2 deviate from the stoichiometric quantities of photogenerated electrons and holes.Hematite (α-Fe2O3) has a 2.1 eV bandgap and is one of the most abundant and low-cost semiconductor materials capable of absorbing a substantial amount of solar light. The doping of α-Fe2O3 with cations such as Mg2+, Zn2+ and Cu2+, or with anions such as N3−, can induce p-type conduction.5–9 It was recently reported that N and Zn-codoped Fe2O3 exhibits an improved cathodic photocurrent during O2 reduction and higher band potentials compared to undoped Fe2O3.10 The negative band edge shift was expected to be due to a surface dipole moment induced by N-doping and Zn-doping. Morikawa et al. have reported that N-doped Ta2O5 films exhibit a negative band edge shift of 0.9 V compared to undoped Ta2O5.11 This N–Ta2O5 generates hydrogen under visible light irradiation via the photocathodic water splitting reaction,12 while no photoreaction occurs at the undoped Ta2O5 anode. Density functional theory calculations by Jinnouchi et al. suggest that the surface dipole moments generated by nitrogen doping would result in the negative band edge shift.13 Hikita et al. also reported band edge shift due to surface dipole moment. They demonstrated modulation of the flat band potential of SrTiO3 photoanodes by as much as 1.3 V, by generating subsurface electrostatic dipoles near a Nb-doped SrTiO3/aqueous electrolyte interface via the formation of a surface LaAlO3 layer.14 In the same manner, a sputter-deposited N,Zn-codoped Fe2O3 film is expected to exhibit a similar shift in band edge energy. Actually, the edge shift was observed for doping N alone, Zn alone, and N,Zn-codoping.8,10 The conduction band minimum (CBM) of N,Zn–Fe2O3 lies higher than the standard electrode potential for H2 generation from water.
Although p-type Fe2O3 has potential applications as a photocathode for H2 generation from water, the reported efficiencies of p-type Fe2O3 electrodes during H2 generation have been minimal. Several reasons have been suggested for this low activity, including poor carrier mobility,15 short photogenerated charge carrier lifetimes,16 the leaking of electrons into the electrolyte,17 and photochemical dissolution under reductive conditions.18 These problems can all be attributed to defect states on the surface and bulk of the Fe2O3. However, to the best of our knowledge, one cannot directly observe the surface defects on a Fe2O3 photoelectrode. Using standard XPS, it is difficult to resolve the close Fe2+ and Fe3+ peaks. In addition, the outermost electrode surface tends to undergo air oxidation prior to XPS observations. Non-destructive hard X-ray photoelectron spectroscopy (HAXPES) in conjunction with wide angular resolution and intense light irradiation at the SPring-8 (Super Photon ring-8 GeV) synchrotron radiation facility (Hyogo, Japan) has, however, resulted in higher energy resolution and greater depth profiles (ca. 30 nm) than conventional XPS, based on the analysis of the take-off angle dependence of the photoelectron spectra.19–21 In the present work, we attempted to observe defect states by HAXPES to investigate the causes of the low activity of Fe2O3 photoelectrodes.
The formation of a heterojunction in the photoelectrode is expected to prevent the formation of defect states during the PEC process. In addition, the potential of the VB and CB edges around the interface of the heterojunction will exhibit a gradient due to the diffusion of charge carriers between two semiconductors having different Fermi levels. This band bending can enhance both photogenerated charge separation and carrier transfer, thus improving the PEC performance.22–27 Moreover, the formation of a surface junction layer is expected to passivate the surface state28,29 and protect the unstable semiconductor surface from side reactions such as reductive dissolution or photocorrosion.30,31 Therefore, if a rigid junction structure is constructed using a stable semiconductor having an appropriate band alignment, it should be possible to improve the photoelectrode performance.
In the present work, significant enhancement of hydrogen generation by water splitting over a photocathode based on p-type Fe2O3 was realized following the introduction of heterojunctions. Overall solar water splitting was also demonstrated using an unassisted tandem (two electrodes) system in combination with a SrTiO3−x photoanode. Here p-type N,Zn–Fe2O3 (ref. 10) was employed as a photoabsorber, in conjunction with an n-type TiO2 coating to ensure both passivation and charge separation. Pt, acting as a cocatalyst for H2 production, was deposited on the TiO2 layer and a Cr2O3 layer was inserted to provide enhanced hole transfer between a bottom transparent conductive oxide (TCO) layer and N,Zn–Fe2O3.
Experimental section
Materials
A tin(IV) oxide/indium tin oxide (ITO) double layered transparent conducting glass (TCO; Geomatec Co., Ltd) was used as the substrate for the electrodes. Commercially available Fe2O3, ZnO, TiO2, Cr and Pt sputtering targets were obtained from Kojundo Chemical Laboratory Co.Preparation
A N,Zn–Fe2O3 film with a thickness of ca. 190 nm was deposited by radio frequency (RF) reactive magnetron sputtering of the Fe2O3 and ZnO targets with an Ar/N2 (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) plasma.10 The input power values used for sputtering of Fe2O3 and ZnO were 600 and 35 W, respectively. The sputtering substrate was SnO2/ITO layered glass with or without a 10 nm thick Cr layer deposited by RF magnetron sputtering with an Ar plasma. After deposition of the N,Zn–Fe2O3 layer, the electrodes were annealed at 823 K under a N2/O2 (4:1 v/v) gas flow for 2 h.
:1 v/v) plasma.10 The input power values used for sputtering of Fe2O3 and ZnO were 600 and 35 W, respectively. The sputtering substrate was SnO2/ITO layered glass with or without a 10 nm thick Cr layer deposited by RF magnetron sputtering with an Ar plasma. After deposition of the N,Zn–Fe2O3 layer, the electrodes were annealed at 823 K under a N2/O2 (4:1 v/v) gas flow for 2 h.
Subsequently, a 10 or 60 nm thick TiO2 layer was deposited on the surface of the N,Zn–Fe2O3 thin film by RF reactive magnetron sputtering of a TiO2 target with an Ar/O2 (4:1 v/v) plasma. The deposited electrodes were then annealed at 748 K under an O2 gas flow for 2 h.
The Pt cocatalyst was loaded onto either TiO2-coated or bare N,Zn–Fe2O3 specimens by RF magnetron sputtering. The amount of Pt applied was adjusted to obtain a thickness of 1 nm.
The n-SrTiO3 photoanode was fabricated using a method described in a previous publication.32 Briefly, (100)-oriented SrTiO3 (Shinkosha Co.) was reduced by heating at 1073 K for 2 h in a quartz tube furnace in a N2/H2 (97:3 v/v) gas flow, followed by cooling for 7 h. The transparent SrTiO3 crystals took on a dark blue coloration.
Photoelectrochemical measurements
PEC assessments of photocathode materials were conducted using an electrochemical analyzer (ALS2325, ALS Co., Ltd), in conjunction with a three-electrode configuration. The photocathode, a silver–silver chloride (Ag/AgCl) electrode and a platinum electrode were used as the working, reference and counter electrodes, respectively. In these trials, the exposed TCO layer of the photocathode was covered with silicone rubber, and all potentials were converted to the reversible hydrogen electrode (RHE) reference scale using the equation E (vs. RHE) = E (vs. Ag/AgCl) + 0.20 V + 0.059 V × pH. A sealed Pyrex® glass cell was employed as the reactor and Ar-saturated aqueous 0.2 M K2SO4 (pH 5.8) or 0.5 M NaHCO3–Na2CO3 (1:1; pH 9.7) solutions were used as the electrolytes. The electrode was irradiated with a light intensity equivalent to one sun (AM 1.5; 100 mW cm−2) using a solar simulator (HAL-320, Asahi Spectra Co.). Prior to exposure, the light intensity was adjusted using a CS-20 instrument (Asahi Spectra Co.). The sample irradiation area was limited to an area of 10 × 10 mm via a slit. Linear sweep voltammetry was conducted at a scan rate of 50 mV s−1 under chopped light irradiation. The PEC water splitting reaction, in conjunction with the application of an electrical bias, was conducted while measuring the photocurrent generated under continuous irradiation at a fixed electrode potential of −0.5 V vs. Ag/AgCl. After incubation for 30 min to allow the products to equilibrate between the liquid and gas phases, the gaseous reaction products were analyzed by gas chromatography (GC; GC-2014, Shimadzu Co.), employing a thermal conductivity detector (TCD), an active carbon column (Shincarbon ST, Shinwa Chemical Industries Co.) and Ar as the carrier gas. Incident photon to current efficiency (IPCE) spectra were acquired under monochromatic light generated by a 300 W xenon lamp (MAX-303, Asahi Spectra Co.), using band-pass filters to obtain specific wavelengths.
Water splitting reaction with the tandem cell
The anode and cathode electrodes were immersed in aqueous 0.2 M KHCO3 in a sealed Pyrex® cell. The electrolyte solution was purged with Ar prior to each trial. Both the electrodes were connected to a digital multimeter (PC7000, Sanwa) to record the current in a two-electrode configuration without the application of an external potential bias. The anode was subsequently exposed to light at one sun intensity, while the cathode was irradiated by the light transmitted through the anode. The illuminated area during these trials was 10 × 10 mm. Following an incubation period of 30 min to allow the products to reach gas–liquid equilibrium, the gaseous products were analyzed by TCD-GC.Characterization
Scanning electron microscopy (SEM) observations were conducted using an S5500 microscope (Hitachi High-Tech). Scanning transmission electron microscopy (STEM) together with energy dispersive X-ray spectroscopy (EDX) was performed with a JEM-2100F microscope (Jeol Co.). Samples for these observations were cut out using a focused ion beam (FIB; NB5000, Hitachi High-Tech Co.). Prior to cutting the samples, the film surfaces were coated with Pt and W layers to protect the specimens from the FIB. The crystal structures of the films were analyzed using X-ray diffraction (XRD; Ultima IV, Rigaku Co.) with Cu Kα radiation, and the optical properties of the films were assessed by UV-vis absorption spectroscopy (UV-3600, Shimadzu Co.). The Raman spectra were measured with a NRS-3300 (Jasco) using a 532 nm excitation light. The amounts of Fe and Zn dissolved in the reaction solution during the PEC water splitting reaction were determined by inductively coupled plasma optical emission spectrometry (ICP-OES, CIRIOS 120EOP, Rigaku). The positions of the valence band maximum of each semiconductor were estimated by photoelectron spectroscopy in air (PESA; AC2, Riken Keiki). These PESA measurements were conducted after immersion of the specimens in 0.5 M NaHCO3–Na2CO3 (1:1; pH 9.7) to adjust the surface pH. The irradiated photon energy (PE) was converted to the RHE reference scale using the equation E (V vs. RHE) = PE − 4.44 V + 0.059 V × pH. XPS data were acquired using a Quantera SXM (Ulvac-Phi Co.) spectrometer, and HAXPES (operating at 7.94 keV) was performed at the BL47XU beamline of SPring-8. The energy and angular distributions of the photoelectrons were determined with a hemispherical analyzer (R4000-HV, VG-Scienta). The objective lens of the HAXPES instrument had an effective acceptance angle of approximately ±30°, and the stability of the system was ascertained using the Au 4f7/2 photoelectron peak of a Au film on a Si substrate. The overall stability of the photoelectron energy was found to be within 50 meV. The angular distribution of the photoelectrons was measured at photoelectron take-off angles of 35 ± 30°, where the take-off angle of the direction perpendicular to the surface was defined as 90°.
Results and discussion
Structural properties and elemental composition of N,Zn–Fe2O3
In this work, N,Zn–Fe2O3 was generated by co-sputtering Fe2O3 and ZnO in a mixed plasma composed of N2 and Ar, followed by post-annealing. The XRD patterns of the resulting N,Zn–Fe2O3 on TCO exhibited peaks assignable to (110) and (300)-oriented hematite but shifted to lower angles compared to those produced by pristine Fe2O3 on TCO (Fig. 1). This shift to lower angles indicates that larger N3− and Zn2+ ions were successfully substituted at O2− and Fe3+ sites in the hematite lattice, respectively. All other diffraction peaks were attributed to SnO2 and ITO from TCO, and no peaks of impurity phase were observed. On the other hand, the Raman spectrum of N,Zn–Fe2O3 exhibited a peak around 670 cm−1 as well as typical hematite peaks (Fig. S1†). The 670 cm−1 peak position was close to those of A1g mode of ZnFe2O4 (ref. 33) and Fe3O4.34 Therefore, an amorphous or very small ZnFe2O4 phase might influence the conduction properties, though the amount of Zn was small compared to that in the previous report of Zn-doped Fe2O3.6 | ||
| Fig. 1 XRD peaks obtained from N,Zn–Fe2O3 (red lines) and pristine Fe2O3 (blue lines) on SnO2/ITO. No other peaks attributable to hematite were detected between 10 and 80 degrees. (a) For (110) and (b) for (300). | ||
The XPS spectrum generated by N,Zn–Fe2O3 is compared with those of Zn–Fe2O3 and N–Fe2O3 in Fig. 2. Here, the N 1s peak of N,Zn–Fe2O3 is shifted to a higher binding energy compared to that of N–Fe2O3, while the Zn 2p peak of N,Zn–Fe2O3 appears at a lower binding energy relative to that of Zn–Fe2O3. These results indicate that the electrons in the Zn atoms would be compensated for by N atoms in N,Zn–Fe2O3. Therefore, it can be expected that the anionic N and cationic Zn dopants in N,Zn–Fe2O3 are likely to be located at the nearest neighboring positions.10 Based on the intensity of the XPS peaks, the N and Zn concentrations in N,Zn–Fe2O3 were estimated to be 1.2 and 0.5%, respectively (Table 1). Although these dopant levels were greater than those in typical semiconductors such as Si or GaAs, these N and Zn concentrations have been optimized to generate high photocurrents for O2 reduction.10 Various p-type Fe2O3 materials, including Zn–Fe2O3,6 Cu–Fe2O3 (ref. 7) and N–Fe2O3,8 also require similar heavy doping levels in order to function optimally as electrodes. These high N and Zn concentrations evidently do not degrade the host material band because the gap of 2.0 eV is not changed by the doping.10 This doping is believed to result in an acceptor level above the VB of Fe2O3 that is composed of Fe 3d and O 2p orbitals, which in turn positively shifts the Fermi level of Fe2O3. As a result, the energy difference between the VB and the Fermi level in the doped material is smaller than in pristine Fe2O3, as confirmed by the VB data obtained by HAXPES (Fig. S2†). N,Zn–Fe2O3 also exhibited a negative slope in its Mott–Schottky (M–S) plot (Fig. S3†), indicating stable p-type conductivity. Therefore, the cathodic photocurrent enhancement resulting from N and Zn doping is attributed to improved p-type conductivity. The determination of the flatband-potential of N,Zn–Fe2O3 in the M–S plot would be difficult due to the gradient of dopant concentration or surface defect.35 The VB maximum was estimated to be 1.5 V vs. RHE from PESA36–38 (Fig. S4†). Since the band gap of N,Zn–Fe2O3 was 2.1 eV, the CB minimum can be estimated to be −0.6 V vs. RHE.39 Therefore, water reduction by N,Zn–Fe2O3 is thermodynamically possible. Since the CB minimum of n-type Fe2O3 has been reported to be +0.6 V vs. RHE, our results suggest that the band alignment was shifted by N and Zn doping. Similar upward shifts of the CB have been demonstrated in other doped semiconductors, such as N–Ta2O5,11 Mg–TiO2 (ref. 40) and N–Cu2O.41 Although the mechanism of the upward shift in band alignment has not yet been clarified, it could be induced by surface dipoles13 or structural changes40,41 resulting from the negative charges in the bulk generated by doping.
 | ||
| Fig. 2 (a) N 1s and (b) Zn 2p XPS spectra obtained from N,Zn–Fe2O3 (red lines), Zn–Fe2O3 (blue lines) and N–Fe2O3 (green lines). | ||
| Sample | N | Oc | Fe | Zn |
|---|---|---|---|---|
| a The compositions were estimated from the intensity of the photoelectron spectra by irradiation with soft X-rays (Al Kα, 1487 eV). b The electrode was used for photocurrent measurement at +0.1 V vs. RHE for 1 h in 0.2 M K2SO4 aqueous electrolyte. c The O concentration measured by XPS contains O2 adsorbed on the N,Zn–Fe2O3 surface. | ||||
| As-prepared | 1.2% | 78.5% | 19.8% | 0.5% |
| 1 h-measuredb | 0.8% | 78.9% | 19.8% | 0.6% |
Photoelectrochemical properties of bare N,Zn–Fe2O3
Fig. 3a (gray line) presents the current–potential data for the N,Zn–Fe2O3-based electrodes in a 0.5 M Na2CO3–NaHCO3 (1:1) buffer electrolyte (pH 9.7) under chopped AM 1.5 irradiation (100 mW cm−2). The bare N,Zn–Fe2O3 electrode generated a spike-like cathodic photocurrent below +1.4 V vs. RHE, an anodic dark current at values more positive than +1.6 V (which was correlated with the VB edge of the p-type N,Zn–Fe2O3), and a cathodic dark current at values more positive than +0.3 V, due to electron leakage. Accordingly, the cathodic photocurrent at a constant potential of +0.1 V vs. RHE was observed to decay immediately after the irradiation (Fig. 3b, gray line). Following irradiation for 1 h, a H2 quantity of 0.015 μmol cm−2 was determined in the gas phase, in conjunction with a low faradaic efficiency of 23%. Subsequently, the spike-like cathodic photocurrent declined and the dark current at a negative voltage also deteriorated (Fig. 3c, orange line). The UV/vis absorption of the material also decreased slightly (Fig. S5,† green line). This decrease is attributed to dissolution of the Fe2O3 in the electrolyte, as confirmed by ICP-OES analysis of the electrolyte (Fig. 4, green line). In contrast, almost no dissolution occurred in 0.2 M K2SO4 (Fig. S5,† red line), although an unstable photocurrent and low PEC H2 evolution rates were observed (Fig. S6†), similar to the results obtained in the 0.5 M Na2CO3–NaHCO3 (1:1) buffer. These results indicate that the bare N,Zn–Fe2O3 electrode was probably deactivated prior to dissolution.
 | ||
| Fig. 3 (a) Current–potential characteristics and (b) photocurrent transients at +0.1 V vs. RHE in a 0.5 M Na2CO3–NaHCO3 (1:1) buffer electrolyte (pH 9.7) under one sun (100 mW cm−2, AM 1.5) illumination for bare N,Zn–Fe2O3 (gray lines), Pt/TiO2/N,Zn–Fe2O3 (blue lines) and Pt/TiO2/N,Zn–Fe2O3/Cr2O3 (red lines). (c) Current–potential characteristics for bare N,Zn–Fe2O3 before (gray line) and after (orange line) the 1 h PEC stability test at +0.1 V vs. RHE, and of the as-prepared Pt/N,Zn–Fe2O3 electrode (green line). | ||
 | ||
| Fig. 4 ICP-OES Fe and Zn data obtained for a bare N,Zn–Fe2O3 electrode in a 0.5 M Na2CO3–NaHCO3 (1:1) solution before (blue lines) and after (green lines) the 3 h PEC stability test, and those after the 3 h PEC stability test for a Pt/TiO2/N,Zn–Fe2O3 electrode (red lines). The PEC stability tests were performed under solar simulation light (AM 1.5, one sun, 100 mW cm−2) at +0.1 V vs. RHE. | ||
To investigate the deactivation in detail, the electronic states of the Fe atoms were analyzed using HAXPES. In order to distinguish dissolution from deactivation, the bare N,Zn–Fe2O3 electrode was measured before and after the stability test in 0.2 M K2SO4. Fig. 5 presents the angular-resolved Fe 2p spectra. The monitored angle was varied from 3° to 54°, corresponding to depths ranging from 3 to 26 nm.42 Although the Fe 2p binding energy peaks were shifted in the positive direction following the 1 h PEC stability test, both spectra exhibit two peaks around 710.5 and 709.5 eV, attributed to Fe3+ and Fe2+ species, respectively.43 In the as-prepared sample, although intrinsic Fe2+ species presumably originating from oxygen vacancies and/or charge compensation by N3− and Zn2+ doping were observed, the Fe2+/Fe3+ ratio was not dependent on the take-off angle. However, in the case of the deactivated sample, this ratio was increased at lower take-off angles. A fitting analysis also demonstrated that the difference between the data acquired at angles of 3° and 54° was statistically significant (Fig. S7†). Therefore, it appears that surface Fe3+ ions were reduced during the PEC measurements. Photogenerated electrons presumably reduced Fe3+ to Fe2+ without reducing H+ to produce H2 at the surface. These reduced Fe species would then generate electron trap states, leading to the spike-like photocurrent curve. In addition, the reduced Fe species would be dissolved in Na2CO3–NaHCO3 buffer electrolytes as [Fe(CO3)2]2−. Deactivation of the dopant was also observed. In the case of the Zn 2p spectra acquired at 89°, equivalent to a depth of 32 nm, the shoulder at 1020.2 eV (attributed to doped Zn adjacent to N based on XPS analysis: see Fig. 2) disappeared following the PEC measurements (Fig. 6). In addition, soft XPS showed that the N concentration at the surface decreased from 1.2 to 0.8% (Table 1). These results indicate self-oxidative deactivation, that is, photogenerated holes oxidized N anions to N2 (2N3− + 6h+ → N2).44 The observed increase in the O concentration from 78.5 to 78.9% can possibly be attributed to the oxidation of vacant sites accompanied by N elimination, as the result of exposure to air during the XPS sample preparation. The migration of photogenerated holes was slow in the bare N,Zn–Fe2O3 photocathode formed on the TCO, so that these holes were available for self-oxidation even at the surface. Hence, the shifts in the Fe 2p and Zn 2p binding energy values following stability testing were due to the Fermi level shift caused by the deactivation of the N and Zn dopants.
 | ||
| Fig. 5 Take-off angle dependent Fe 2p spectra acquired from (a) the bare as-prepared N,Zn–Fe2O3 electrode and (b) the N,Zn–Fe2O3 electrode following the 1 h stability test. Spectra were obtained by hard X-ray (7938.3 eV) irradiation and monitoring of the take-off photoelectrons at various angles. | ||
 | ||
| Fig. 6 Zn 2p spectra acquired from the bare as-prepared N,Zn–Fe2O3 electrode before (red line) and after (blue line) the 1 h stability test. Spectra were obtained by hard X-ray (7938.3 eV) irradiation and monitoring of the take-off photoelectrons at 89°. The spectrum for the electrode after the 1 h stability test is shifted along the x axis by +0.35 eV because the Fermi level was shifted positively during the stability testing. | ||
To improve the hydrogen evolution rate, the PEC self-redox reaction must be prevented by the promotion of electron transfer from the surface of N,Zn–Fe2O3 to the active sites for H2 evolution, and by the promotion of hole transfer from N,Zn–Fe2O3 to the TCO layer. Pt nanoparticles were deposited on N,Zn–Fe2O3 by sputtering to act as a co-catalyst for H2 evolution. The presence of these Pt nanoparticles was confirmed by TEM observations (Fig. S8†). The current–potential curve for Pt/N,Zn–Fe2O3, as acquired using chopped light illumination in a 0.5 M Na2CO3–NaHCO3 (1:1) buffer electrolyte, exhibited both an anodic dark current at values more positive than 1.5 V and a cathodic dark current between 1.2 and 0 V, including a peak at 0.4 V (Fig. 3c). Loading with Rh and Au nanoparticles also resulted in similar dark currents. The significant dark current seen in these trials suggests that new leakage states were generated following the metal deposition. Although we have no direct evidence, this phenomenon could possibly be explained by an adsorbate-induced surface state,45 resulting from the rearrangement of dangling bonds on the surface. The close proximity of Pt atoms to the N,Zn–Fe2O3 surface to form bonds would split the dangling-bonds within the bandgap into bonding and anti-bonding states. The cathodic peak observed at 0.4 V vs. RHE is therefore attributed to electron leakage to these anti-bonding states. There have been no reports of metallic nanoparticles showing a beneficial effect for hydrogen production by water splitting with Fe-based p-type semiconductors.5 Therefore, in order to effectively employ a metal co-catalyst, the dangling bonds at the N,Zn–Fe2O3 surface should be passivated.
Coating with TiO2 as a passivation and electron transfer layer
The surface of the N,Zn–Fe2O3 photocathode was covered with a TiO2 layer prior to deposition of the Pt nanoparticles (Pt/TiO2/N,Zn–Fe2O3). TEM and STEM combined with EDX for elemental mapping were used to assess a cross-section of Pt/TiO2/N,Zn–Fe2O3, as shown in Fig. 7. The TiO2 layer was confirmed to completely cover the N,Zn–Fe2O3 photocathode, thus preventing the direct contact of N,Zn–Fe2O3 with Pt and the electrolyte. Although the XRD peak attributable to TiO2 was not observed in TiO2/N,Zn–Fe2O3/SnO2/ITO/glass because of the overlap of diffraction from the TCO underlayer, TiO2/glass showed XRD peaks attributable to anatase and rutile phases (Fig. S9†). The UV/vis absorption spectrum of TiO2/N,Zn–Fe2O3 (Fig. S10†) was consistent with the summation of the spectra of TiO2 and N,Zn–Fe2O3 below 400 nm.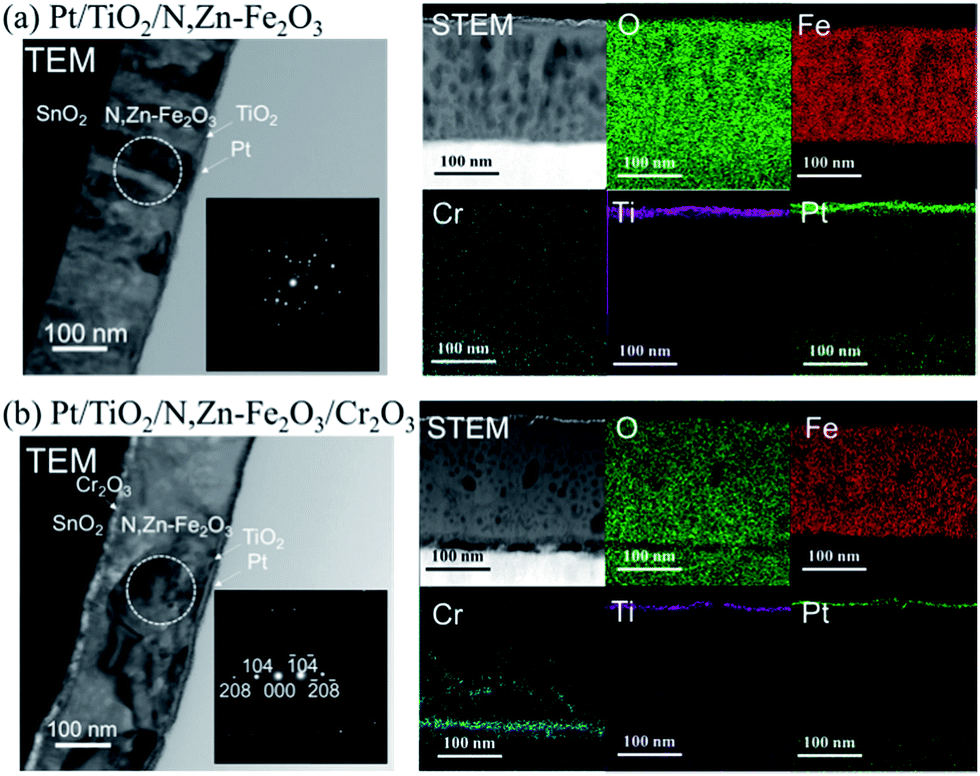 | ||
| Fig. 7 Cross-sectional TEM and STEM images and STEM-EDX elemental maps for (a) Pt/TiO2/N,Zn–Fe2O3 and (b) Pt/TiO2/N,Zn–Fe2O3/Cr2O3. Insets in the TEM images show selected area electron diffraction patterns of the regions indicated by the circles. | ||
Fig. 3a (blue line) shows the current–potential curve for Pt/TiO2/N,Zn–Fe2O3 in a 0.5 M Na2CO3–NaHCO3 (1:1) solution under chopped one sun (100 mW cm−2, AM 1.5) irradiation. An anodic photocurrent was observed at +0.9 V vs. RHE and higher, and this current is believed to have originated from TiO2 excitation because it disappeared under visible light (λ > 420 nm) irradiation (Fig. S11†). A higher cathodic photocurrent than that obtained from the bare N,Zn–Fe2O3 was generated below +0.9 V. In this case, the dark current for Pt/N,Zn–Fe2O3 resulting from Pt adsorption disappeared, suggesting that the TiO2 layer passivated dangling bonds on the N,Zn–Fe2O3 surface, resulting in a significant enhancement of the photocurrent. Fig. 3b (blue line) presents the time course of the photocurrent at a constant potential of +0.1 V vs. RHE. Although the photocurrent decayed slowly, from 50 to 5 μA cm−2, during a 1 h irradiation, 0.33 μmol cm−2 of H2 was generated with a faradaic efficiency of 100%, and no dissolution was evident from the ICP-OES analysis (Fig. 4, red line). These data demonstrate that the self-redox reaction was completely prevented by the TiO2 layer coating. To confirm the origin of the cathodic photocurrent, the IPCE spectrum of Pt/TiO2/N,Zn–Fe2O3 was acquired (Fig. 8). The IPCE values were found to be higher than those obtained from the bare N,Zn–Fe2O3 and exhibited good correlation with the UV-vis absorption spectrum of N,Zn–Fe2O3. This result clearly indicates that the enhanced cathodic photocurrent originated from excitation of N,Zn–Fe2O3.
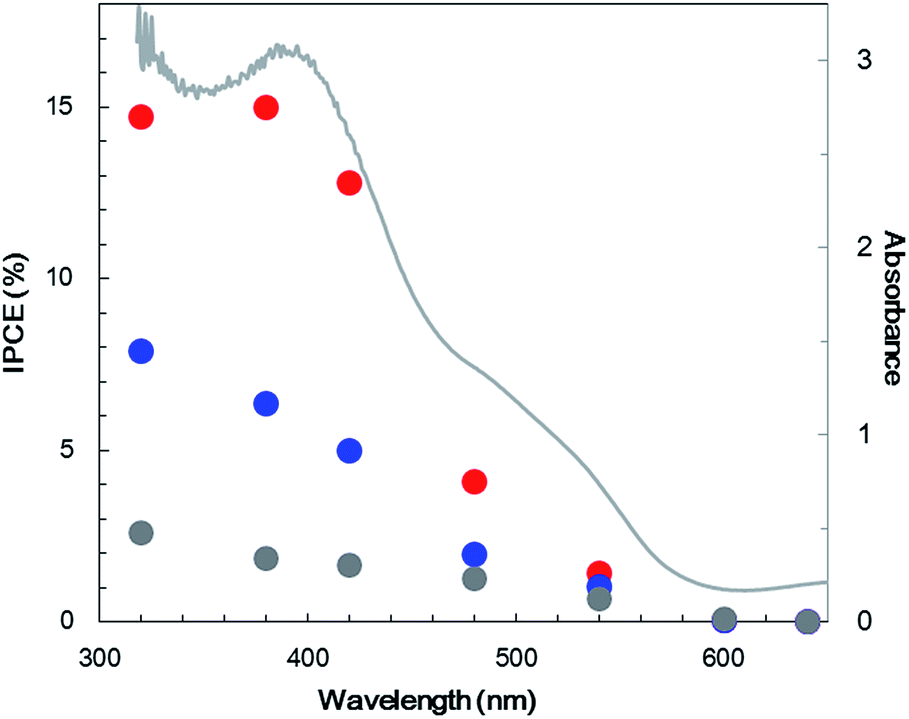 | ||
| Fig. 8 IPCE spectra (data points; left axis) for bare N,Zn–Fe2O3 (gray), Pt/TiO2/N,Zn–Fe2O3 (blue) and Pt/TiO2/N,Zn–Fe2O3/Cr2O3 (red) and the UV-visible absorption spectrum (line; right axis) for N,Zn–Fe2O3. IPCE data were acquired at 0.1 V vs. RHE with monochromatic light irradiation. | ||
The enhancement of the photocurrent can be explained by the formation of a p–n junction. Since the position of the CB minimum of TiO2 is reported to be −0.2 V vs. RHE,46 the band diagram before contact can be described as in Fig. 9a. Following the application of the n-type TiO2 layer (with a higher Fermi level than N,Zn–Fe2O3), some of the holes in N,Zn–Fe2O3 should diffuse across the junction and combine with electrons to form positive ions in the TiO2 region. Similarly, some electrons in TiO2 would be expected to diffuse through the material and combine with holes to form negative ions in the N,Zn–Fe2O3 region. These carrier diffusions should equalize the Fermi levels.47 As a consequence, the N,Zn–Fe2O3 band edge will be shifted to a higher energy at the bulk side and the band edge of N,Zn–Fe2O3 should bend downward towards the interface. In contrast, the TiO2 band should shift to a lower energy, resulting in upward band bending at the interface, as shown in Fig. 9b. The increased CB level of N,Zn–Fe2O3 at the bulk side increases the driving force for electron transfer to the Pt co-catalyst. Moreover, the band-bending between N,Zn–Fe2O3 and TiO2 facilitates charge separation.
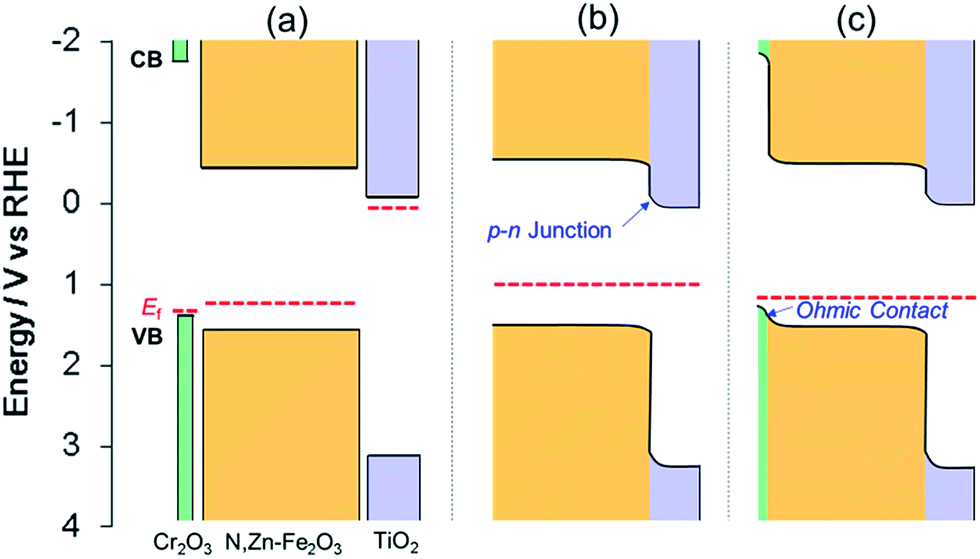 | ||
| Fig. 9 Band energy diagrams for TiO2, N,Zn–Fe2O3 and Cr2O3 (a) before contact and after contact between (b) TiO2/N,Zn–Fe2O3 and (c) TiO2/N,Zn–Fe2O3/Cr2O3. The VB maxima for Cr2O3 and N,Zn–Fe2O3 were estimated from photoelectron spectroscopy measurements in air (Fig. S4†). The band position for TiO2 was obtained from the literature.46 Fermi levels (EF) were estimated from the onset potential of each electrode. | ||
However, it was also evident that the photocurrent of Pt/TiO2/N,Zn–Fe2O3 decayed over time. This photocurrent decay is attributed to self-oxidation due to insufficient hole transfer in N,Zn–Fe2O3. The TiO2 coating improved the electron transfer to the Pt as the result of band bending at the surface side of N,Zn–Fe2O3. However, the surface band bending induced by the TiO2 may have been insufficient to generate hole transfer to the back side contact, because the photogenerated holes had to transfer through the N,Zn–Fe2O3 layer.
Insertion of Cr2O3 as a hole transfer layer
In an attempt to improve the hole transfer in the bulk, N,Zn–Fe2O3 was made by inserting a Cr2O3 layer between N,Zn–Fe2O3 and TCO. The Cr2O3 layer was prepared by depositing Cr followed by the deposition of N,Zn–Fe2O3 and annealing under an O2 flow. The crystal structure was confirmed to consist of α-Cr2O3 by XRD analysis (Fig. S12†). The current–potential curve for the resulting Pt/TiO2/N,Zn–Fe2O3/Cr2O3 is shown in Fig. 3a (red line). The cathodic photocurrent was enhanced by the insertion of the Cr2O3 layer and the offset potential was shifted positively. The enhanced photocurrent was confirmed to originate from the photoabsorption of N,Zn–Fe2O3 based on the IPCE spectrum (Fig. 8). The dark current observed in the current–potential curve was reduced to 1 μA when applying a constant potential of +0.1 V for 1 min under dark conditions. At a constant potential of +0.1 V vs. RHE, a stable photocurrent of ca. 300 μA cm−2 was obtained under one sun (100 mW cm−2, AM 1.5) irradiation, as shown in Fig. 3b (red line). Although the photocurrent decayed slightly after irradiation for 1 h, it was recovered after turning off the light and electrical bias for one minute. This decay is therefore attributed to a gradual accumulation of electrons and holes over prolonged irradiation, as is sometimes observed for nanocrystalline photoelectrodes with high resistivity.30 By repeating this process, the electrode was able to exhibit a stable photocurrent over more than 6 h (Fig. 10a), and the faradaic efficiency for H2 evolution was found to be 100%. It is also important to note that this reaction produced H2 and O2 at a molar ratio of 2:1, which represents a stoichiometric overall solar water splitting reaction (Fig. 10b).
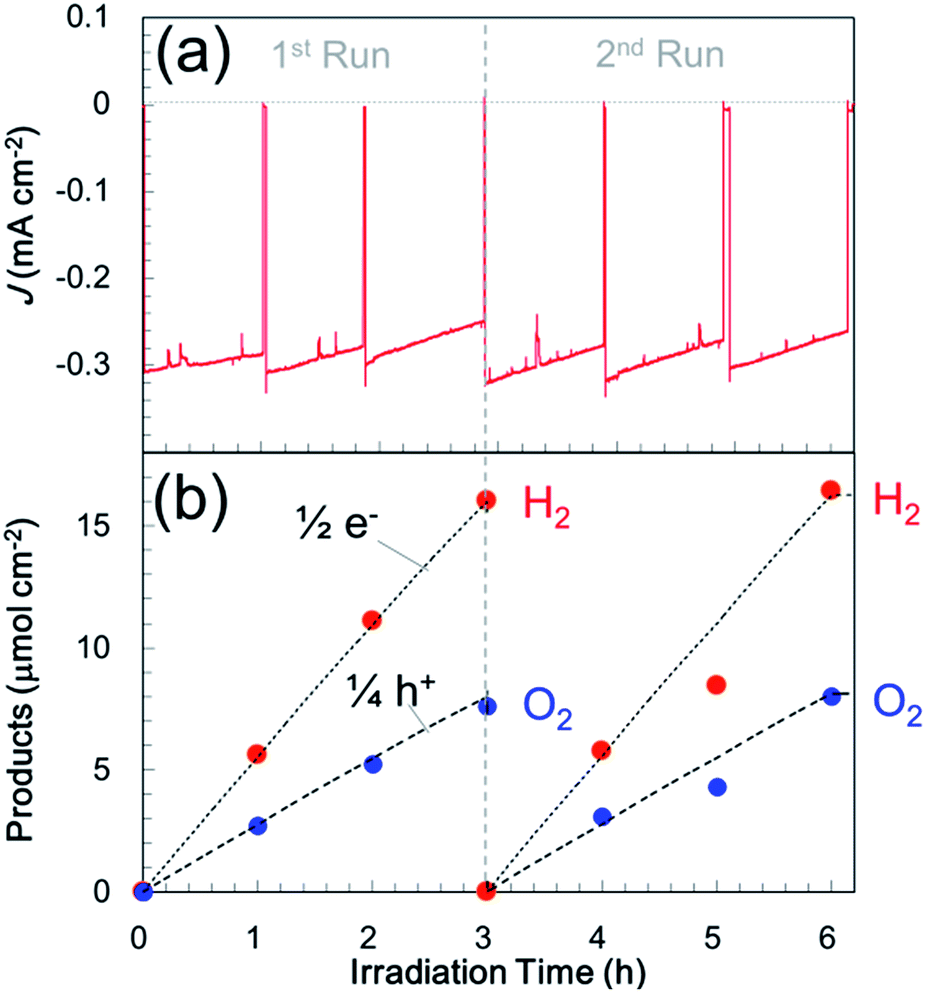 | ||
| Fig. 10 (a) Photocurrent transients for Pt/TiO2/N,Zn–Fe2O3/Cr2O3 in a three-electrode configuration at +0.1 V vs. RHE in a 0.5 M Na2CO3–NaHCO3 (1:1) buffer electrolyte (pH 9.7) under one sun (100 mW cm−2, AM 1.5) light illumination. Irradiation and application of a bias were briefly ceased at each measurement point to allow the products to achieve gas–liquid equilibrium, and the electrolyte and gas phase were exchanged every 3 h. (b) Time course for gas evolution in the sealed cell, which shows a stoichiometric reaction for overall solar water splitting. | ||
STEM-EDX mapping of Pt/TiO2/N,Zn–Fe2O3/Cr2O3 (Fig. 7b) showed that some of the Cr atoms diffused to the grain boundaries of the Fe2O3 particles. This diffused Cr was found not to contribute to the enhanced photocurrent, because Pt/TiO2/pristine-Fe2O3/Cr2O3 exhibited a negligibly small cathodic photocurrent (Fig. S13†). TEM images (Fig. 7) clarified that the Cr formed a layered structure between the TCO and N,Zn–Fe2O3. The voids observed between the TCO and Cr2O3 are not intrinsic but rather were generated during the FIB cutting process. TEM observations of the Fe2O3 structure indicated a significant effect of the Cr2O3. In the absence of the Cr2O3 underlayer, the Fe2O3 crystals were oriented perpendicularly to the film plane, while in the presence of the Cr2O3 layer larger Fe2O3 crystals were found to be oriented parallel to the film plane. These results are consistent with the larger scale TEM images (Fig. S14†). The larger Fe2O3 crystallite sizes were also confirmed by selected area electron diffraction, which showed fewer diffraction spots (Fig. 7). From XRD patterns (Fig. 11), N,Zn–Fe2O3 on TCO was determined to be oriented in the (110) and (300) directions, while the average crystallite diameter was estimated to be 46 nm using the Scherrer equation. The XRD pattern of N,Zn–Fe2O3/Cr2O3 showed that the orientation changed to the (012), (104) and (116) directions, and that the crystallite diameter increased to 89 nm following the introduction of the Cr2O3 layer. Crystals tend to grow in the direction of lower interface energy. The lattice parameter of Cr2O3 is very close to that of N,Zn–Fe2O3;48 therefore, the interfacial energy between N,Zn–Fe2O3 and the substrate was reduced by the Cr2O3 coating on TCO, such that the N,Zn–Fe2O3 crystal growth was in-plane. Grain coarsening could have contributed to the enhancement of the photocurrent because of the lower resistivity at the grain boundaries.
 | ||
| Fig. 11 XRD patterns obtained from (a) N,Zn–Fe2O3/SnO2/ITO, (b) N,Zn–Fe2O3/Cr2O3/SnO2/ITO, and (c) SnO2/ITO. Stars indicate the diffraction peaks attributed to α-Fe2O3 (PDF# 00-001-1053). | ||
Moreover, the Cr2O3 layer promoted charge transfer from N,Zn–Fe2O3 to TCO. Cr2O3 is known as a wide bandgap (Eg ≈ 3 eV) semiconductor.49 The Cr2O3 film deposited on the TCO electrode exhibited a typical cathodic photocurrent (Fig. S15†), indicating that the Fermi level of Cr2O3 was located near the VB edge. PESA demonstrated that the energy of the VB edge of Cr2O3 was ca. 0.2 eV higher than that of N,Zn–Fe2O3 (Fig. S4†). Therefore, the band alignment can be described as shown in Fig. 9a. The positive shift in the offset potential resulting from the insertion of the Cr2O3 layer indicates that the Cr2O3 facilitated the upward band-bending of N,Zn–Fe2O3 toward the Cr2O3 with an ohmic contact, as shown in Fig. 9c. As a result, the cathodic photocurrent was significantly enhanced and stabilized by preventing self-oxidation in the bulk. The TiO2 layer was also helpful in facilitating electron transfer, since N,Zn–Fe2O3 cannot directly donate electrons to H+ or Pt because of electron traps at surface states that lead to self-reduction. In fact, the photocurrents generated by N,Zn–Fe2O3/Cr2O3 and Pt/N,Zn–Fe2O3/Cr2O3 were observed to completely decay within a few minutes.
The existence of heterojunctions was suggested by electrochemical impedance spectroscopy (EIS) data. The Nyquist diagrams obtained by EIS under dark conditions are shown in Fig. 12a. Only single large semicircles were generated by the bare N,Zn–Fe2O3, Pt/TiO2/N,Zn–Fe2O3 and Pt/TiO2/N,Zn–Fe2O3/Cr2O3. Since the impedance spectrum for Fe2O3 includes many parameters, such as the resistance and capacitance of both the bulk and surface states, it is difficult to determine the origins of the resistance and capacitance via the fitting of only one semicircle.50 However, we can compare the total resistances of the electrodes, because the radius of the arc relative to the Z′ axis corresponds to the total resistance. The radius of the Pt/TiO2/N,Zn–Fe2O3 plot was smaller than that of the bare N,Zn–Fe2O3, which indicates that the total resistance in the electrode was decreased by the TiO2 coating and the Pt deposition, in spite of the addition of an interfacial resistance across the junctions. It is believed that the p–n junction would have helped lower the charge transfer resistance. Moreover, the smallest radius was observed in the case of Pt/TiO2/N,Zn–Fe2O3/Cr2O3, suggesting that the lowest charge-transfer resistance resulted from ohmic contact at the N,Zn–Fe2O3/Cr2O3 junction and grain coarsening of N,Zn–Fe2O3. Similar reductions in the radius resulting from heterojunctions have been previously observed.24,27,51
 | ||
| Fig. 12 (a) Nyquist plots from electrochemical impedance spectroscopy for N,Zn–Fe2O3 (red circles), Pt/TiO2/N,Zn–Fe2O3 (blue circles) and Pt/TiO2/N,Zn–Fe2O3/Cr2O3 (green circles), as obtained at a bias of 0.5 V vs. RHE with frequencies varying from 100 kHz to 0.5 Hz under dark conditions. (b) Open-circuit photovoltage (Voc) decays for N,Zn–Fe2O3 (red line), Pt/TiO2/N,Zn–Fe2O3 (blue line) and Pt/TiO2/N,Zn–Fe2O3/Cr2O3 (green line). The electrodes were illuminated with one sun light (AM 1.5, 100 mW cm−2) before measuring the Voc decay in the dark. The Voc values are the differences from open-circuit potentials prior to irradiation. | ||
Charge-separation characteristics were evaluated by open-circuit photovoltage (Voc) analyses. The time profiles of Voc before and after ceasing the photoirradiation are shown in Fig. 12b. In the “light on” region, the Voc values were in the following order: N,Zn–Fe2O3 < Pt/TiO2/N,Zn–Fe2O3 < Pt/TiO2/N,Zn–Fe2O3/Cr2O3. Since the Voc values are closely related to the recombination of charges,52 higher Voc values suggest lower rates of recombination. The recombination of photogenerated electrons and holes could be observed upon turning off the photoirradiation. In the “light off” region, all the electrodes exhibited a negative decay immediately after the cessation of irradiation, and a slower decay should correlate with effective charge separation.53–55 In the case of N,Zn–Fe2O3 and Pt/TiO2/N,Zn–Fe2O3, the Voc decayed in 1 and 5 s, respectively. The slower recombination decay in Pt/TiO2/N,Zn–Fe2O3 compared to the bare N,Zn–Fe2O3 suggests that the electrons accumulated in TiO2 tend to prevent back electron transfer to N,Zn–Fe2O3 by band bending around the p–n junction. The positive change after the negative decay is believed to be connected with the recovery of the species deactivated by the self-redox reaction during photoirradiation. The Voc decay is extended by both TiO2 coating and Cr2O3 insertion, which indicates that the holes accumulated in Cr2O3 also prevented back hole transfer to N,Zn–Fe2O3 by band bending at the N,Zn–Fe2O3/Cr2O3 junction. Therefore, heterojunctions are essential to enhancing and stabilizing the photocurrent.
Overall water splitting with a tandem cell
The p-type Pt/TiO2/N,Zn–Fe2O3/Cr2O3 photocathode has the potential to achieve overall water splitting in combination with an n-type semiconductor photoanode. We employed n-type SrTiO3−x for the photoanode because its CBM is located at −0.4 V,56 which is sufficiently negative to transfer electrons extracted from water molecules to numerous photocathodes. It has been shown to be appropriate to demonstrate a Z-scheme (two-step photoexcitation) reaction in the tandem configuration.57 In the present case, n-type SrTiO3−x is also beneficial, owing to a small absorption overlap with N,Zn–Fe2O3 (Fig. S10†), and a relatively negative onset potential that facilitates electron transfer to the Fe2O3-based photocathode (Fig. S16†). PEC tandem cells were constructed by connecting p-type Pt/TiO2/N,Zn–Fe2O3/Cr2O3 and n-type SrTiO3−x electrodes. The band model for the photocell is shown in the inset of Fig. 13. When these tandem electrodes were irradiated with solar simulation light (AM 1.5, 100 mW cm−2), the UV components were absorbed by the forward SrTiO3−x electrode and the residual light (λ > 400 nm) reached N,Zn–Fe2O3 at the back. Photogenerated holes in SrTiO3−x oxidized water to produce oxygen, while the photogenerated electrons in N,Zn–Fe2O3 reduced water to hydrogen. This tandem cell system exhibited a stable photocurrent with an average value of 114 μA cm−2 during a 3.5 h irradiation period without the application of an external voltage. Fig. 13 shows the amounts of photoelectrons and gaseous products generated as functions of the irradiation time. The H2:O2 molar ratio was 2:1, and the amounts of these gases produced were equal to one half and one quarter of the total number of electrons and holes calculated from the photocurrent, respectively. This result indicates that the overall water splitting reaction was stoichiometric, with a faradaic efficiency of 100%. The quantity of evolved H2 was 2.1 μmol cm−2 after irradiation for 1 h and the solar conversion efficiency was 0.14%. As control experiments, the irradiation of only SrTiO3−x in the same system or in a two-electrode system composed of SrTiO3−x and Pt wire was assessed (Fig. S17†). These trials produced only 0.25 and 0.18 μmol cm−2 of H2, respectively. Therefore, it is evident that Pt/TiO2/N,Zn–Fe2O3/Cr2O3 significantly enhanced the H2 evolution when employed as a photocathode for two-step photoexcitation in the tandem configuration.
 | ||
| Fig. 13 Time course of gas evolution for a two-electrode tandem system consisting of Pt/TiO2/N,Zn–Fe2O3/Cr2O3 and SrTiO3−x under one sun (AM 1.5) irradiation with no electrical bias. The reaction was carried out in a 0.5 M Na2CO3–NaHCO3 (1:1) aqueous solution. The light was irradiated from the SrTiO3−x side. Irradiation was paused at each measurement point to allow the products to achieve gas–liquid equilibrium. | ||
Conclusion
A p-type N,Zn-codoped Fe2O3 (hematite) photocathode for PEC solar hydrogen production by water splitting was realized. To overcome the issues of low H2 generation rates accompanied by self-redox reaction of the p-type Fe2O3, a TiO2 overlayer was applied over the surface of N,Zn–Fe2O3. The TiO2 acted not only as a passivation layer, but also as a charge separation layer, by forming a p–n junction with N,Zn–Fe2O3. Furthermore, insertion of a thin Cr2O3 layer between the N,Zn–Fe2O3 and TCO layers significantly enhanced the cathodic photocurrent during H2 production by facilitating band-bending in N,Zn–Fe2O3, forming an ohmic contact, and reducing grain boundaries in N,Zn–Fe2O3. As a result, the Pt/TiO2/N,Zn–Fe2O3/Cr2O3 photocathode generated a stable photocurrent during water reduction of 300 μA cm−2 under one sun (100 mW cm−2, AM 1.5) irradiation at +0.1 V vs. RHE. The faradaic efficiency of the water reduction was 100% and the device was found to be stable over a span of 6 h. Furthermore, by connecting an n-type SrTiO3−x photoanode to the Pt/TiO2/N,Zn–Fe2O3/Cr2O3 photocathode, overall water splitting without the application of an external electrical bias was demonstrated. This is the first example of overall stoichiometric water splitting using an iron-based photocathode. The present strategy of applying an overcoat with a multi-heterojunction could be expanded to many unstable and low-efficiency p-type photocathodes.Acknowledgements
The authors wish to thank S. Sato, S. Kosaka, N. Takahashi, K. Kitazumi, Y. Kimoto, N. Isomura, Y. Kato, K. Abiko and E. Ikenaga for their valuable assistance during these experiments. The HAXPES analyses were performed at the BL47XU beamline at SPring-8, with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2013B1018, 2014A1009 and 2014B1018). This work was supported by the Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C) of the Japan Science and Technology Agency (JST).Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- S. Ida, K. Yamada, T. Matsunaga, H. Hagiwara, Y. Matsumoto and T. Ishihara, J. Am. Chem. Soc., 2010, 132, 17343–17345 CrossRef CAS PubMed.
- C. Y. Lin, Y. H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482–3487 RSC.
- P. Bornoz, F. F. Abdi, S. D. Tilley, B. Dam, R. van de Krol, M. Graetzel and K. Sivula, J. Phys. Chem. C, 2014, 118, 16959–16966 CAS.
- C. Leygraf, M. Hendewerk and G. A. Somorjai, J. Catal., 1982, 78, 341–351 CrossRef CAS.
- W. B. Ingler Jr, J. P. Baltrus and S. U. M. Khan, J. Am. Chem. Soc., 2004, 126, 10238–10239 CrossRef PubMed.
- W. B. Ingler Jr and S. U. M. Khan, Int. J. Hydrogen Energy, 2005, 30, 821–827 CrossRef.
- T. Morikawa, K. Kitazumi, N. Takahashi, T. Arai and T. Kajino, Appl. Phys. Lett., 2011, 98, 242108 CrossRef.
- X. Qi, G. She, M. Wang, L. Mu and W. Shi, Chem. Commun., 2013, 49, 5742–5744 RSC.
- T. Morikawa, T. Arai and T. Motohiro, Appl. Phys. Express, 2013, 6, 041201 CrossRef.
- T. Morikawa, S. Saeki, T. Suzuki, T. Kajino and T. Motohiro, Appl. Phys. Lett., 2010, 96, 142111 CrossRef.
- T. M. Suzuki, S. Saeki, K. Sekizawa, K. Kitazumi, N. Takahashi and T. Morikawa, Appl. Catal., B, 2017, 202, 597–604 CrossRef CAS.
- R. Jinnouchi, A. V. Akimov, S. Shirai, R. Asahi and O. V. Prezhdo, J. Phys. Chem. C, 2015, 119, 26925–26936 CAS.
- Y. Hikita, K. Nishio, L. C. Seitz, P. Chakthranont, T. Tachikawa, T. F. Jaramillo and H. Y. Hwang, Adv. Energy Mater., 2016, 6, 1502154 CrossRef.
- J. H. Kennedy and K. W. Frese, J. Electrochem. Soc., 1978, 125, 709–714 CrossRef CAS.
- N. J. Cherepy, D. B. Liston, J. A. Lovejoy, H. Deng and J. Z. Zhang, J. Phys. Chem. B, 1998, 102, 770–776 CrossRef CAS.
- R. L. Spray, K. J. McDonald and K. S. Choi, J. Phys. Chem. C, 2011, 115, 3497–3506 CAS.
- B. C. Faust and M. R. Hoffmann, Environ. Sci. Technol., 1986, 20, 943–948 CrossRef CAS PubMed.
- E. Ikenaga, M. Kobata, H. Matsuda, T. Sugiyama, H. Daimon and K. Kobayashi, J. Electron Spectrosc. Relat. Phenom., 2013, 190, 180–187 CrossRef CAS.
- G. Panaccione and K. Kobayashi, Surf. Sci., 2012, 606, 125–129 CrossRef CAS.
- M. Imura, S. Tsuda, T. Nagata, H. Takeda, M. Liao, A. Yang, Y. Yamashita, H. Yoshikawa, Y. Koide, K. Kobayashi, T. Yamaguchi, M. Kaneko, N. Uematsu, T. Araki and Y. Nanishi, J. Appl. Phys., 2013, 114, 033505 CrossRef.
- Y. Lin, Y. Xu, M. T. Mayer, Z. I. Simpson, G. McMahon, S. Zhou and D. Wang, J. Am. Chem. Soc., 2012, 134, 5508–5511 CrossRef CAS PubMed.
- M. H. Lee, K. Takei, J. Zhang, R. Kapadia, M. Zheng, Y. Z. Chen, J. Nah, T. S. Matthews, Y. L. Chueh, J. W. Ager and A. Javey, Angew. Chem., Int. Ed., 2012, 51, 10760–10764 CrossRef CAS PubMed.
- Y. Hou, F. Zuo, A. Dagg and P. Feng, Angew. Chem., Int. Ed., 2013, 52, 1248–1252 CrossRef CAS PubMed.
- F. Meng, J. Li, S. K. Cushing, M. Zhi and N. Wu, J. Am. Chem. Soc., 2013, 135, 10286–10289 CrossRef CAS PubMed.
- N. Guijarro, M. S. Prevot and K. Sivula, Phys. Chem. Chem. Phys., 2015, 17, 15655–15674 RSC.
- Q. Huang, F. Kang, H. Liu, Q. Li and X. Xiao, J. Mater. Chem. A, 2013, 1, 2418–2425 CAS.
- T. Hisatomi, F. Le Formal, M. Cornuz, J. Brillet, N. Tetreault, K. Sivula and M. Gratzel, Energy Environ. Sci., 2011, 4, 2512–2515 CAS.
- R. Liu, Z. Zheng, J. Spurgeon and X. Yang, Energy Environ. Sci., 2014, 7, 2504–2517 CAS.
- A. Paracchino, V. Laporte, K. Sivula, M. Gratzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- M. J. Choi, J.-Y. Jung, M.-J. Park, J.-W. Song, J.-H. Lee and J. H. Bang, J. Mater. Chem. A, 2014, 2, 2928–2933 CAS.
- A. Kumar, P. G. Santangelo and N. S. Lewis, J. Phys. Chem., 1992, 96, 834–842 CrossRef CAS.
- S. Diodati, L. Pandolfo, A. Caneschi, S. Gialanella and S. Gross, Nano Res., 2014, 7, 1027–1042 CrossRef CAS.
- J. F. Lin, J. J. Wu, J. Zhu, Z. Mao, A. H. Said, B. M. Leu, J. G. Cheng, Y. Uwatoko, C. Q. Jin and J. S. Zhou, Sci. Rep., 2014, 4, 6 Search PubMed.
- Y. Matsumoto, M. Omae, K. Sugiyama and E. Sato, J. Phys. Chem., 1987, 91, 577–581 CrossRef CAS.
- M. Uda, Y. Nakagawa, T. Yamamoto, M. Kawasaki, A. Nakamura, T. Saito and K. Hirose, J. Electron Spectrosc. Relat. Phenom., 1998, 88–91, 767–771 CrossRef CAS.
- J. Jasieniak, M. Califano and S. E. Watkins, ACS Nano, 2011, 5, 5888–5902 CrossRef CAS PubMed.
- M. Moriya, T. Minegishi, H. Kumagai, M. Katayama, J. Kubota and K. Domen, J. Am. Chem. Soc., 2013, 135, 3733–3735 CrossRef CAS PubMed.
- W. A. Smith, I. D. Sharp, N. C. Strandwitz and J. Bisquert, Energy Environ. Sci., 2015, 8, 2851–2862 CAS.
- C. Zhang, S. Chen, L. e. Mo, Y. Huang, H. Tian, L. Hu, Z. Huo, S. Dai, F. Kong and X. Pan, J. Phys. Chem. C, 2011, 115, 16418–16424 CAS.
- Y. Nakano, S. Saeki and T. Morikawa, Appl. Phys. Lett., 2009, 94, 022111 CrossRef.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 2011, 43, 689–713 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828–11829 CrossRef CAS PubMed.
- S. Hasegawa, J. Phys.: Condens. Matter, 2000, 12, R463–R495 CrossRef CAS.
- L. Kavan, M. Grätzel, S. E. Gilbert, C. Klemenz and H. J. Scheel, J. Am. Chem. Soc., 1996, 118, 6716–6723 CrossRef CAS.
- L. Li, P. A. Salvador and G. S. Rohrer, Nanoscale, 2014, 6, 24–42 RSC.
- S. A. Chambers, Y. Liang and Y. Gao, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 13223–13229 CrossRef CAS.
- H. Cao, X. Qiu, Y. Liang, M. Zhao and Q. Zhu, Appl. Phys. Lett., 2006, 88, 241112 CrossRef.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- M. Wang, L. Sun, J. Cai, P. Huang, Y. Su and C. Lin, J. Mater. Chem. A, 2013, 1, 12082–12087 CAS.
- L. C. C. Elliott, J. I. Basham, K. P. Pernstich, P. R. Shrestha, L. J. w. Richter, D. M. DeLongchamp and D. J. Gundlach, Adv. Energy Mater., 2014, 4, 1400356 CrossRef.
- T. Li, Y. Lee and H. Teng, Energy Environ. Sci., 2012, 5, 5315–5324 CAS.
- L. Wei, Y. Na, Y. Yang, R. Fan, P. Wang and L. Li, Phys. Chem. Chem. Phys., 2015, 17, 1273–1280 RSC.
- J. Du, X. Meng, K. Zhao, Y. Li and X. Zhong, J. Mater. Chem. A, 2015, 3, 17091–17097 CAS.
- J. M. Bolts and M. S. Wrighton, J. Phys. Chem., 1976, 80, 2641–2645 CrossRef CAS.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HAXPES spectra, photoelectrochemical properties, photoelectron spectra in air, UV-vis absorbance spectra, TEM images and X-ray diffraction patterns. See DOI: 10.1039/c7ta00431a |
| This journal is © The Royal Society of Chemistry 2017 |