
Control of the molecular geometry and nanoscale morphology in perylene diimide based bulk heterojunctions enables an efficient non-fullerene organic solar cell†
R.
Singh‡
a,
J.
Lee‡
a,
M.
Kim
a,
P. E.
Keivanidis
 *b and
K.
Cho
*a
*b and
K.
Cho
*a
aDepartment of Chemical Engineering, Pohang University of Science and Technology, Pohang, 790−784, Korea. E-mail: kwcho@postech.ac.kr
bDepartment of Mechanical Engineering and Materials Science and Engineering Cyprus University of Technology, Limassol 3041, Cyprus. E-mail: p.keivanidis@cut.ac.cy
First published on 10th November 2016
Abstract
Herein we present the design of three perylene diimide (PDI) derivatives with different molecular geometries; namely the monomeric PDI1, the bay-linked PDI2 dimer, and the bay-linked PDI4 tetramer with a 9,9′-spirobifluorene core that are utilized as electron acceptors in non-fullerene organic solar cells (OSCs). In all cases the PTB7-Th polymer is used as the electron donor. Among the three PTB7-Th:PDI systems, the highest power conversion efficiency (PCE) is obtained by the PDI4-based OSC device that exhibits a maximum PCE = 6.44% followed by the PDI2-based (PCE = 5.32%) and PDI1-based (PCE = 2.48%) devices. The detailed study of the photoluminescence quenching, morphology and temperature-dependent charge transport properties of the three systems reveal that the highest PCE of PTB7-Th:PDI4 is a consequence of the three-dimensional (3D) molecular architecture of PDI4 that tunes energetic disorder in the PDI phase and contributes to the improvement of electron transport. Transient photovoltage characterization experiments further identify that the actual effect coming from the 3D molecular geometry of the PDI4 acceptor on PCE is the minimization of non-geminate charge recombination losses. This study provides updated guidelines for optimizing further the molecular structure of 3D small molecular electron acceptors that can be used in highly efficient non-fullerene OSCs.
Introduction
Organic solar cells (OSCs) based on an electron donor and an electron acceptor are a promising cost-effective alternative for the utilization of solar energy. During the last two decades, OSCs have attracted much research interest because of their advantages of solution processability, and capability to fabricate flexible large-area modules and being low cost and lightweight.1–4 Due to the rapid development of donor polymers and acceptors for use in active layers, the record power conversion efficiencies (PCEs) of OSCs have surpassed 12%.5–7 Fullerene derivatives are most commonly used in solution-processed bulk heterojunction (BHJ) organic solar cells due to their high electron affinity, good electron transport, and capability of forming favorable nanoscale phase separation. However, these molecules have their own physical and structural insufficiencies, such as (a) limited spectral breadth, (b) inferior ambient stability, (c) difficulty in functionalization and tuning of their electronic properties, and (d) a relatively high retail cost.8–11 Therefore, it is highly desirable to develop novel non-fullerene acceptors in a scalable manner, which possess a strong absorption ability in the visible and NIR regions, adjustable energy levels, tunable electronic properties, and a nanoscale interpenetrating network morphology with donor polymers.At present, perylene diimide derivatives (PDIs) are attracting significant attention because of their several appealing properties for OSCs, including low cost, chemical robustness, strong electron-accepting ability, ease of functionalization, and high electron mobility.12–18 Unlike the case of fullerene-based acceptors, the use of PDI derivatives in OSC devices requires a subtle tuning of the PDI aggregation level. Earlier studies suggested that severe PDI aggregation leads to the trapping of photogenerated electrons within the PDI crystallites.19 Nonetheless, in the complete absence of PDI aggregates both macroscopic electron transport and collection are disabled.20
In the solid state, the planar molecular structure of disk-shaped PDI derivatives expedites the electronic coupling of adjacent PDI monomers resulting in the formation of dimers or even higher-order columnar aggregates that are stabilized by π–π and electrostatic (quadrupolar) interactions.15,21 Following light absorption by the PDI phase, PDI excitons convert rapidly to PDI excimers22,23 that can also contribute to photocurrent generation, as long as the size of the PDI aggregate is kept smaller than the PDI excimer diffusion length.24 Under these conditions, PDI excimers can reach the PDI/polymer interface where they can dissociate by charge transfer. The photogenerated charges can then undergo transport in the PDI phase via the small-sized interconnected PDI aggregates that are necessary for facilitating charge transport.24 Control over the packing and intermolecular interaction of PDIs is important for realizing highly efficient non-fullerene OSCs.9,15,20,25 Practically, when monomeric PDI-based BHJ layers are employed, it is difficult to spontaneously enable the formation of suitable active layer morphologies with small-sized interconnected aggregates in the PDI phase for ensuring a balanced charge transport. This can be achieved when appropriate plasticizing additives such as 1,8-diiodooctane (DIO) are used that result in shrinking of the PDI domains.26 With this approach, the best PCE for polymer/monomeric–PDI organic solar cells was obtained, reaching the level of 3.7%.24 In the same vein, recent studies found that the strong π–π interaction in the PDI aggregation can be in principle disrupted when bis-adduct PDI derivatives are used for which, a torsion is introduced in the conjugated backbone of PDI molecules.27–29 In light of these generic guidelines, many efforts have been devoted to developing dimeric, trimeric, or tetrameric PDI adducts with twisted three-dimensional (3D) structural conformations. The rationale behind these attempts was based on the suggestion that, by increasing the dimensionality of the chemical structure, the detrimental intermolecular interactions can be suppressed and a multidimensional interconnectivity would achieve isotropic charge transport.30,31 Despite these efforts, the reported PCEs of 3D PDI-based non-fullerene solar cells when mixed with different polymers are less than 8%.16,30–35 Only very recently, Heeger's and Nuckolls's groups have restructured the PDI core and the PDI functional groups to improve the charge transport properties and to increase the degree of non-planarity by synthesizing twisted or helical shaped PDIs, which can yield a PCE of 8.42%.36,37
Despite the ongoing research on the development of PDI multiadducts and the realization of high-efficiency OSC devices, the exact impact of increasing the dimensionality of the molecular structure in a PDI multiadduct on the performance of an OSC device is not yet fully understood. The main anticipated effects of the molecular architecture are as follows: (i) minimizing the PDI aggregate size and increasing the dissociation efficiency of the initially photogenerated neutral excited states, (ii) tuning effectively the energetic disorder of the PDI phase and improving its electron transport properties, and (iii) minimizing recombination losses of the free charges for favouring charge extraction.
In order to resolve which of the above corresponds to the actual effect of the PDI molecular architecture on OSC performance, we have designed three PDI-based electron acceptors, including monomeric PDI, bis-PDI and tetra-PDI with different geometries of the PDI core (Fig. 1). The corresponding OSC performance was checked by combining high efficiency polymer poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b;4,5-b′]dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene-)-2-carboxylate-2-6-diyl] (PTB7-Th). The active layer morphology and packing motifs of all materials studied are systematically investigated both in their corresponding neat and blend films, by using atomic force microscopy (AFM), transmission electron microscopy (TEM), and grazing incidence wide angle X-ray scattering (GIWAXS). The processes of charge transport and non-geminate charge carrier recombination are studied by means of the space charge limited current (SCLC) model and transient photovoltage (TPV) and transient photocurrent (TPC) measurements, respectively.
 | ||
| Fig. 1 Molecular structure of PDI1, PDI2 and PDI4 as non-fullerene small molecule acceptors and the PTB7-Th polymer as an electron donor. | ||
Results and discussion
Design and synthesis
In this study, three different PDI molecules namely a monomeric PDI (PDI1), a bay-linked PDI dimer (PDI2), and a bay-linked PDI tetramer with a 9,9′-spirobifluorene core (PDI4) were facilely prepared according to the reported methods (Scheme S1†).31,38,39 The chemical structures of the three PDIs were carefully determined by NMR spectroscopy, MALDI-TOF mass spectroscopy, or elemental analysis (ESI†). The three PDIs were readily soluble in common organic solvents such as dichloromethane, chloroform (CF), THF, or chlorobenzene (CB) at room temperature due to the solubilizing alkyl substituents.Conformation of PDIs
To understand the geometric difference between PDI1, PDI2, and PDI4, theoretical calculations were performed using the density functional theory (DFT) at the B3LYP/6-31G(d) level. As shown in Fig. S1,† the PDI1 molecule has a highly planar backbone conformation, while the PDI2 molecule has a non-planar backbone conformation with a 65° torsion angle between the two constituent PDI subunits. The non-planar conformation of PDI2 is a consequence of repulsion between the two C–H bonds at the inner bay position of adjacent PDI units.40 Moreover, the bay-linked molecular structure of PDI2 affords an about 7° rotational twist (core-contortion) between the two naphthalene imide subunits in the monomeric PDI, concomitant with strongly improved solubility and slipped stack J-aggregation properties.41,42 In PDI4, the torsion angle between the PDI plane and the fluorene plane is about 56°, and the four bay-linked PDI subunits are oriented along the four cardinal directions due to the orthogonal structure of the spirobifluorene core. Similar to PDI2, the four monomeric PDI subunits of PDI4 have a core-contortion. The highly twisted and 3D coordinated molecular geometry of PDI4 could possess appropriate intermolecular connectivity without a severe molecular aggregation, which in turn influenced the BHJ morphology in OSCs (Fig. S1†).Electrochemical properties
The electrochemical properties of the three PDI molecules were characterized by cyclic voltammetry (Fig. S2a†) and the results are summarized in Table S1.† From the potential of the first reduction peak, the LUMO was estimated to be −3.80, −3.88, and −3.78 eV for PDI1, PDI2, and PDI4, respectively, assuming the absolute energy level of FeCp2+/0 to be 4.8 eV below the vacuum level.43,44 The energy diagrams of the three PDIs and PTB7-Th as photoactive materials, the interlayers, and the electrodes used in our study of the photovoltaic characteristics are shown in Fig. S2b.† The trends of HOMOs and LUMOs agree well with the DFT results (Fig. S3†). Owing to the electron-donating ability of the spirobifluorene core, PDI4 has a slightly higher lying LUMO level compared to the lower-adduct analogues, which is beneficial for improving the open-circuit voltage (Voc) of OSCs.45Optical properties
UV-Vis absorption and time-integrated photoluminescence (PL) spectra were recorded for the three PDI molecules both in dilute solution and in the solid state (Fig. 2b and S4†). Extreme care was taken for preparing dilute solution in the nM molar concentration regime so that any possibility of intermolecular interactions between the PDI derivatives can be safely ruled out. Table S1† summarizes the overview of the corresponding UV-Vis and PL data. Both UV-Vis and PL spectral profiles of the PDI1 derivative confirm that in the solid state, PDI1 forms H-aggregates responsible for the photo-induced activation of PDI excimers.15,22,23 In solution, the typical mirror-image related UV-Vis absorption and PL spectral profiles of the PDI1 monomer can be seen. Instead, in the solid state the UV-Vis absorption profile broadens due to the appearance of low- and high-energy absorption features. Similarly, the PL spectrum of the solid-state PDI1 red-shifts and it spectrally broadens, indicating the activation of PDI excimers within the H-aggregate.22 The UV-Vis spectra of the PDI2 and PDI4 derivatives in dilute solution are significantly different from the expected monomer-like absorption spectral profile shown for PDI1, looking similar to their corresponding solid-state UV-Vis absorption spectra. Although in the high dilution regime the PDI multiadducts are isolated, the coupling of the transition dipole moments in each monomer of the multiadducts is found to be significant. Likewise, the PL spectra of both PDI2 and PDI4 derivatives in dilute solution exhibit the typical monomeric PDI excimer-like spectral shape, comparable to their corresponding PL spectra in the solid state. The striking similarity of the UV-Vis and PL spectra between the dilute solution and solid state indicates an effective excitonic coupling between the covalently linked PDI monomers in each of the two PDI2 and PDI4 multiadducts. In contrast, several previous studies on different PDI multiadducts found the typical monomer-like UV-Vis spectral profile in dilute solution.35,46 Essentially, in a PDI multiadduct the magnitude of intramolecular coupling is expected to vary depending on the relative orientation of the PDI transition dipoles in the multiadduct. At present the UV-Vis and PL spectra of PDI1, PDI2 and PDI4 suggest that the three PDI derivatives in the solid state form aggregates either due to direct π–π stacking interactions (PDI1) or due to electrostatic (quadrupolar) interactions (PDI2, PDI4).15 The magnitude of π–π stacking interactions in the solid state form of the three PDI derivatives will be further discussed below, in the X-ray diffraction section of our paper.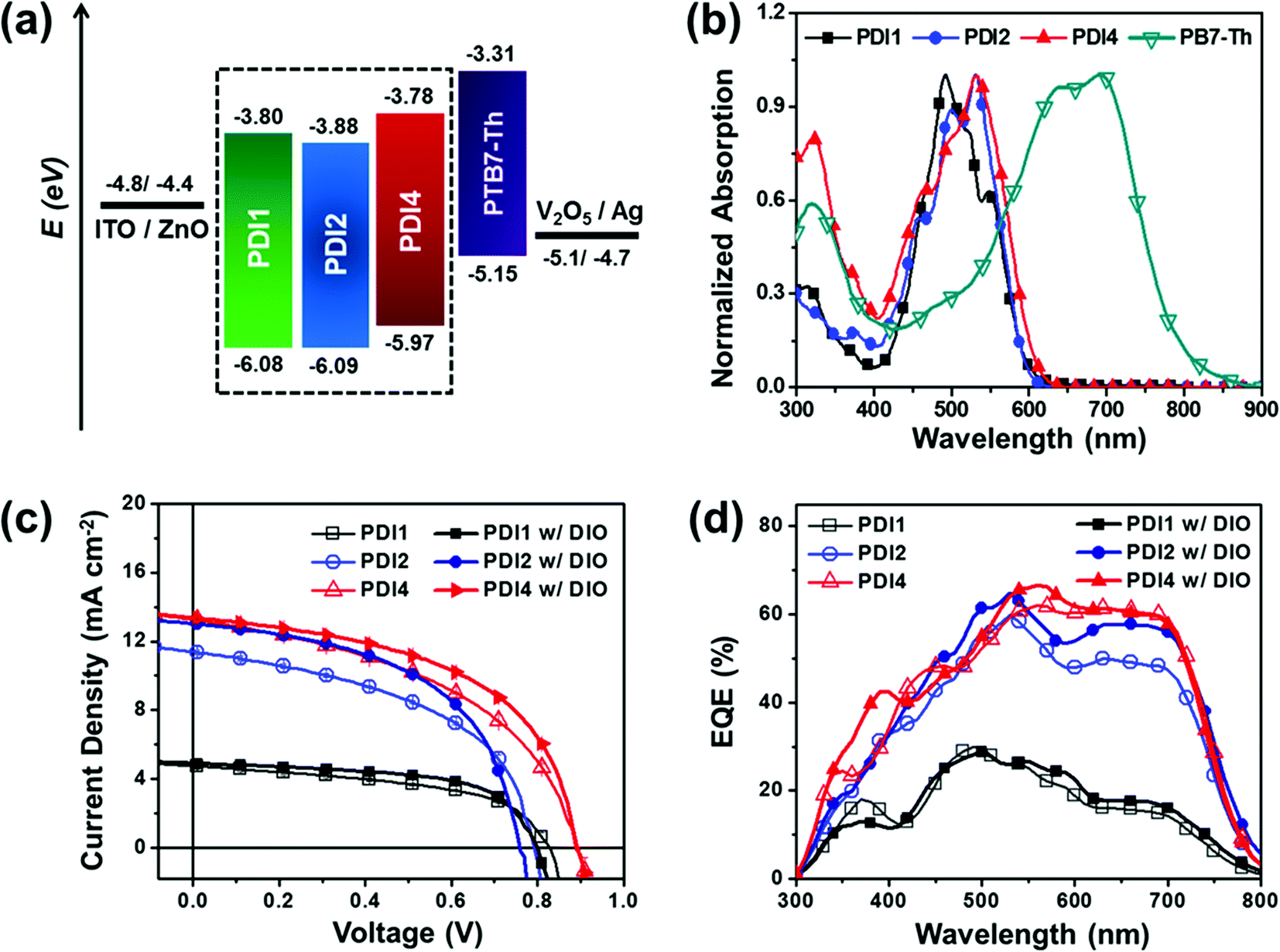 | ||
| Fig. 2 (a) Energy level diagram of the inverted device structure with active materials, (b) normalized UV-Vis absorption spectra of the three acceptors (PD1, PDI2 and SF-PDI4) and PTB7-Th polymer, and (c) J–V curves and (d) EQE spectra of PTB7-Th:PD1, PTB7-Th:PDI2 and PTB7-Th:PDI4 blend film w/o and w/DIO based BHJ OSCs. | ||
Concerning the absorption properties of PTB7-Th, Fig. 2b illustrates that the main PTB7-Th absorption is in the range of 500–800 nm, which complements well the absorption spectra of the three PDIs. No resonance electronic energy transfer is expected from PDI to PTB7-Th, regardless of the spectral overlap between the PDI excimer luminescence and the PTB7-Th absorption.26
OSC fabrication and characterization
In order to evaluate the photovoltaic properties, OSCs were fabricated with an inverted structure, ITO/ZnO(40 nm)/PTB7-Th:PDIs(95–100 nm)/V2O5(2 nm)/Ag(100 nm), reported in our previous work.25 The optimal weight ratio of donor/acceptor was found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S5†) and an active layer spin-cast from chlorobenzene was formed with a thickness of ∼100 nm. The photovoltaic performance of the solar cells fabricated under different conditions is summarized in Tables 1 and S2,† and the photocurrent density–voltage (J–V) characteristics and the corresponding external quantum efficiency (EQE) spectra of the devices are shown in Fig. 2c and d. The device results showed that the PCEs of the three PDI-based OSCs increased to 2.09%, 4.51% and 5.66% for PDI1, PDI2, and PDI4, respectively. The PDI4-based OSCs showed a higher Voc value (≈0.90 V) followed by the PDI1-based (≈0.82 V) and PDI2-based (≈0.78 V) OSCs, which is consistent with the trends of LUMO levels measured by the CV and DFT calculations (Fig. S2 and S3†). A large difference is observed in the short-circuit current density (Jsc) of the three PDI-based OSCs: 4.76 mA cm−2, 11.38 mA cm−2, and 13.25 mA cm−2 for PDI1, PDI2, and PDI4, respectively, which consequently results in different PCEs. The obtained trend in the generated photocurrent is in agreement with the corresponding trend of the hole-transfer induced PL quenching of the PDI excimer luminescence (see Table S6 in the ESI†) of these blends. However, a discrepancy is found in the corresponding trend of the electron-transfer induced PL quenching of the polymer luminescence, implying that charge recombination is operative in these systems. Both PL quenching and charge recombination will be discussed in detail in the next section of the paper.
:acceptor (D:A) ratio was 1:1 (w/w%). The active layer was deposited from solution in chlorobenzene w/o and w/DIO. The averages are from over eight devices
:1 (Fig. S5†) and an active layer spin-cast from chlorobenzene was formed with a thickness of ∼100 nm. The photovoltaic performance of the solar cells fabricated under different conditions is summarized in Tables 1 and S2,† and the photocurrent density–voltage (J–V) characteristics and the corresponding external quantum efficiency (EQE) spectra of the devices are shown in Fig. 2c and d. The device results showed that the PCEs of the three PDI-based OSCs increased to 2.09%, 4.51% and 5.66% for PDI1, PDI2, and PDI4, respectively. The PDI4-based OSCs showed a higher Voc value (≈0.90 V) followed by the PDI1-based (≈0.82 V) and PDI2-based (≈0.78 V) OSCs, which is consistent with the trends of LUMO levels measured by the CV and DFT calculations (Fig. S2 and S3†). A large difference is observed in the short-circuit current density (Jsc) of the three PDI-based OSCs: 4.76 mA cm−2, 11.38 mA cm−2, and 13.25 mA cm−2 for PDI1, PDI2, and PDI4, respectively, which consequently results in different PCEs. The obtained trend in the generated photocurrent is in agreement with the corresponding trend of the hole-transfer induced PL quenching of the PDI excimer luminescence (see Table S6 in the ESI†) of these blends. However, a discrepancy is found in the corresponding trend of the electron-transfer induced PL quenching of the polymer luminescence, implying that charge recombination is operative in these systems. Both PL quenching and charge recombination will be discussed in detail in the next section of the paper.
:acceptor (D:A) ratio was 1:1 (w/w%). The active layer was deposited from solution in chlorobenzene w/o and w/DIO. The averages are from over eight devices
| D:A |
V oc (V) | J sc (mA cm−2) | FF (%) | PCEave(max) (%) | μ h (cm2 V−1 s−1) | μ e (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|
| PTB7-Th:PDI1 | 0.82 ± 0.01 | 4.76 ± 0.12 | 52.1 ± 1.2 | 2.04 (2.09) | 3.19 × 10−5 | 6.57 × 10−7 |
| PTB7-Th:PDI1 w/DIO | 0.80 ± 0.01 | 4.88 ± 0.27 | 60.2 ± 1.4 | 2.35 (2.48) | 1.31 × 10−4 | 8.73 × 10−7 |
| PTB7-Th:PDI2 | 0.78 ± 0.01 | 11.38 ± 0.10 | 49.8 ± 0.2 | 4.39 (4.51) | 3.49 × 10−5 | 2.01 × 10−6 |
| PTB7-Th:PDI2 w/DIO | 0.76 ± 0.005 | 13.01 ± 0.16 | 53.3 ± 0.6 | 5.27 (5.32) | 9.36 × 10−5 | 4.12 × 10−6 |
| PTB7-Th:PDI4 | 0.90 ± 0.01 | 13.25 ± 0.14 | 47.6 ± 0.6 | 5.58 (5.66) | 1.69 × 10−5 | 4.17 × 10−6 |
| PTB7-Th:PDI4 w/DIO | 0.90 ± 0.005 | 13.36 ± 0.05 | 53.6 ± 0.3 | 6.32 (6.44) | 3.18 × 10−5 | 6.79 × 10−6 |
On the basis of the 1:1 blend ratio as an optimal blend composition, DIO was used to further improve the cell performance and the photovoltaic performance of the solar cells fabricated with various DIO concentrations is summarized in Table S3.† Both types of solar cells prepared with (w/) and without (w/o) DIO yielded broad EQE spectra from 300 to 800 nm. It is noted that at a DIO concentration of 0.5–0.7%, the Jsc and FF simultaneously increased for the three PDI-based OSCs. Particularly for the case of the PDI4-based OSC device the use of DIO significantly improved the FF, delivering the maximum PCE of 6.44%, and indicating efficient charge collection. For all OSC devices prepared, the short-circuit current densities calculated from the EQE spectra agreed well with the corresponding trends in the direct photo J–V measurements under simulated solar illumination (Table S4†).
Charge transport properties
SCLC measurements were performed for investigating the bulk electron and hole mobilities of the blend films (Table 1 and Fig. S6†). The electron and hole mobility values were determined by fitting dark J–V curves on the basis of the Mott–Gurney equation24 as presented in the ESI.† The electron mobility (μe) values of the devices prepared w/o DIO increased from PTB7-Th:PDI1 (6.57 × 10−7 cm2 V−1 s−1) to PTB7-Th:PDI2 (2.01 × 10−6 cm2 V−1 s−1) to PTB7-Th:PDI4 (4.17 × 10−6 cm2 V−1 s−1), indicating that the twisted 3D molecular geometry of the PDIs could facilitate electron transport. The addition of DIO improves both electron and hole mobility values for the three PDI-based devices, most likely due to the positive impact of DIO that enables the optimum intermolecular packing of the polymer backbone via the plasticization of the branched side-chains of the polymer.26 Surprisingly, the electron mobility of the best performing PTB7-Th:PDI4 w/DIO system is comparable to that of the previously reported best PDI monomeric OSC device based on a polymer:PDI combination.24 Therefore, the replacement of the planar monomeric PDI with a 3D-twisted PDI multiadduct results in a marginal improvement in the electron transport, although the increase in the photocurrent generation is significant. In order to better resolve this interesting observation, we performed detailed characterization experiments on the film morphology and molecular structure of the studied systems.Film morphology and photoluminescence quenching
AFM height images of the neat PDI1, PDI2, and PDI4 films are presented in Fig. 3a–c. The neat PDI1 film exhibited highly aggregated features with the domain sizes of the aggregates based on the PDI1 molecules having a width of 0.58 ± 0.12 μm and a length of 0.27 ± 0.08 μm, and with a root-mean-square (rms) roughness of 4.43 nm. In contrast, in the corresponding AFM images of the neat PDI2 and PDI4 films, no aggregate formation could be seen and the films were found to be fairly smooth with a rms roughness of 0.37 nm and 0.36 nm, respectively. The surface roughness of the neat PTB7-Th was found to be 0.62 nm. The surface texture of PTB7-Th, PDI1, PDI2, and PDI4 neat films w/DIO was also studied (Fig. S8†). By adding the DIO component, the surface roughness of the neat PDI1 film drastically increased to 34.3 nm, whereas a slight increase in that of the PDI2 (0.43 nm) and PDI4 (0.93 nm) was recorded. For the case of the neat PTB7-Th film the use of DIO doubled its surface roughness (1.23 nm). The boiling point of DIO (Tb = 332 °C) is higher than that of the chlorobenzene host solvent (Tb = 131 °C), which would allow sufficient time for PDIs and PTB7-Th to aggregate. | ||
| Fig. 3 AFM height (5 μm × 5 μm) images of blend films based on neat PDI1, PDI2 and PDI4 (a–c) and PTB7-Th:PDI1, PTB7-Th:PDI2 and PTB7-Th:PDI4 blend films without DIO (d and e) and with DIO (g–i). | ||
For the PTB7-Th:PDI1 blend film, the height AFM image (Fig. 3d) showed the formation of largely aggregated PDI1 phases, similar to those observed for other monomeric PDI derivatives blended with polymer donors.20 In contrast, the AFM images of the PTB7-Th:PDI2 and PTB7-Th:PDI4 blend films suggest a well-mixed state of the donor and acceptor components at the mesoscopic length scale with small surface roughness values of 0.73 nm and 0.48 nm for the PTB7-Th:PDI2 and PTB7-Th:PDI4 blend films, respectively (Fig. 3e and f). The addition of the DIO component in the blends affects the layer morphology significantly due to the effect of DIO on the organization of the PTB7-Th polymer and PDIs as shown in Fig. 3g–i. In the presence of DIO, the donor and acceptor components in the PTB7-Th:PDI1 blend film tend to segregate into smaller domains (Fig. 3g) and a better mixing of the donor and acceptor components was achieved. For the case of the PTB7-Th:PDI2 and PTB7-Th:PDI4 blend films, the use of DIO leads to the formation of larger domains with increased surface roughness values of 2.43 nm and 2.71 nm, respectively. The addition of DIO in the PTB7-Th:PDI4 films attained a nanoscale segregated morphology with improved molecular interconnectivity that is required for efficient charge transport, hence explaining the optimum electron mobility found in the SCLC characterization experiments (see Table 1).20,47 The weak tendency of PDI2 and PDI4 to undergo π–π stacking interactions in the solid state was further verified by transmission electron microscopy (TEM) imaging. TEM images of neat PDI1, PDI2, and PDI4 films were recorded (Fig. S9†) to probe the formation of PDI aggregates in the bulk. Largely aggregated PDI domains were clearly visible in the neat film of PDI1 whereas, for the case of PDI2 and PDI4 neat films, only weak contrast images were obtained, most likely due to the incapability of these PDI derivatives to self-organize into larger aggregate species.
The correlation between the PCE trend and BHJ morphological properties of the OSC devices was explored by collecting photoluminescence (PL) intensity measurements from the blend films and comparing these results with those obtained from the relevant PTB7-Th and three PDI neat films as shown in Fig. S10 (ESI†). The calculated PL quenching efficiencies are summarized in Table S6 (ESI†). Among the studied films, the PTB7-Th:PDI1 exhibits the lowest PL quenching efficiency confirming the coarser phase separation between PTB7-Th and PDI1 that results in a low Jsc and PCE. Both PDI2 and PDI4 based BHJ films exhibit high PL quenching efficiencies (>90%) that correlate reliably with the better intermixing of the polymer and PDI components in the BHJ films. Particularly for the case of the PTB7-th:PDI4 system, the obtained PL quenching is the highest, reaching ∼95% (hole transfer quenching, excimers from PDI4).
Grazing incidence wide angle X-ray scattering
Grazing incidence wide angle X-ray scattering (GIWAXS) was used to understand the effect of the chemical structure on the molecular packing and aggregation properties of the neat PDIs and of the PTB7-Th:PDI blend films (Fig. 4, 5 and S11†), and the detailed information extracted from GIWAXS is summarized in Tables S7, S8 and S10.† 2D GIWAXS patterns of neat PDI1, PDI2, PDI4, and PTB7-Th are shown in Fig. 4a–d. In the XRD image of the monomeric PDI1 film, several distinct diffraction peaks appear with a preferential crystalline orientation. Perylene diimide-based molecules form various crystalline structures in thin films, depending on the alkyl chain conformation and chemical substitutes.48 The PDI1 film shows an intense and narrow diffraction peak at q = 0.35 Å−1 that corresponds to a d-spacing of ∼17.84 Å, which indicates that the long axis of the PDI1 molecule might be tilted from the substrate plane because the d-spacing is shorter than the longitudinal length of the PDI1 molecules (∼22.8 Å) (see Fig. S1 in the ESI†).49 On the other hand, in PDI2 and PDI4, few broad peaks appear with a weaker intensity and less pronounced molecular orientation, showing a weaker order of molecular stacking compared to the PDI1 molecule. In addition, the 1st diffraction peaks in PDI2 and PDI4 films shifted toward lower q values compared to that in PDI1 due to the larger molecular size of PDI2 and PDI4. The diffraction scans for the neat PTB7-Th show the in-plane (100) reflection at qxy = 0.29 Å−1 (d = 21.7 Å) and the broad out-of-plane (010) reflection at qz = 1.52 Å−1 (d = 4.13 Å), which indicates the typical face-on orientation of PTB7-Th crystals.50 For the neat PDI1 film, the 1st peak in the in-plane scan at q = 0.35 Å−1 (d = 17.84 Å) corresponds to alkyl chain stacking indicating strong aggregation with a coherence length of ∼321.1 Å, whereas the 1st peak in the in-plane scan of PDI2 (q = 0.32 Å−1, d = 19.6 Å and a coherence length of ∼68.9) and of PDI4 (q = 0.30 Å−1, d = 20.9 Å and a coherence length of ∼81.7) is weak and broad suggesting that the aggregation of PDIs is drastically reduced, which is consistent with the AFM and TEM images (shown in Fig. 3 and S9†). The coherence length was estimated from the full width half maxima (FWHM) of peaks by using the Scherrer equation.51 A strong 1st peak in out-of-plane corresponding to alkyl chain stacking and a peak at qxy = 1.77 Å−1 corresponding to π–π stacking25 suggest a dominating edge-on orientation in PDI1. However, the difference in the in-plane and the out-of-plane scan of PDI2, in which the 1st peak (∼0.32 Å−1) in out-of-plane is stronger than that in in-plane and stronger π–π stacking peak (∼1.3 Å−1) in in-plane than that in out-of-plane, suggests that the preferential orientation is weakly edge-on. On the other hand, PDI4 films showed random orientations with the identical in-plane and out-of-plane profiles.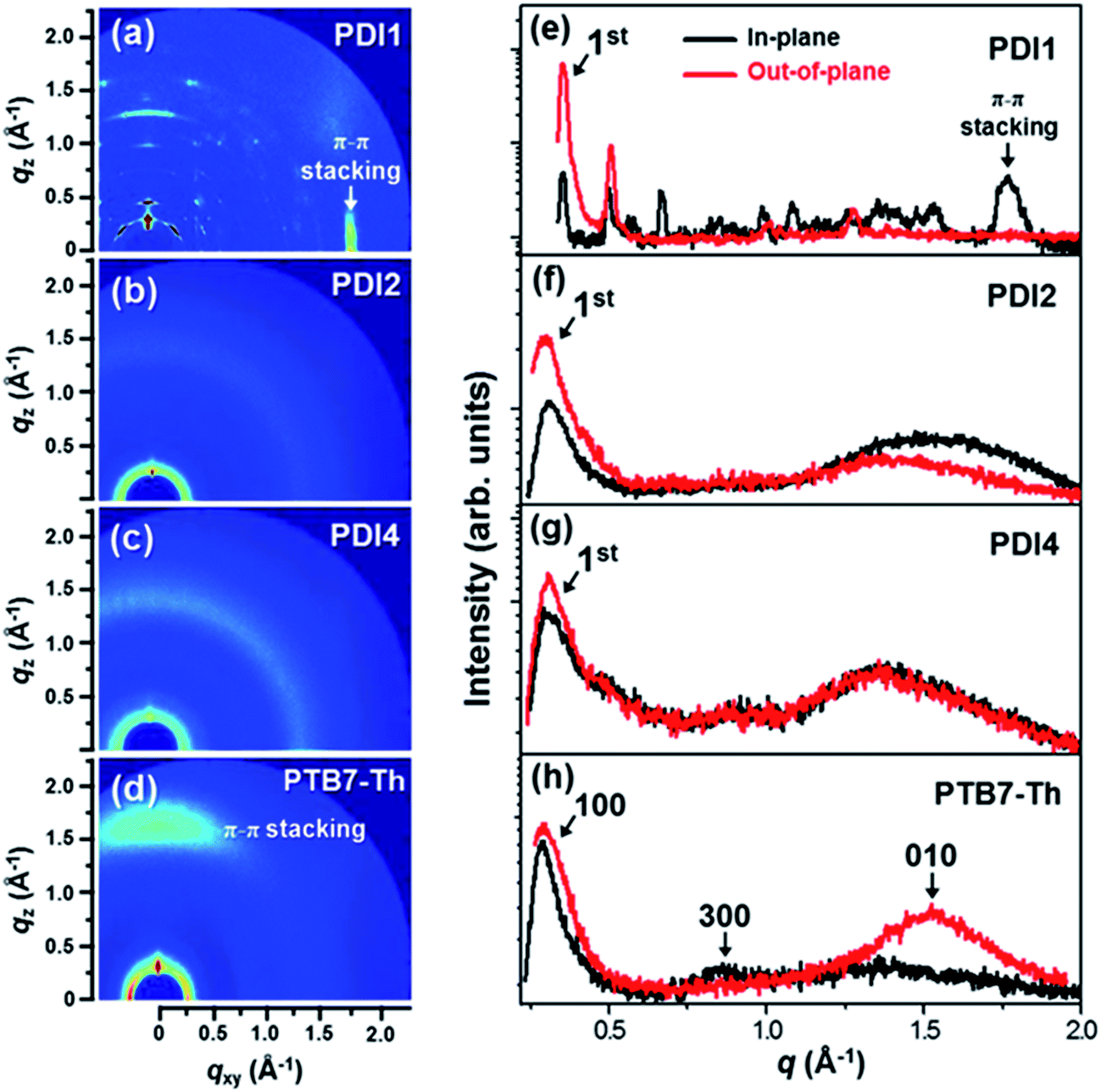 | ||
| Fig. 4 (a–d) GIWAXS images and (e–h) in-plane and out-of-plane scans of the PDI1, PDI2, PDI4 and PTB7-Th neat thin films. | ||
 | ||
| Fig. 5 In-plane and out-of-plane scans of the PTB7-Th:PDI1, PTB7-Th:PDI2 and PTB7-Th:PDI4 blend films (a–c) w/o DIO and (d–f) w/DIO. GIWAXS images of the respective films are shown as an inset in the graphs. | ||
In the in-plane scan of PTB7-Th:PDI1 shown in Fig. 5, the (100) reflection at qxy = 0.28 Å−1 is associated with the stacking of polymer backbones while the remaining peaks are associated with the PDI1. The PDI1 peak at qxy = 0.34 Å−1 (coherence length of 305.7 Å) indicates a strong aggregation of PDIs in PTB7-Th:PDI1 and after the addition of the DIO additive, 1st peak of PDI1 becomes more intense with a small shift in qxy (0.346 Å−1) indicating an increase in the coherence length (455.8 Å) and a small decrease in the d-spacing (18.15 Å). These results are in agreement with the harshly phase-separated morphology in the PTB7-Th:PDI1 film. However, the PTB7-Th:PDI2 film and PTB7-Th:PDI4 film exhibit strong diffraction peaks for lamellar stacking of PTB7-Th polymers, albeit weak diffraction peaks from PDI molecules. We deconvoluted the 1st diffraction peaks for comparison of the coherence length in the PTB7-Th:PDI blend films. The PTB7-Th:PDI2 film and PTB7-Th:PDI4 film show a weak increase in the coherence length even using DIO (from 84.3 Å to 100.9 Å for PDI2 and from 93.1 Å to 106.6 Å for PDI4).
Charge recombination dynamics
To further investigate the variation in the photovoltaic properties of the PDI-based OSCs, light intensity-dependent Jsc, transient photovoltage (TPV) and transient photocurrent (TPC) measurements were performed to look into the charge carrier recombination losses. The Jsc of all fabricated devices is plotted as a function of incident light intensity (Iexc) and the data are fitted with the power law equation Jsc ∝ Iexcα (ESI, Fig. S13†).52 All OSCs exhibit a quasilinear dependence of Jsc on Iexc, with coefficients of α = 0.915 for PTB7-Th:PDI1, α = 0.918 for PTB7-Th:PDI1 with DIO, α = 0.962 for PTB7-Th:PDI2, α = 0.975 for PTB7-Th:PDI2 with DIO, α = 0.966 for PTB7-Th:PDI4 and α = 0.981 for PTB7-Th:PDI4 with DIO. The minor deviations of the obtained α values from linearity could indicate that the Jsc of the PDI-based OSCs without DIO is limited by charge recombination losses,53 however such a deviation could also be attributed to space charge limited effects54 or to inefficient charge extraction caused by an improper choice of charge carrier-collecting electrodes.20,55In order to unambiguously monitor the process of free charge carrier recombination dynamics the data of the TPV and TPC measurements were analyzed. For the TPC measurement, a device was connected in series with a 50 Ω resistor, over which the laser pulse-induced charge decay dynamics were recorded with a digital oscilloscope and a differential capacitance technique was used to calculate the steady-state charge carrier density (n).56 The charge carrier lifetime was obtained from the TPV measurements by the exponential fitting of the photovoltage decay.57Fig. 6a shows the charge carrier lifetime as a function of carrier density for the optimized OSCs (w/o DIO). The corresponding results of OSCs w/DIO can be found in Fig. S14 (ESI†). Under 1 sun simulated solar illumination conditions, the PTB7-Th:PDI1, PTB7-Th:PDI2 and PTB7-Th:PDI4 devices w/o DIO show carrier lifetimes of 4.38 μs, 1.48 μs and 1.71 μs, respectively. The PTB7-Th:PDI1 devices exhibit longer carrier lifetimes as compared to PTB7-Th:PDI2 and PTB7-Th:PDI4 which is mainly due to the significantly lower carrier densities.56 At a similar charge carrier density for comparable devices, the carrier lifetime of PTB7-Th:PDI2 is found to be τPTB7-Th:PDI2(n=8×1017 cm−3) = 1.67 μs, and for the PTB7-Th:PDI4 devices, it is found to be τPTB7-Th:PDI4(n=8×1017 cm−3) = 3.37 μs, indicating that the PTB7-Th:PDI4-based device has a significantly slower recombination rate with respect to PTB7-Th:PDI2-based OSCs. The steep reduction of τ(n) with increasing charge carrier density shown in Fig. 6a further suggests the dominance of trap-based recombination in the studied PTB7-Th:PDI devices.58 The data shown in Fig. 6a were fitted with the power-law equation, τ(n) = τ0n−λ, where n is the charge carrier density, τ(n) corresponds to the charge carrier lifetime at n, and λ + 1 represents the apparent non-geminate recombination order.59 The value of λ is highest for the PTB7-Th:PDI1 system (λ = 2.83), decreasing to λ = 1.86 for the PTB7-Th:PDI2 system, and reaching λ = 1.65 for the PTB7-Th:PDI4 system. The highest λ value for PTB7-Th:PDI1 suggests the dominance of trap-based recombination, which can be correlated with larger PDI domains appeared in the AFM and TEM images (Fig. 3 and S9†), as compared to PTB7-Th:PDI2 and PTB7-Th:PDI4-based OSCs. Previous reports have discussed on the presence of trap states in the PDI phase of PDI monomer-based BHJ films.25 At present we find that non-geminate recombination losses are severe in PTB7-Th:PDI1 OSCs but they are reduced significantly in the PTB7-Th:PDI2 and PTB7-Th:PDI4 OSCs. Fig. 6b shows the recombination coefficient (k)58 as a function of charge carrier density for the PTB7-Th:PDI1, PTB7-Th:PDI2 and PTB7-Th:PDI4 OSCs w/o DIO. In a comparison of the PTB7-Th:PDI2 and PTB7-Th:PDI4-based OSCs, PTB7-Th:PDI4 OSCs exhibit a slightly lower recombination coefficient. With the DIO additive, the values of λ are reduced for PTB7-Th:PDI2 (λ = 1.67) and PTB7-Th:PDI4 (λ = 1.54) OSCs implying that the density of the trap states slightly decreases, whereas for the case of PTB7-Th:PDI1 (λ = 2.82) OSCs it remains approximately the same (Fig. S14 in the ESI†). Therefore, from a practical point of view the main consequence of increasing the dimensionality of the PDI core is the suppression of non-geminate charge recombination losses. We consider this an important finding for the future development of 3D-structured non-fullerene based electron acceptors.
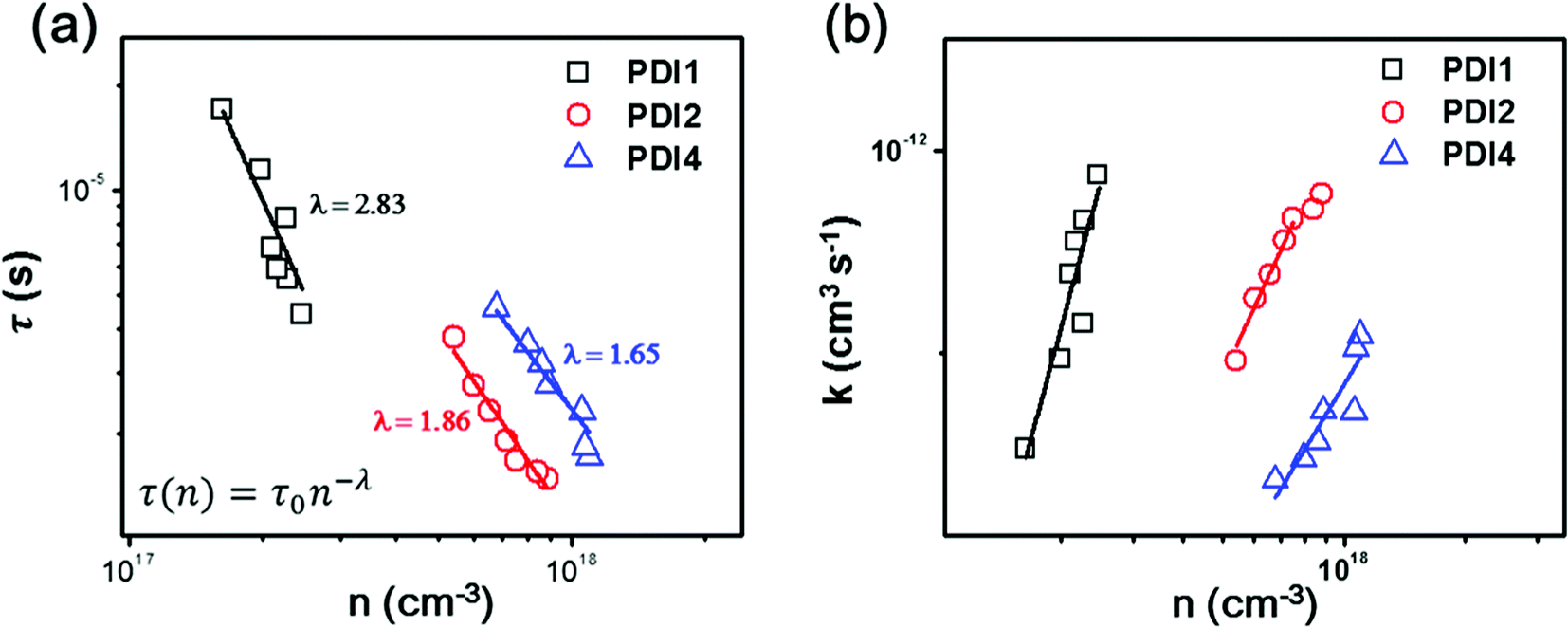 | ||
| Fig. 6 (a) Charge carrier lifetime (τ) derived from TPV measurements as a function of charge carrier density (n) obtained from TPC and (b) charge recombination coefficient (k) versus n for PTB7-Th:PDI1, PTB7-Th:PDI2 and PTB7-Th:PDI4 solar cells w/o DIO. In (a) the open scattered points are representing experimental data points, while the solid line demonstrates the fitting according to the inset equation. | ||
Low-temperature dependent charge carrier mobility
Temperature-dependent charge carrier mobility measurements are used to evaluate the difference in the trap distribution of three optimized PDI-based BHJ blend systems. Thus, temperature dependent electron and hole mobility values were determined through their dark J–V characteristics in an identical fashion as above. In Fig. 7a and b the hole and electron mobility values of the devices are plotted as a function of temperature within the range of 200–300 K, and fitted with the Arrhenius equation μ(T) = μ0exp(−ΔE/kT); here ΔE corresponds to the activation energy, which is closely related to the width of the energetic disorder or the trap distribution.60,61 From Fig. 7a and b, ΔE values of 0.260 eV, 0.168 eV and 0.161 eV are obtained for PTB7-Th:PDI1, PTB7-Th:PDI2 and PTB7-Th:PDI4 hole only devices, and 0.290 eV, 0.226 eV and 0.225 eV are obtained for PTB7-Th:PDI1, PTB7-Th:PDI2 and PTB7-Th:PDI4 electron-only devices, respectively. Interestingly, the width of the energetic disorder of the electron-only devices is not as broad as the corresponding width found for the hole-only devices. Evidently, the activation energy for the hole and electron transport is smaller in PDI2 and PDI4-based devices which also supports the reduced trap-based recombination in their respective OSC devices.61 Nonetheless, the difference between the activation energy of the three electron-only devices is small, suggesting that the energetic disorder in the lowest unoccupied molecular orbitals of the PDI derivatives depends weakly on the molecular structure of the PDI adduct.
 | ||
| Fig. 7 Temperature dependent (a) hole and (b) electron mobility of PTB7-Th:PDI (PDI1, PDI2, and PDI4) devices. The solid line represents the fitting curve with the Arrhenius equation to calculate the activation energy (ΔE). | ||
Conclusions
We have systematically compared the morphological and photovoltaic properties of three geometrically different PDI-based acceptors PDI1, PDI2, and PDI4 for elucidating the effect of increasing dimensionality of the PDI molecular structure on the performance of PDI-based OSCs devices, fabricated when PTB7-Th is used as the polymer donor. Our study reveals that the formation of excimers in PDI multiadducts is independent of the onset of π–π stacking in the solid state, as verified by the spectroscopic characterization of the three derivatives in dilute solution. Although by increasing the dimensionality of the PDI core the tendency of PDI molecules for intermolecular π–π stacking is reduced, the characteristic spectral signature of the PDI excimer persists due to the occurrence of intramolecular electrostatic interactions that favours the electronic coupling of the transition dipole moments of the PDI subunits in the multiadduct. Therefore, the participation of PDI excimers in photocurrent generation always needs to be taken into account regardless of the π–π stacking strength in these PDI-based OSCs.The use of the orthogonal structure of a 9,9′-spirobifluorene core for the design of the PDI4 derivative gave an optimum 3D molecular architecture that delivered a maximum PCE of 6.44%. Upon replacing the PDI1 monomer with the PDI2 and PDI4 multiadducts, the increased dimensionality of the PDI core improved the PL quenching efficiency of the initially formed PDI excimers and the electron transport in the PTB7-Th:PDI2 and PTB7-Th:PDI4 blends. Nonetheless, this improvement could not fully justify the high device performance of the PTB7-Th:PDI4 system. The actual effect of the increased 3D molecular architecture of the PDI4 derivative was identified by transient photovoltage characterization experiments that confirmed the suppression of non-geminate charge recombination losses in PTB7-Th:PDI4 devices. In light of these findings, the fine-tuning in the chemical structure of PDI derivatives with a 3D core will further optimize the PDI aggregate content in BHJ blend films and improve the photovoltaic properties of PDI-based, non-fullerene OSCs.
Experimental section
The UV-Vis absorption and PL spectra of the fabricated films were recorded with a Perkin Elmer, Lambda 1050 spectrometer and with a Horiba Jobin Yvon NanoLog spectrofluorimeter, respectively. The morphologies of the neat and blend films were determined using an atomic force microscope (MultiMode 8 Scanning Probe Microscope VEECO Instruments Inc.) in tapping mode and transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2200FS (with Image Cs-corrector). 2D-GIWAXS measurements were performed at the 3C beamline at the Pohang Accelerator Laboratory (PAL) in Korea. The photon energy is 10.6408 keV (λ = 1.1651 Å). The process of OSC fabrication is described in the ESI.† The current–voltage (J–V) characteristics were measured using a Keithley 4200 power source under AM 1.5G illumination at an intensity of 1 suns (Oriel 1 kW solar simulator) with a PVM 132 reference cell certified by NREL. The external quantum efficiency was measured using a photomodulation spectroscopy setup (Merlin, Oriel) with monochromatic light from a xenon lamp. The power density of the monochromatic light was calibrated using a Si photodiode certified by the National Institute for Standards and Technology. Photoexcitation intensity dependent J–V curves were measured with the same solar simulator setup by varying the intensity from 0.05 to 1 sun. TPV and TPC were measured using a TDC3054C digital oscilloscope connected with high-speed reamplifiers: SR560 and DHPCA-100. The samples were excited by using a 3 ns pulse laser at 532 nm (OBB, NL4300, and OD401) under AM 1.5G illumination at an intensity of 0.3–1.9 suns. For low temperature measurements, the devices were held in the standard sample holder (purchased from JANIS along with the cryostat) in a liquid-nitrogen cryostat (JANIS VPF-100). The low temperature (200–300 K) of the devices was controlled by connecting the cryostat to a Lake Shore, 335 temperature controller unit, and dark J–V characteristics were measured using a Keithley 2636 source meter.Acknowledgements
R. Singh and J. Lee contributed equally to this work. This work was supported by a grant (Code No. 2011-0031628) from the Center for Advanced Soft Electronics under the Global Frontier Research Program of the Ministry of Science, ICT & Future Planning, Korea. The authors thank the Pohang Accelerator Laboratory for providing the synchrotron radiation sources at the 3C beamline used for this study.References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- S. B. Darling and F. You, RSC Adv., 2013, 3, 17633 RSC.
- M. Kim, J. H. Park, J. H. Kim, J. H. Sung, S. B. Jo, M.-H. Jo and K. Cho, Adv. Energy Mater., 2015, 5, 1500802 CrossRef.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- P. Sonar, J. P. Fong Lim and K. L. Chan, Energy Environ. Sci., 2011, 4, 1558–1574 CAS.
- G. Ren, E. Ahmed and S. A. Jenekhe, Adv. Energy Mater., 2011, 1, 946–953 CrossRef CAS.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef CAS PubMed.
- C. W. Struijk, A. B. Sieval, J. E. J. Dakhorst, M. van Dijk, P. Kimkes, R. B. M. Koehorst, H. Donker, T. J. Schaafsma, S. J. Picken, A. M. van de Craats, J. M. Warman, H. Zuilhof and E. J. R. Sudhölter, J. Am. Chem. Soc., 2000, 122, 11057–11066 CrossRef CAS.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- A. Sharenko, C. M. Proctor, T. S. van der Poll, Z. B. Henson, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2013, 25, 4403–4406 CrossRef CAS PubMed.
- P. E. Keivanidis, I. A. Howard and R. H. Friend, Adv. Funct. Mater., 2008, 18, 3189–3202 CrossRef CAS.
- Y. Cai, L. Huo, X. Sun, D. Wei, M. Tang and Y. Sun, Adv. Energy Mater., 2015, 5, 1500032 CrossRef.
- M. C. R. Delgado, E.-G. Kim, D. A. d. S. Filho and J.-L. Bredas, J. Am. Chem. Soc., 2010, 132, 3375–3387 CrossRef PubMed.
- K. C. Song, R. Singh, J. Lee, D. H. Sin, H. Lee and K. Cho, J. Mater. Chem. C, 2016, 4, 10610–10615 RSC.
- J. J. Dittmer, E. A. Marseglia and R. H. Friend, Adv. Mater., 2000, 12, 1270–1274 CrossRef CAS.
- R. Singh, E. Giussani, M. M. Mrόz, F. Di Fonzo, D. Fazzi, J. Cabanillas-González, L. Oldridge, N. Vaenas, A. G. Kontos, P. Falaras, A. C. Grimsdale, J. Jacob, K. Müllen and P. E. Keivanidis, Org. Electron., 2014, 15, 1347–1361 CrossRef CAS.
- F. Würthner, C. Thalacker and A. Sautter, Adv. Mater., 1999, 11, 754–758 CrossRef.
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem. – Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed.
- J. M. Giaimo, J. V. Lockard, L. E. Sinks, A. M. Scott, T. M. Wilson and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 2322–2330 CrossRef CAS PubMed.
- R. Singh, E. Aluicio-Sarduy, Z. Kan, T. Ye, R. C. I. MacKenzie and P. E. Keivanidis, J. Mater. Chem. A, 2014, 2, 14348 CAS.
- E. Aluicio-Sarduy, R. Singh, Z. Kan, T. Ye, A. Baidak, A. Calloni, G. Berti, L. Duo, A. Iosifidis, S. Beaupre, M. Leclerc, H. J. Butt, G. Floudas and P. E. Keivanidis, ACS Appl. Mater. Interfaces, 2015, 7, 8687–8698 CAS.
- R. Singh, R. Shivanna, A. Iosifidis, H. J. Butt, G. Floudas, K. S. Narayan and P. E. Keivanidis, ACS Appl. Mater. Interfaces, 2015, 7, 24876–24886 CAS.
- S. Rajaram, R. Shivanna, S. K. Kandappa and K. S. Narayan, J. Phys. Chem. Lett., 2012, 3, 2405–2408 CrossRef CAS PubMed.
- J. Zhao, Y. Li, H. Lin, Y. Liu, K. Jiang, C. Mu, T. Ma, J. Y. Lin Lai, H. Hu, D. Yu and H. Yan, Energy Environ. Sci., 2015, 8, 520–525 CAS.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed.
- S.-Y. Liu, C.-H. Wu, C.-Z. Li, S.-Q. Liu, K.-H. Wei, H.-Z. Chen and A. K. Y. Jen, Adv. Sci., 2015, 2, 1500014 CrossRef.
- J. Lee, R. Singh, D. H. Sin, H. G. Kim, K. C. Song and K. Cho, Adv. Mater., 2016, 28, 69–76 CrossRef CAS PubMed.
- W. Chen, X. Yang, G. Long, X. Wan, Y. Chen and Q. Zhang, J. Mater. Chem. C, 2015, 3, 4698–4705 RSC.
- Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- N. Liang, K. Sun, Z. Zheng, H. Yao, G. Gao, X. Meng, Z. Wang, W. Ma and J. Hou, Adv. Energy Mater., 2016, 6, 1600060 CrossRef.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS PubMed.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C. Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y. L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- Y. Huang, J. Hu, W. Kuang, Z. Wei and C. F. Faul, Chem. Commun., 2011, 47, 5554–5556 RSC.
- W. Jiang, C. Xiao, L. Hao, Z. Wang, H. Ceymann, C. Lambert, S. Di Motta and F. Negri, Chem. – Eur. J., 2012, 18, 6764–6775 CrossRef CAS PubMed.
- D. Zhao, Q. Wu, Z. Cai, T. Zheng, W. Chen, J. Lu and L. Yu, Chem. Mater., 2016, 28, 1139–1146 CrossRef CAS.
- Z. Xie, V. Stepanenko, B. Fimmel and F. Würthner, Mater. Horiz., 2014, 1, 355 RSC.
- F. Würthner, C. Thalacker, A. Sautter, W. Schärtl, W. Ibach and O. Hollricher, Chem. – Eur. J., 2000, 6, 3871–3886 CrossRef.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed. Engl., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- J. Lee, S. B. Jo, M. Kim, H. G. Kim, J. Shin, H. Kim and K. Cho, Adv. Mater., 2014, 26, 6706–6714 CrossRef CAS PubMed.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- K. A. Kistler, C. M. Pochas, H. Yamagata, S. Matsika and F. C. Spano, J. Phys. Chem. B, 2012, 116, 77–86 CrossRef CAS PubMed.
- H.-C. Liao, C.-C. Ho, C.-Y. Chang, M.-H. Jao, S. B. Darling and W.-F. Su, Mater. Today, 2013, 16, 326–336 CrossRef CAS.
- S.-G. Liu, G. Sui, R. A. Cormier, R. M. Leblanc and B. A. Gregg, J. Phys. Chem. B, 2002, 106, 1307–1315 CrossRef CAS.
- M. Al-Hussein, H. C. Hesse, J. Weickert, L. Dössel, X. Feng, K. Müllen and L. Schmidt-Mende, Thin Solid Films, 2011, 520, 307–313 CrossRef CAS.
- W. Huang, E. Gann, L. Thomsen, C. Dong, Y.-B. Cheng and C. R. McNeill, Adv. Energy Mater., 2015, 5, 1401259 CrossRef.
- H. G. Kim, B. Kang, H. Ko, J. Lee, J. Shin and K. Cho, Chem. Mater., 2015, 27, 829–838 CrossRef CAS.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2015, 9, 35–41 CrossRef CAS.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
- L. J. A. Koster, V. D. Mihailetchi, H. Xie and P. W. M. Blom, Appl. Phys. Lett., 2005, 87, 203502 CrossRef.
- T. Ye, R. Singh, H.-J. Butt, G. Floudas and P. E. Keivanidis, ACS Appl. Mater. Interfaces, 2013, 5, 11844–11857 CAS.
- B. Yang, Y. Yuan and J. Huang, J. Phys. Chem. C, 2014, 118, 5196–5202 CAS.
- S. B. Jo, M. Kim, D. H. Sin, J. Lee, H. G. Kim, H. Ko and K. Cho, Adv. Energy Mater., 2015, 5, 1500802 CrossRef.
- D. Spoltore, W. D. Oosterbaan, S. Khelifi, J. N. Clifford, A. Viterisi, E. Palomares, M. Burgelman, L. Lutsen, D. Vanderzande and J. Manca, Adv. Energy Mater., 2013, 3, 466–471 CrossRef CAS.
- A. Foertig, J. Kniepert, M. Gluecker, T. Brenner, V. Dyakonov, D. Neher and C. Deibel, Adv. Funct. Mater., 2014, 24, 1306–1311 CrossRef CAS.
- N. I. Craciun, J. Wildeman and P. W. M. Blom, Phys. Rev. Lett., 2008, 100, 056601 CrossRef CAS PubMed.
- A. Pivrikas, N. S. Sariciftci, G. Juška and R. Österbacka, Prog. Photovolt. Res. Appl., 2007, 15, 677–696 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta08870h |
| ‡ Dr R. Singh and J. Lee contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |