
Click-based porous cationic polymers for enhanced carbon dioxide capture†
Alessandro
Dani
*ab,
Valentina
Crocellà
b,
Claudio
Magistris
c,
Valentina
Santoro
d,
Jiayin
Yuan
a and
Silvia
Bordiga
 *b
*b
aDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, OT Golm, D-14476 Potsdam, Germany. E-mail: alessandro.dani@mpikg.mpg.de
bDepartment of Chemistry, NIS and INSTM Reference Centre, University of Turin, Via Quarello 15, 10135 Torino, Italy. E-mail: silvia.bordiga@unito.it
cDepartment of Chemistry and NIS Interdepartmental Centre, University of Turin, Via P. Giuria 7, 10125 Torino, Italy
dDepartment of Molecular Biotechnology and Health Sciences, University of Turin, Via Nizza 52, 10126 Torino, Italy
First published on 9th November 2016
Abstract
Imidazolium-based porous cationic polymers were synthesized using an innovative and facile approach, which takes advantage of the Debus–Radziszewski reaction to obtain meso-/microporous polymers following click-chemistry principles. In the obtained set of materials, click-based porous cationic polymers have the same cationic backbone, whereas they bear the commonly used anions of imidazolium poly(ionic liquid)s. These materials show hierarchical porosity and a good specific surface area. Furthermore, their chemical structure was extensively characterized using ATR-FTIR and SS-NMR spectroscopies, and HR-MS. These polymers show good performance towards carbon dioxide sorption, especially those possessing the acetate anion. This polymer has an uptake of 2 mmol g−1 of CO2 at 1 bar and 273 K, a value which is among the highest recorded for imidazolium poly(ionic liquid)s. These polymers were also modified in order to introduce N-heterocyclic carbenes along the backbone. Carbon dioxide loading in the carbene-containing polymer is in the same range as that of the non-modified versions, but the nature of the interaction is substantially different. The combined use of in situ FTIR spectroscopy and micro-calorimetry evidenced a chemisorption phenomenon that brings about the formation of an imidazolium carboxylate zwitterion.
Introduction
Porous organic polymers are well-established among the field of porous materials.1–5 More than ten years of research allowed for the discovery of many materials that differ in structure, porosity, functional group crystallinity, and long-range order. Extensive research for the tuning of these properties leads to target-specific porous polymers, useful to meet a variety of applications, such as gas or molecule storage,6–8 separation, drug delivery,9 electronics,10,11 and catalysis.12,13 Most of these materials are rich in phenyl and alkyne moieties,14,15 while only some of them possess ionic groups.7,16 One of the promising goals in the field of porous polymers is to introduce task-specific functional groups into the chemical structure of the porous network and target the topology and the structure of the material to a well-defined application.1,17 The synthetic pathways of these materials are usually time-demanding and require noble metal-based catalysts, since the common approaches involve Suzuki, Sonogashira–Hagihara cross-coupling and Yamamoto-type Ullmann cross-coupling reactions.18 In recent years, “click-chemistry” has been exploited for the development of these materials, leading to advantageous and competitive syntheses in terms of time and costs.19–23 A current challenge for porous polymers is to build up new structures, taking advantage of facile synthetic strategies, e.g. click chemistry-based ones, to transfer functional moieties into the polymers to define relevant industrial applications.Among various kinds of porous polymers, poly(ionic liquid)s (PILs) are gaining more and more interest over the past years because of the high density of ionic liquid species in the macromolecular architecture, which leads to a broad range of applications.24–29 Usually, porous PILs are synthesized in a bottom-up approach via common radical polymerization, using cross-linker monomers and/or templates.30–33 Some recent studies describe the introduction of imidazolium or pyridinium ionic liquid functionalities inside a microporous polymer, basically by the synthesis of different imidazolium or pyridinium-functionalized monomers, respectively, which are connected together by tetrahedral building units, using different palladium-based cross-coupling catalysis.12,13,16,34 Furthermore, two other groups reported about the closure of the imidazolium ring during the formation of the polymer network, in a two-step reaction. In this case, the first step was the synthesis of the Schiff base polymer network; once the material was isolated, the imidazolium ring was closed in the second step.7,35
In this work, we describe how to obtain a set of “click-chemistry” based imidazolium porous poly(ionic liquid)s. These porous cationic polymers are obtained taking advantage of the modified Debus–Radziszewski imidazolium synthesis, which follows the main principles of the “click-chemistry” defined by Sharpless et al.36,37 The Debus–Radziszewski reaction for the production of imidazole dates back to more than a century ago,38,39 because of the high yield and the very mild conditions, this reaction remains the benchmark for industrial imidazole production.40 A modified version of this reaction developed by Esposito et al. allows for the direct synthesis of imidazolium ionic liquids. This reaction already proved efficiency towards the synthesis of amino acid-derived imidazolium ionic liquids, linear poly(ionic liquid)s, and to cross-link polymers with dangling amino groups.41–43 Its efficiency was also proven starting from aromatic amines and aldehydes as reactants.44,45
Herein we exploited the Debus–Radziszewski imidazolium synthesis for the homocondensation of tetrakis(4-aminophenyl)methane, one of the most common tetrahedral building units used for the synthesis of microporous polymers. This click reaction leads to a network that expands in all the three dimensions, and the intrinsic steric hindrance of the monomers confers porosity to the resulting polymer. The reaction is irreversible because of the covalent closure of the thermodynamically stable imidazolium ring and runs under kinetic rather than thermodynamic control; therefore the resulting material is amorphous in nature. The as-obtained polymeric architecture faces the imidazolium cation linked through positions 1 and 3 to the main chain, differing from the common PILs that bear imidazolium as dangling groups. This imidazolium main chain polymeric architecture was described only in a few studies and showed increased thermal stability of the polymer and different chemical properties arising from the conjugation of the imidazolium with two phenyl rings.46,47
Ionic liquids and poly(ionic liquid)s are well-known materials for carbon dioxide adsorption.48–50 We would like to take both the advantages of their intrinsic affinity towards this molecule and transfer it to microporous polymers, which are already known for their good gas adsorption properties.51 The as-synthesized click reaction-based porous cationic polymers (hereafter referred to as CB-PCPs) show one of the highest carbon dioxide adsorption capacities ever reported among porous PILs, 2 mmol g−1 at 273 K and 1 bar. CB-PCPs are imidazolium-functionalized porous polymers, obtained via facile click-synthesis, and bearing all the common anions used in the field of PILs for carbon dioxide adsorption. N-Heterocyclic carbenes (NHC) are introduced into CB-PCPs, and the microporosity is tuned by varying the synthetic parameters. This set of CB-PCPs permit to understand how carbon dioxide adsorption properties are related to the structure and the porosity of the materials. In addition, a deep physico-chemical study performed with in situ FTIR spectroscopy and adsorption micro-calorimetry, allows one to distinguish different interactions between carbon dioxide and ionic polymers, or NHC bearing polymers.
Syntheses
Click reaction-based porous cationic polymers (CB-PCPs) were synthesized according to the procedure reported in Scheme 1. Briefly, tetrakis(4-aminophenyl)methane was dissolved in a mixture of water and acetic acid. In a second vial, formaldehyde and methyl glyoxal were dissolved in water and acetic acid. These two solutions were mixed together and the reaction proceeded immediately, as evidenced by the solution color variation from yellow to dark brown, due to the development of the conjugated aromatic system; then the solution was heated at 80 °C for 12 hours (Scheme 1 part a) to complete the reaction. The acetic acid is used as the catalyst and also ended up as the acetate anion in the final product, named CB-PCP 1. The proposed mechanism of the modified Debus–Radziszewski imidazolium synthesis is reported in Fig. S1.† Other CB-PCPs having different anions are obtained by the anion exchange of CB-PCP 1 polymer solution with different salts. We used sodium tetrafluoroborate, bis(trifluoromethane)sulfonimide lithium salt, potassium hexafluorophosphate, and sodium trifluoromethanesulfonate to obtain, respectively, CB-PCP 2, 3, 4 and 5 (Scheme 1, part b). NHC was introduced by reacting the polymer CB-PCP 1 with potassium tert-butoxide in anhydrous THF for 72 hours; the obtained sample is named CB-PCP 6 (Scheme 1, part c). All the polymers were purified according to their physical aggregation status: if in solution, they were dialyzed against water; if as a gel they were washed with water. Finally, powdery CB-PCPs 1–5 were obtained by freeze-drying, whereas the moisture-sensitive CB-PCP 6 was washed with anhydrous solvents and freeze-dried from 1,4-dioxane. A detailed description of syntheses can be found in the ESI.† A successful anion exchange offers indirect evidence of the successful closure of the imidazolium ring during the synthesis. In fact, only a cationic polymer is able to permanently bind various anions along the structure, without their leaching during the washing step. This new click-based synthetic approach makes PCPs and porous PILs even more green and sustainable.19 The main strengths of this reaction route are the following: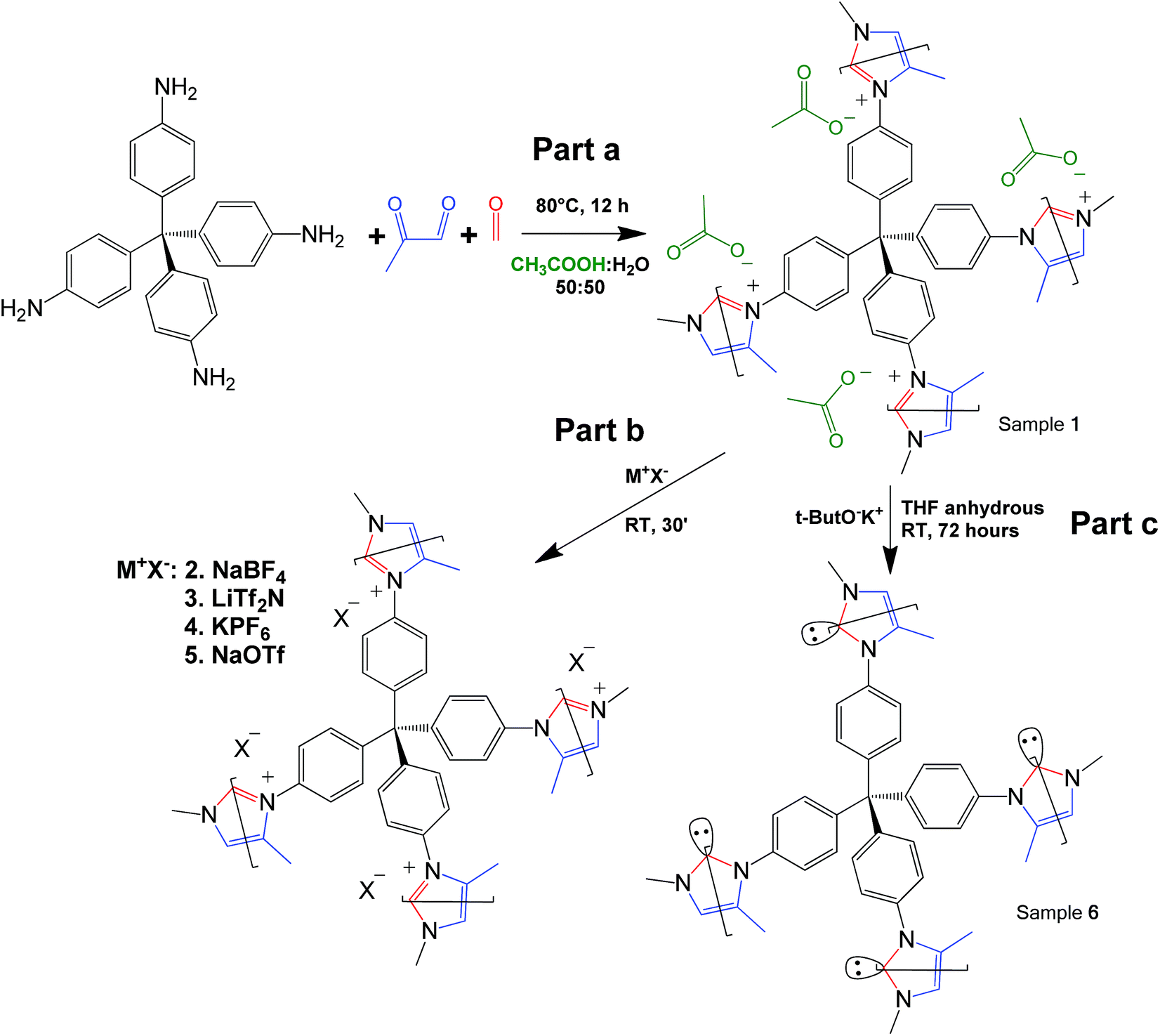 | ||
| Scheme 1 Part a: the synthetic route towards the preparation of the sample CB-PCP 1 starting from tetrakis(4-aminophenyl)methane in a one-step reaction. Part b: the anion exchange between the samples CB-PCP 2, 3, 4 and 5. Part c: the synthetic route towards the preparation of the sample CB-PCP 6 introducing NHC from the imidazolium ring. | ||
• It leads to high yield of the final polymer (almost 100%).
• It only forms water as a sub-product.
• It has a high thermodynamic driving force arising from the closure of the imidazolium ring.
• Reaction products are stable and the reaction is not reversible.
• 85% efficiency in atomic economy is obtained.52
Reaction takes place under very mild conditions (80 °C for 12 hours).
• Reagents are easily available except for the tetra-amine, which requires some synthetic steps.
• Reaction uses water and acetic acid as solvents.
• Final products are solids (CB-PCPs 1a, 1b, and 2–5) and they are easily purified from the residual monomers by washing with water.
The modified Debus–Radziszewski reaction itself works at room temperature and is rather fast (about 1 hour) when performed at a molecular level or in the case of linear polymers.41,42 In contrast, in the case of cross-linked polymers, it is necessary to increase both temperature and the reaction time due to the higher viscosity.
Vibrational properties of CB-PCPs
The attenuated total reflection-infrared (ATR-IR) spectrum of sample 1, reported in Fig. 1 curve (b), was collected as the first evidence of the successful polymerization. The spectrum is characterized by a series of complex overlapping absorptions. In particular, the two bands located at 1504 and 811 cm−1 can be ascribed to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) stretching of the conjugated aromatic system and to the τ(C–H) out-of-plane bending, respectively.53 These bands are also well evident in the spectrum of the tetrakis(4-aminophenyl)methane monomer, reported as a reference in Fig. 1 curve (a). The first, clear evidence of the occurred polymerization is the absence, in the spectrum of the CB-PCP, of the two peaked signals at 3156 and 3395 cm−1 related to the ν(N–H) stretching modes of the amino groups of the monomer. In fact, in the 3500–3000 cm−1 region, only a broad and ill-defined band is present, ascribable to the ν(O–H) stretching vibrations of physisorbed water. Unfortunately, the peculiar absorption band of the imidazolium ring, at around 1160 cm−1, appears too weak to be detected, due to the presence of the broad set of signals in the 1500–800 cm−1 spectral range. For this reason, this specific absorption cannot be used as evidence of the successful polymerization. The band at 1705 cm−1 is ascribable to the ν(CO) stretching mode of the amide moiety of the chain terminals, generated by the reaction between the amino groups of the monomer and the excess of acetic acid. This chemical behavior is also well evident in the mass spectra of the dimer reported herein later. Both the symmetric and antisymmetric stretching modes of the acetate anion can be observed in the spectrum of sample 1, at 1409 and 1566 cm−1, respectively.54 These bands well evident also in the spectrum of sodium acetate, are reported as reference in Fig. 1 curve (c).
C) stretching of the conjugated aromatic system and to the τ(C–H) out-of-plane bending, respectively.53 These bands are also well evident in the spectrum of the tetrakis(4-aminophenyl)methane monomer, reported as a reference in Fig. 1 curve (a). The first, clear evidence of the occurred polymerization is the absence, in the spectrum of the CB-PCP, of the two peaked signals at 3156 and 3395 cm−1 related to the ν(N–H) stretching modes of the amino groups of the monomer. In fact, in the 3500–3000 cm−1 region, only a broad and ill-defined band is present, ascribable to the ν(O–H) stretching vibrations of physisorbed water. Unfortunately, the peculiar absorption band of the imidazolium ring, at around 1160 cm−1, appears too weak to be detected, due to the presence of the broad set of signals in the 1500–800 cm−1 spectral range. For this reason, this specific absorption cannot be used as evidence of the successful polymerization. The band at 1705 cm−1 is ascribable to the ν(CO) stretching mode of the amide moiety of the chain terminals, generated by the reaction between the amino groups of the monomer and the excess of acetic acid. This chemical behavior is also well evident in the mass spectra of the dimer reported herein later. Both the symmetric and antisymmetric stretching modes of the acetate anion can be observed in the spectrum of sample 1, at 1409 and 1566 cm−1, respectively.54 These bands well evident also in the spectrum of sodium acetate, are reported as reference in Fig. 1 curve (c).
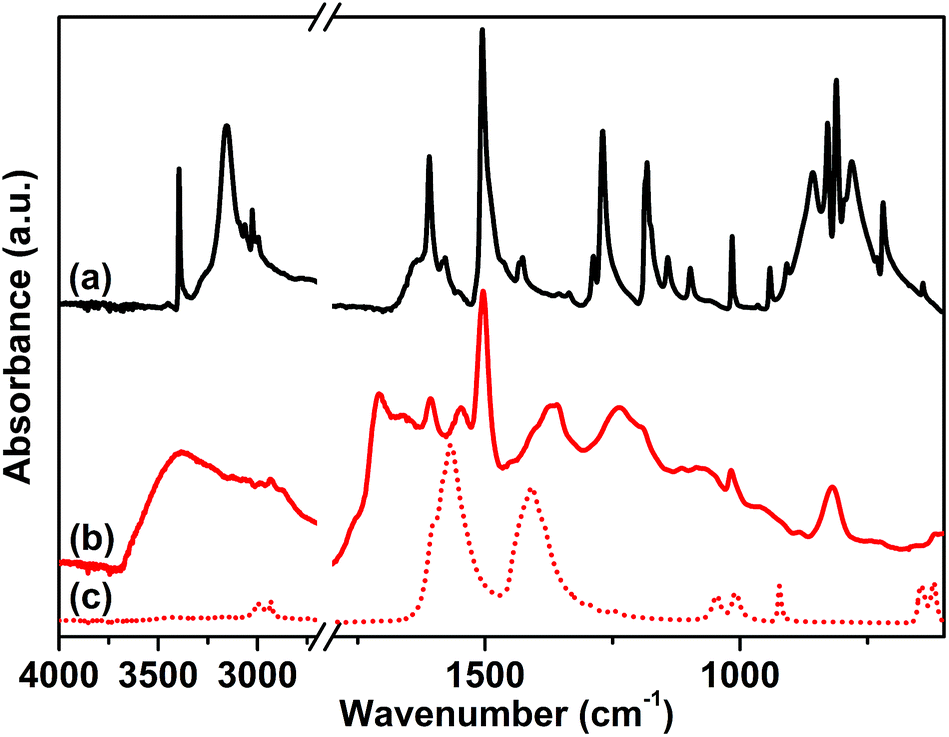 | ||
| Fig. 1 ATR-IR spectra of tetrakis(4-aminophenyl)methane (a), sample 1 (b) and sodium acetate (c) collected in air. | ||
The anion exchange with various salts was performed, as previously described, in order to replace the acetate anion and to obtain a set of different CB-PCPs. The ATR-IR spectra of all these materials are reported in Fig. 2 and compared with the spectra of the corresponding salts. In all cases, the vibrational modes of the polymeric network are clearly overlapped with the spectral features of the employed anion. In particular, the following can be observed:
 | ||
| Fig. 2 ATR-IR spectra of sample 2 (a), NaBF4 (b), sample 3 (c), LiTf2N (d), sample 4 (e), KPF6 (f), sample 5 (g), NaOTf (h) and sample 6 (i) collected in air. Starred bands (*) in polymers 1–5 were assigned to anions. | ||
(i) The ATR-IR spectrum of sample 2 (Fig. 2 curve a) displays a medium broad band at 1060 cm−1, assigned to the asymmetric vibrational modes of the BF4− anion,55 and a signal at around 3152 cm−1, overlapped with the broad band of physisorbed water, due to the combination of the imidazolium ring stretching mode with the BF4− stretching vibrations.56,57 The reference spectrum of NaBF4 (Fig. 2 curve b) exhibits a single band at 1005 cm−1 related to the BF4− vibrational modes and this signal is red-shifted with respect to sample 2 due to the strong force field of the Na+ cation interacting with BF4−.
(ii) In the case of sample 3 the ATR-IR spectrum (Fig. 2 curve c) exhibits all the Tf2N− ion peculiar vibrational modes (see asterisks) at 747 cm−1 (ν(S–N)), 798 cm−1 (ν(C–S) + ν(S–N)), 1188 cm−1 (ν(CF3)) and 1356 cm−1 (ν(SO3)),58 overlapped with that of the polymer network, as highlighted by the comparison with the reference LiTf2N spectrum (Fig. 2 curve d).
(iii) A strong band at 841 cm−1, due to the ν(P–F) stretching mode of the PF6− anion, is evident in the ATR-IR spectrum of sample 4 (Fig. 2 curve e). In the KPF6 reference spectrum (Fig. 2 curve f), this band is red-shifted to 805 cm−1 due to the interaction between the ion pairs. In fact, the strong force field of the small K+ cation affects the length of the P–F bonds of the PF6− anion.
(iv) Sample 5 presents, in its ATR-IR spectrum (Fig. 2 curve g), three new intense bands at 1245 cm−1, 1153 and 1027 cm−1 ascribable to the ν(CF3) and ν(SO3) modes characteristic of the TfO− anion, as proved by the comparison with the reference spectrum of NaOTf reported in Fig. 2 curve (h).59
The NHC formation in sample 6 is pointed out by the appearance of a new strong adsorption band at 1605 cm−1, arising from the stretching of the NHC ring (Fig. 2 curve i).7
Elemental analysis
The elemental analysis of sample 1 was performed in order to obtain quantitative information on the elemental structure of the material. The percentages of C, H and N, reported in Table S1,† are, respectively, 74.78%, 5.55% and 9.11%. These values are in perfect agreement with the calculated ones, pointing out a polymer structure having an elemental composition corresponding to the suggested structure.Solid-state NMR
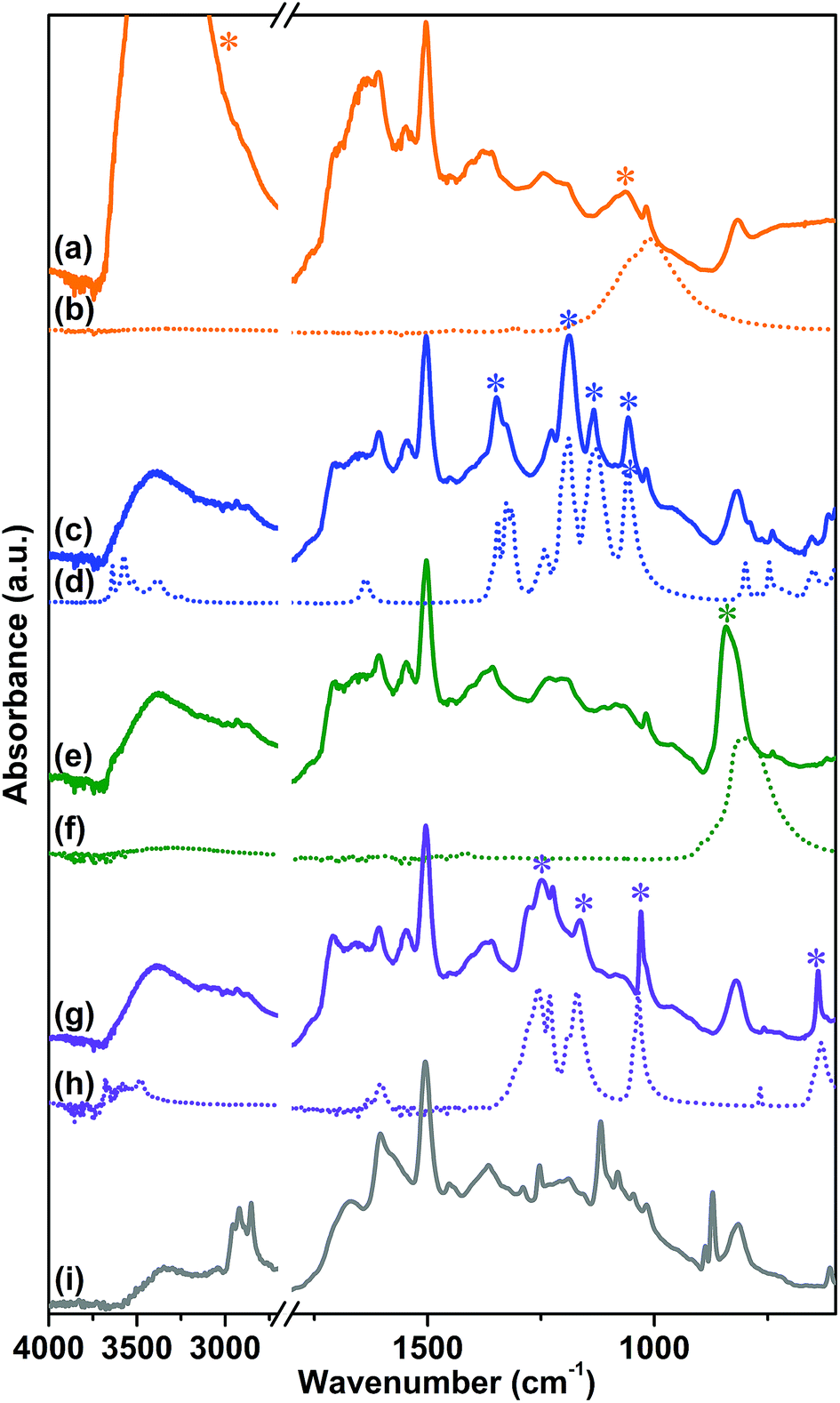
In order to prove the structure of CB-PCP 1 and the presence of the imidazolium inside the polymeric network, 1H (Magic-Angle Spinning) MAS and 13C Cross Polarization Magic-Angle Spinning (CP-MAS) solid-state NMR spectroscopy measurements were performed. Fig. 3a shows the 1H MAS solid-state NMR of sample 1 in which two families of protons are clearly distinguishable. The one at the lower chemical shift is related to the –CH3 protons of the dangling methyl group in position 4 on the imidazolium ring and also to the –CH3 protons of the acetate anion, whereas the other one is related to the aromatic protons along the polymeric backbone. Fig. 3b and c report, respectively, the 13C CPMAS solid-state NMR spectra and a polymer scheme with the corresponding assignations. In the 100–150 ppm range, the carbons constituting the phenyl and the imidazolium rings are well evident. | ||
| Fig. 3 1H MAS (a) and 13C CP-MAS (b) solid-state NMR spectra of sample 1. (c) Scheme of sample 1 that evidences the assignment of 13C CP-MAS spectrum signals. | ||
Three bands are well distinguishable in Fig. 3b; however, a specific assignment is not possible due to the overlap between the signals of the phenyl ring and the imidazolium ring carbons. In particular, the band at 62 ppm is related to the quaternary carbon C1, the signal at 24.9 ppm is ascribed to the methyl group carbons C11 and C9, whereas the 13C carbonyl signal C10 of the acetate anion is visible as a small and broad band at around 200 ppm. It is worth noting that the main inconvenience to be avoided during the synthesis of CB-PCP-1 is the formation of a polyimine network, in which the closure of the imidazolium ring by formaldehyde is not fully performed. In this specific case, two 13C strong bands between 150 and 200 ppm, related to imine (CN) 13C should be present, as reported by Thiel et al.35 The CB-PCP-113C CP-MAS solid-state NMR spectrum does not show the two bands related to the imine moiety, indicating the successful formation of the imidazolium network. The weak band at 160 ppm can be ascribed to the acetamide moiety present at the chain terminal.60
High resolution mass spectroscopy
Since sample 1 is obtained in solution after the reaction, we tried to confirm the structure by means of high-resolution mass spectroscopy (HR-MS).61 Unfortunately, the mass weight of the crosslinked polymeric chain of the sample was too high and the electrospray ionization (ESI) failed to transfer the sample from the solution to the gas phase. Therefore, we synthesized a partially linear polymer (sample 1s), using a lower ratio of aldehydes with respect to the amino monomer (details about this synthesis are reported in the ESI† section), and analysed the reaction solution using HR-MS. From the HR-MS spectra reported in Fig. S2,† in the reaction solution, the presence of the dimer having an m/z ratio of 809.4156 is evident, and furthermore, a distribution of species ascribable to the dimer bearing the acetamide moiety on the amino groups can be observed (Table S2†). The formation of the acetamide moiety is induced by the presence of an excess of acetic acid in the solution that reacts with the free amino group of the dimer. The trimer and tetramer formed during the reaction are also visible in the HR-MS spectra (Fig. S3 and S4†), along with their distribution bearing the acetamide moiety on the amino group (Tables S3 and S4†), proving the growth of the polymeric chain. Polymeric chains longer than a tetramer are difficult to be observed because of the high molecular weight, which prevents the de-solvation and the ionization operated by the ESI source.Further evidence of the closure of the imidazolium ring is furnished by MSn fragmentation obtained for the dimer having an m/z ratio of 809.4156, which is reported, together with the proposed chemical structure, in Fig. S5 part a–d.† The fragmentation shows the loss of the four phenylamino groups, the first two in the form of radicals, and the second two in the form of aniline. The imidazolium ring is not fragmented as it is the strongest part of the molecule due to the system of conjugated bonds. MSn fragmentation is also performed on the dimer with one acetamide moiety (m/z of 851.4181 shown in Fig. S6†), highlighting both the losses of free phenylamino groups and phenylamino groups with the acetamide moiety. These two signals exhibit different intensities due to statistical distribution.
Textural and morphological properties of CB-PCPs
The pore structures of CB-PCPs were investigated by means of N2 adsorption at 77 K. The adsorption isotherms of samples 1 to 6 are reported in Fig. 4. All the isotherms possess characteristic behavior, already observed in the case of other polymeric networks.62,63 More in detail, the adsorption isotherms of samples 1–5 exhibit a pronounced knee at low relative pressures indicating the presence of micropores, and, moreover, the isotherm profile constantly rises after the micropore filling, not reaching a plateau. Still, a peculiar hysteresis, in which the desorption branch of the isotherm is not close to the adsorption branch at low relative pressures, can be observed for all the polymers. These phenomena, particularly evident for samples 3, 4 and 6, are mostly attributed to a swelling of the polymeric matrix due to the elastic deformations occurring during nitrogen adsorption.62,63 For all these reasons, the porosity of these materials is better described by the desorption branches of the isotherms. In fact, the swelling of the materials being proportional to the gas pressure, a fraction of pores is available for N2 adsorption only at high values of the relative pressure.62,63 For sample 2, the desorption branch exhibits a weak step at 0.5 p/p0, typical of materials with slit pores, whereas, in the case of samples 1 and 5, the hysteresis is less evident than for the other polymers. These differences are probably derived from the various anions, which can slow down the swelling of the material operated by N2 during the adsorption step. | ||
| Fig. 4 N2 adsorption isotherms at 77 K for samples: 1 (red curve), 2 (orange curve), 3 (blue curve), 4 (green curve), 5 (violet curve) and 6 (dark grey curve). | ||
The Brunauer–Emmett–Teller (BET) specific surface areas (SSAs) of the different CB-PCPs are reported in Table 1. The microporosity of these materials arises from the inefficient packing of the sterically hindered tecton monomers that induces empty spaces inside the network and therefore high accessibility to the formed imidazolium moieties. A fraction of mesopores is also present, derived from the fragmentation of the network. Since the adsorption isotherms exhibit mixed behaviour, typical of both mesoporous and microporous materials, the values of both BET and Langmuir SSA are reported in Table 1. In all the materials having the same polymeric backbone, the SSA strongly depends on the anion nature. Samples 1, 2 and 5, having hydrophilic anions, exhibit the highest SSAs, probably due to the higher capacity of swelling in aqueous solution that leads to a better retaining of the porous structure during the freeze-drying process. In contrast, samples 3 and 4, with hydrophobic anions swell less when suspended in water and after the freeze-drying procedure. In fact, they show less porosity as the polymeric chains are more entangled. Sample 6, the CB-PCP bearing carbene, exhibits a remarkably lower SSA (BET = 77 m2 g−1) even if its isotherm retains the profile described for the other materials, pointing out its micro/mesoporous character. The decrease of the SSA is probably ascribable to a partial Wanzlick equilibrium occurring during the carbene synthesis, which leads carbenes to couple with each other.64 Furthermore, the different solvents employed for freeze-drying this material (1,4-dioxane) could confer different swelling properties to the polymeric chains.
| Sample name | SSA BET (m2 g−1) | SSA Langmuir (m2 g−1) | V total (cm3 g−1) | V micro (cm3 g−1) |
|---|---|---|---|---|
| a Total pore volume obtained from NL-DFT analysis of the adsorption isotherm. b Micropore volume obtained from NL-DFT analysis of the adsorption isotherm. | ||||
| 1 | 419 | 570 | 0.203 | 0.131 |
| 2 | 436 | 595 | 0.187 | 0.109 |
| 3 | 176 | 245 | 0.109 | 0.034 |
| 4 | 325 | 441 | 0.146 | 0.088 |
| 5 | 426 | 578 | 0.187 | 0.112 |
| 6 | 77 | 105 | 0.055 | 0.013 |
| 1a | 396 | 530 | 0.128 | 0.120 |
| 1b | 8.7 | 12.9 | — | — |
The pore volume was derived from the adsorption branches of the isotherms, using the non-local density functional theory (NL-DFT) pore model for carbon with slit-pore geometry. The total pore volume is around 0.19 cm3 g−1 for samples bearing hydrophilic anions (i.e. samples 1, 2 and 5) while it is lower for samples with an inferior surface area. The percentage ratio between the micropore volume and the total pore volume is approximately constant (∼60%) for all the samples, except for sample 3 which presents a lower ratio (∼30%). This behavior can be ascribed to the steric hindrance of the large, hydrophobic Tf2N− anion, which can occlude a fraction of the micropores. The total pore volume is lower for sample 6 due to its lower SSA, and it also has a low micropore/total pore volume ratio (∼24%), probably due to the increased crosslinking density arising from the coupling of carbenes.
In order to investigate the effect of the starting solution concentration on the textural properties of the resulting materials, samples 1a and 1b have been synthesized employing solutions two or four times more concentrated, respectively (synthesis details are reported in the ESI†). The N2 adsorption/desorption isotherms of these samples are shown in Fig. S7,† whereas the values of the SSA and pore volume are reported in Table 1. The BET SSA of sample 1a is very similar to the value reported for sample 1 (396 m2 g−1), however, this material is almost totally microporous. This behavior could be derived from polymer gelification that can occur by employing a lower reaction solution volume. The decrease of the available reaction volume induces a reduced swell of the polymer that, in turn, is balanced by the steric repulsion of the tecton monomers, producing an extensive polymeric network mainly constituted of micropores. The further lower reaction volume of sample 1b, generates a bulky polymer with an almost null SSA (Fig. S7†). The pore size distribution plots of all samples are reported in Fig. S8 and S9.†
The sample morphology was investigated by SEM. Representative SEM images of sample 1 are reported in Fig. 5 and in Fig. S10† (lower magnification). The CB-PCP 1 appears in the form of particles of 1–20 μm. The particles are irregular in shape with evident fragmentation, clearly visible in Fig. 5. The microporous structure of these polymers is instead not visible with SEM microscopy. The hierarchical porosity in CB-PCPs allows an easy diffusion of gas molecules inside the polymeric matrix thus reaching the imidazolium active sites. SEM images of the other CB-PCPs are not reported, these materials being morphologically very similar to sample 1.
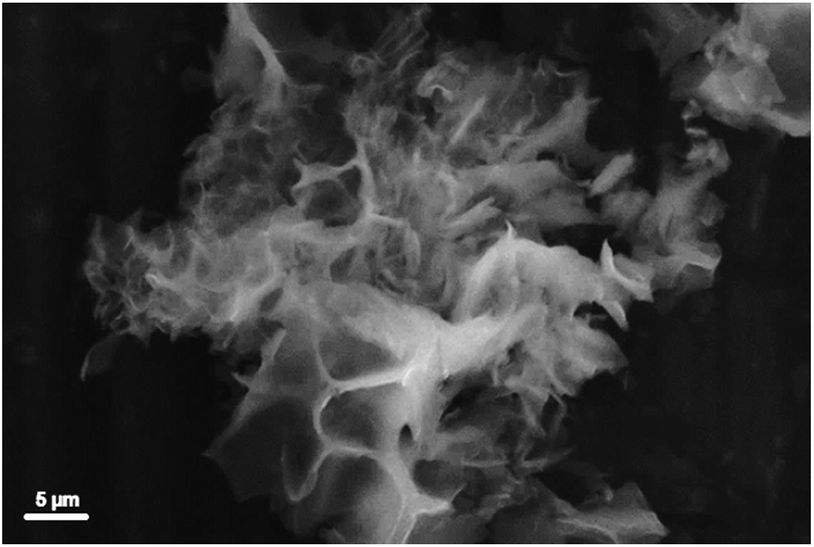 | ||
| Fig. 5 SEM picture of sample 1. | ||
Thermogravimetric analysis
The thermogravimetric analysis of the CB-PCP 1 to 5 samples are reported in Fig. S11.† All the thermogravimetric curves are similar despite the different anions, showing a drastic step of weight loss starting at around 400 °C related to the decomposition of the polymeric network. The decomposition step is not sharp, probably due to the irregular structure of the network, which broadens the thermal energy range at which the polymer chains break. The materials also exhibit one small weight-loss step starting at 110 °C, ascribable to the removal of residual moisture. It is worth noting that the thermal degradation of the CB-PCPs does not depend on the anion nature. This behavior highlights that the thermal degradation pathway is not the de-alkylation of the imidazolium nitrogen, as observed for dangling imidazolium PILs, but rather the decomposition of the whole polymeric network in one step.Carbon dioxide adsorption on CB-PCPs
The presence of the imidazolium ionic liquid moiety, well known to strongly interact with the CO2 molecule, together with a microporous network, makes CB-PCPs promising materials for CO2 capturing. For this reason, a series of volumetric adsorption measurements up to 1 bar and at different temperatures have been performed on the obtained CB-PCPs. Adsorption and desorption isotherms, recorded at 298 K for samples 1–6, are reported in Fig. 6. All samples exhibit the peculiar hysteresis observed also for the N2 adsorption at 77 K, in which the desorption branch of the isotherm is not close to the adsorption branch even at low relative pressures. The CO2 uptake at 298 K and 1 bar is between 1 and 1.2 mmol g−1 for all the materials. In particular, sample 1 shows the highest loading with a value of 1.2 mmol g−1, whereas sample 3 shows the lowest loading of 0.9 mmol g−1. The most pronounced hysteresis, observed in the case of sample 3, can be ascribable to the steric hindrance of the large, hydrophobic Tf2N− anion that limits the access of the CO2 to the imidazolium moiety. Anyhow, all samples show a complete release of CO2 at room temperature. It is worth noting that all the CO2 adsorption isotherms of these samples do not reach a plateau at 1 bar. This feature encourages further studies at higher pressures, extending the interest of these materials to the pre-combustion CO2 capture.51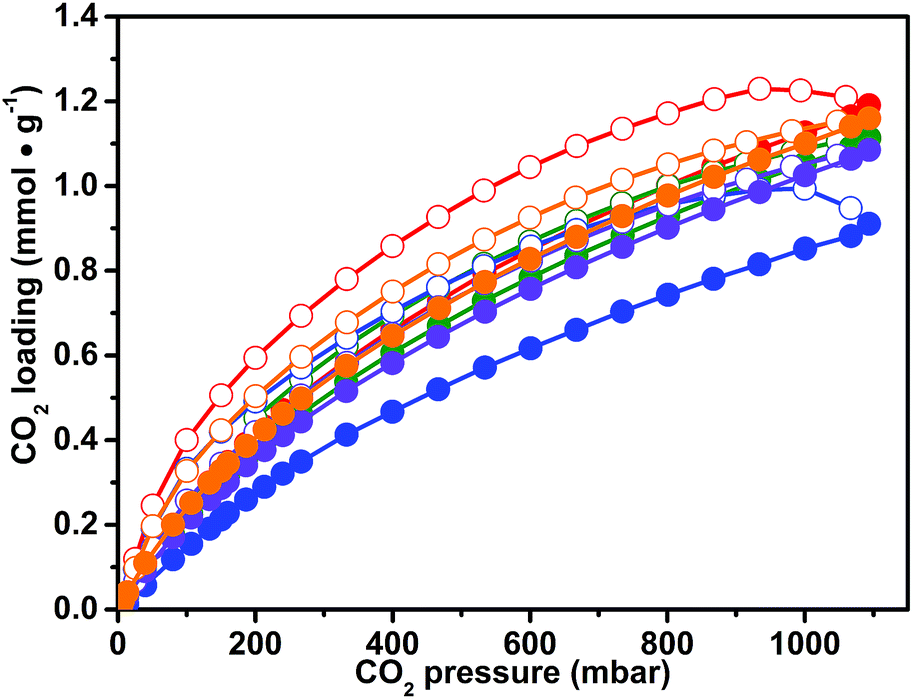 | ||
| Fig. 6 Carbon dioxide adsorption isotherms at 298 K for samples: 1 (red curve), 2 (blue curve), 3 (green curve), 4 (orange curve), and 5 (violet curve). The spheres describe the adsorption branch, while the circles describe the desorption branch. | ||
The CO2 uptake was also evaluated for sample 1 at 273 K and 313 K. The isotherms reported in Fig. S12† highlight only a slight decrease of the adsorption capacity at 313 K with a maximum loading of 0.95 mmol g−1; in contrast, at 273 K, the performance of the material drastically improves, almost doubling the maximum loading to a value of 2.05 mmol g−1. To the best of our knowledge, this value attests the herein described CB-PCPs as the best performing CO2 adsorbent materials in the field of imidazolium PILs. All the previous studies involving PILs for CO2 adsorption are listed in an extensive review from Zulfiqar et al., which encompass PILs with imidazolium, pyridinium or tetraalkylammonium cations and a variety of anions.48 According to this review, the best CO2 adsorption capacity at 1 bar and 273 K (0.46 mmol g−1) was measured in the case of cross-linked mesoporous imidazolium PILs obtained by a silica hard-templating pathway (P(SVImTf2N)).65 In the field of porous imidazolium polymers, it is remarkable to note that the work reported by Zhao et al. in which the cross-linking between the polymeric chains is obtained in a template-free synthesis via complexation between the anion and the cation both present along the main chain (P(CMVImBr1.03-co-AA)); in this case, the reported CO2 adsorption at 1 bar and 273 K was 0.64 mmol g−1.66 Other two studies are also noteworthy, reporting silica-supported tetraalkylammonium PILs (SiO2–P(VBTMA)(BF4)) and linear main-chain anionic PILs, having 1-butyl-3-methylimidazolium as the counterion (PUA-02), both reporting a maximum carbon dioxide adsorption of around 0.4 mmol g−1 at 1 bar and at 303 and 298 K, respectively.67,68 Another recent work, dealing with cross-linked di-vinylimidazolium PILs (PDMBr), reports a CO2 adsorption value of 1.02 mmol g−1 at 1 bar and 273 K,69 whereas the study by Talapaneni et al. dealing with the imidazolium porous polymer obtained by a two-step synthesis (NP-imidazolium) reports adsorption of 1.74 mmol g−1 of CO2 at 1 bar and 273 K.7 This material is very similar to our CB-PCPs having an equal SSA, but it has two more phenyl rings connecting every imidazolium moiety, slightly decreasing the ratio between the imidazolium functional group and the aryl chain and, as a consequence, slightly decreasing the CO2 loading.
In order to allow the comparison with the data reported in the literature, the isotherms of CO2 uptake on CB-PCPs have been reported in mmol of carbon dioxide adsorbed per gram of each material (see Fig. 6). However, to disclose information about CO2 adsorption from the molecular point of view, it is more relevant to take into consideration the mol% of carbon dioxide adsorbed with respect to the imidazolium moiety, as showed in Fig. S13.† From these isotherms, in fact, it is clear that the CO2 uptake depends on the nature of the anion of the CB-PCPs. A previous study reports higher CO2 adsorption for PILs with the acetate anion with respect to other anions,70 and furthermore, some speculations were reported about a possible generation of imidazolium NHC upon heating and outgassing acetate PILs.71,72 On the other hand, in the present case, CB-PCP 1 bearing the acetate anion shows the lowest performance towards CO2 adsorption, if expressed in mol%. Usually, inorganic anions such as PF6− or BF4−, perform better than Tf2N− and TfO− towards CO2 adsorption for PILs developed in a linear fashion.70,73 However, for cross-linked porous PILs, the Tf2N− anion shows the best performance.48,65 Generally speaking, it is difficult to discriminate among the effects of the anion nature, the SSA value and the polymer structure in carbon dioxide capture, due to the complexity of the adsorption process in which the chemical interaction, the diffusion kinetics inside the polymer network and the steric hindrance of the anion have to be considered. In agreement with the data already reported for porous PILs, the CO2 adsorption by CB-PCPs follows this trend: Tf2N− > PF6− ≈ TfO− > BF4− > AcO−.
In comparison to other materials for CO2 capture, our CB-PCPs exhibit better adsorption properties with respect to activated carbon, polycarbazole (PCBZ),74 the porous polymer network (PPN-80),75 hypercrosslinked polymer (HCP-1)76 and can compete with common porous aromatic framework (PAF-1).77 Nevertheless, CB-PCPs still show less CO2 uptake than the top performer materials like the zeolitic imidazolate framework (ZIF-78),78 zeolitic tetrazolate framework (ZTF-1),79 zeolite 13X (ZEO13X),80 and many metal-organic frameworks.81–83Table 2 summarizes the CO2 adsorption at 1 bar for poly(ionic liquid)s and other kinds of materials discussed in the text, in order to have a direct comparison of these sorbents with respect to CB-PCPs.
| Sorbent | CO2 mmol g−1 | Conditions (P, T) | Ref. |
|---|---|---|---|
| Metal-organic framework (Mg-MOF-74) | 8.02 | 1 bar, 298 K | 83 |
| Zeolitic tetrazolate framework (ZTF-1) | 5.59 | 1 bar, 273 K | 79 |
| Metal-organic framework (UTSA-16) | 4.90 | 1 bar, 273 K | 82 |
| Zeolite 13X (ZEO13X) | 4.68 | 1 bar, 298 K | 80 |
| Zeolitic imidazolate framework (ZIF-78) | 3.34 | 1 bar, 273 K | 78 |
| Click-based porous cationic polymer (CB-PCP-1) | 2.05 | 1 bar, 273 K | This work |
| Porous aromatic framework (PAF-1) | 2.05 | 1 bar, 273 K | 77 |
| Poly(ionic liquid) NP-imidazolium | 1.74 | 1 bar, 273 K | 7 |
| Hypercrosslinked polymer (HCP-1) | 1.70 | 1 bar, 298 K | 76 |
| Porous polymer network (PPN-80) | 1.62 | 1 bar, 295 K | 75 |
| Polycarbazole (PCBZ) | 1.13 | 1 bar, 273 K | 74 |
| Poly(ionic liquid) (PDMBr) | 1.02 | 1 bar, 273 K | 69 |
| Poly(ionic liquid) P(CMVImBr1.03-co-AA) | 0.64 | 1 bar, 273 K | 66 |
| Poly(ionic liquid) P(SVImTf2N) | 0.46 | 1 bar, 273 K | 65 |
| Silica-poly(ionic liquid) (SiO2–P(VBTMA) (BF4)) | 0.40 | 1 bar, 303 K | 67 |
| Poly(ionic liquid) (PUA-02) | 0.40 | 1 bar, 298 K | 68 |
Generally speaking, good performances towards CO2 adsorption in porous polymers are obtained from the combination of a high surface area, microporosity, and high concentration of imidazolium active sites along the polymeric backbone. Furthermore, the anion being an integral part of the adsorption properties of these materials, its choice is not straightforward, but depends on the chemical and morphological structure of the polymeric backbone. The weight ratio between the imidazolium moiety and the other parts of the polymer chemical structure is remarkable and this value is very high in our CB-PCPs. Furthermore, the direct conjugation of the imidazolium ring with two phenyl groups can change the distribution of its positive charge, and so affecting the way in which the imidazolium moiety interacts with CO2. It is worth noting that, even though the CB-PCPs have about one-tenth of the SSA of PAF-1 (5640 m2 g−1),15 they show very similar CO2 capture capacities, testifying the relevance of the introduction of highly dispersed and accessible functional groups. In order to evaluate the effect of the porosity of CB-PCPs on carbon dioxide loading, the CO2 adsorption at 298 K and 1 bar has also been performed on morphologically different samples 1a and 1b. The adsorption isotherms are reported in Fig. S14.† Samples 1 and 1a exhibit exactly the same CO2 loading of 1.2 mmol g−1 at 1 bar, moreover, sample 1a shows a less pronounced hysteresis, probably due to the higher uniformity of the material in terms of microporosity.
Sample 1b, with an almost null SSA (see Fig. S7† and Table 1), shows, however, a good adsorption capacity towards CO2, with a maximum loading of 1 mmol g−1. In fact, CO2 can act as a plasticizer for PILs, penetrate in part the bulk structure of non-porous polymers over a long diffusion time.84,85 For this reason, it is clear that the nature of the anions is more relevant than the SSA in determining the CO2 adsorption capacities of these materials.
Carbon dioxide capture in the presence of the carbene moiety
It is well-known that, by means of strong organic bases, it is possible to introduce a N-heterocyclic carbene in the C2 position of the imidazolium ring86–88 This carbene, in turn, can react with CO2 forming a new C–C bond and a carboxylate species directly linked to the imidazolium ring. The so-formed imidazolium carboxylate is thermally unstable and can decompose below 100 °C releasing CO2 and restoring the carbene, which can restart the adsorption/desorption cycle.89–91 The overall process is reported in Scheme 2. The chemical looping is considered as a new frontier in carbon dioxide adsorption.92 This mechanism was already studied for different ionic liquids and poly(ionic liquid)s, as a way for storing CO2 and also for protecting the carbene from decomposition in the presence of moisture.7,66,89,90,93–97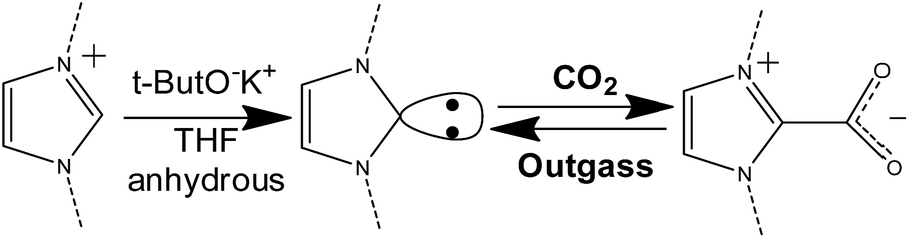 | ||
| Scheme 2 Synthetic step for the introduction of carbenes in the imidazolium ring (left side) and reversible formation of the NHC–CO2 adduct (right side). | ||
In our study, we direct the efforts to unravel the differences, from the physico-chemical point of view, between the CO2 capture by means of the as-synthesized CB-PCPs and the CB-PCPs bearing the NHC. A direct comparison between the CO2 adsorption capacity at 298 K of the CB-PCP bearing the NHC (sample 6) and of its precursor (sample 1) is reported in Fig. 7. The maximum CO2 loading of sample 6 is 1.1 mmol g−1, a perfectly comparable value with the uptake reported for sample 1. Conversely, from a molecular point of view, data reported in mol% of CO2 loading with respect to the imidazolium or the NHC moieties clearly show that the maximum loading drastically decreases after the introduction of the carbene (from 36 mol% for sample 1 to 26 mol% for sample 6). This behavior is ascribable to the lower number of available active sites. In fact, when NHC carbene moieties are formed, their reactivity favours the coupling. This phenomenon is also responsible for the evident decrease of the SSA of sample 6 (see Table 1). The dimers, without free electronic doublets, are not chemically able to bond CO2, thus decreasing the overall adsorption capacity with respect to the imidazolium moiety. However, the lightness of sample 6, arising from a lower molecular weight of the monomeric unit, compensates the loss of active sites, giving the same performance in terms of CO2 loading per mass unit of the adsorbent.
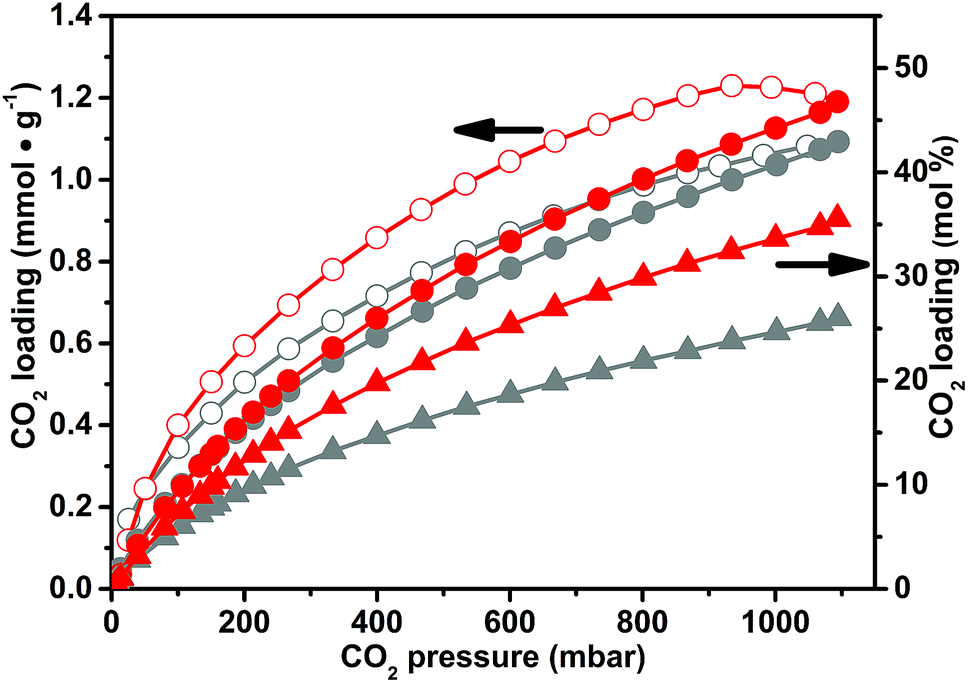 | ||
| Fig. 7 Carbon dioxide adsorption isotherms at 298 K for sample 1 and 6. Spheres and circles describe the adsorption and desorption branches respectively referring to the scale on the left (mmol g−1), while the triangles of the adsorption branch refer to the scale on the right (mol% of CO2 with respect to the imidazolium or NHC moieties). | ||
In situ FTIR spectroscopy was used to follow the reaction between the polymer bearing NHC and carbon dioxide. The effect of CO2 contact on the pre-activated sample 6, is illustrated in Fig. 8. The IR spectrum of sample 6 after activation (black curve) is characterized by the spectral features of the polymeric framework. Upon 1 h contact with 200 mbar of CO2, two new bands appear in the spectrum at 1665 cm−1 and at 1295 cm−1 (dark grey curve), ascribable to the νasym(OCO−) and the νsym(OCO−) of the formed imidazolium carboxylate.66,89,98,99 The formation of a carboxylate species testifies the activation of carbon dioxide, as proved by the deep change in its molecular orbitals.100 It is worth noting that, after the CO2 contact, the evacuation at beam temperature (around 50 °C) (light grey curve) almost restores the spectrum of the material after activation. The same experiment was repeated for sample 1 without any evidence of carboxylate formation upon exposure of the CB-PCP to 200 mbar of CO2.
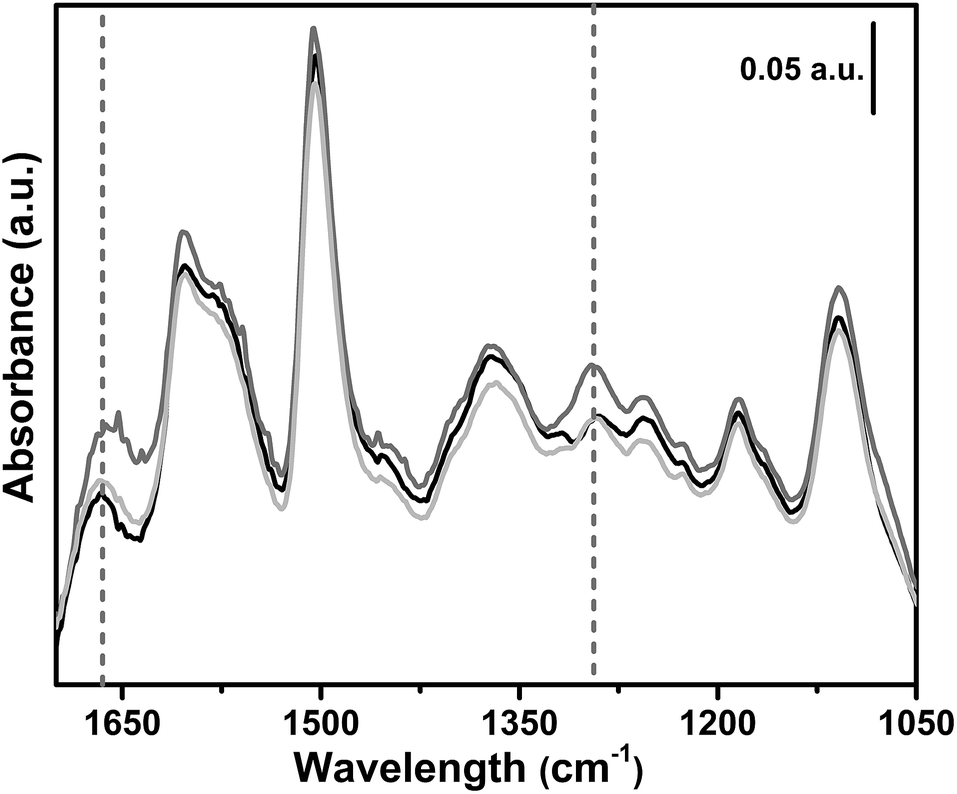 | ||
| Fig. 8 In situ FTIR spectra upon dosage of 200 mbar carbon dioxide on sample 6. Black curve: activated sample. Dark grey curve: 1 h contact with 200 mbar of CO2. Light grey curve: CO2 evacuation. | ||
Micro-calorimetric gas adsorption experiments were performed to further investigate the interaction energy between CO2, the ionic CB-PCP and its carbene counterpart. The differential molar adsorption heats of samples 1 and 6 are reported in Fig. 9 as a function of the carbon dioxide coverage, whereas their corresponding quantitative and calorimetric isotherms are shown in Fig. S15 and S16†, respectively. In the case of sample 1, the differential heat of adsorption at zero coverage is 35 kJ mol−1, then it decreases to 10 kJ mol−1 for higher CO2 coverages, i.e. significantly under the value of CO2 molar liquefaction enthalpy. This low isosteric heat could be explained considering two underlying contributions: the gas adsorption and the structural rearrangement as a consequence of the polymer swelling process.66 Conversely, for sample 6 the differential heat of adsorption is definitely higher, starting from 58 kJ mol−1 at low coverage and decreasing to about 35–40 kJ mol−1 at high coverage. The higher differential heat of interaction of sample 6 is perfectly in line with the formation of the adduct between the NHC and CO2. Still, the comparison between the primary (spheres) and secondary (circles) runs indicates that the CO2 uptake is mostly reversible for sample 6 also.
 | ||
| Fig. 9 Differential molar adsorption heats as a function of the coverage relative to the adsorption at 298 K of CO2 on samples 1 (red curves) and 6 (grey curves). Spheres refer to the primary adsorption and circles to the secondary ones. The dotted horizontal line represents the standard molar enthalpy of liquefaction of CO2 at 298 K. | ||
Conclusions
We described a facile and straightforward synthetic way to obtain click reaction-based micro-/mesoporous cationic polymers. A set of materials with different anions was successfully synthesized and characterized in order to confirm their structure and to study their porosity. It was demonstrated that CB-PCPs exhibit excellent behavior towards carbon dioxide adsorption, either at 298 K and 1 bar, with a CO2 loading above 1 mmol g−1 for all the samples, or even more at 273 K and 1 bar where the loading for CB-PCP 1 is 2 mmol g−1, achieving the highest value for the porous ionic polymer. CB-PCP 1 was also modified in order to introduce the NHC on the imidazolium ring. The performances towards carbon dioxide adsorption of the NHC CB-PCP are of the same level as those of the ionic CB-PCPs, even though they show a different adsorption mechanism. The combination of in situ FTIR spectroscopy and of adsorption micro-calorimetry reveals two adsorption processes of completely different nature: on one hand, a plain physisorption process in the ionic CB-PCPs, and on the other hand, the chemisorption of carbon dioxide in the NHC polymer.Funding sources
The Italian Ministry of Education, University and Research (MIUR). Project PRIN 2010–2011 no. 2010A2FSS9.ERC (European Research Council) Starting Grant. Project number 639720 – NAPOLI.
Acknowledgements
Thanks are due to Prof. Leonardo Marchese and Geo Paul (Università del Piemonte Orientale “A. Avogadro”) for SS-NMR measurements and the useful discussions.References
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17–26 RSC.
- J.-K. Sun, M. Antonietti and J. Yuan, Chem. Soc. Rev., 2016, 45, 6627–6656 RSC.
- H. Ren, T. Ben, E. Wang, X. Jing, M. Xue, B. Liu, Y. Cui, S. Qiu and G. Zhu, Chem. Commun., 2010, 46, 291–293 RSC.
- S. N. Talapaneni, O. Buyukcakir, S. H. Je, S. Srinivasan, Y. Seo, K. Polychronopoulou and A. Coskun, Chem. Mater., 2015, 27, 6818–6826 CrossRef CAS.
- Y. Yuan, F. Sun, H. Ren, X. Jing, W. Wang, H. Ma, H. Zhao and G. Zhu, J. Mater. Chem., 2011, 21, 13498–13502 RSC.
- H. Zhao, Z. Jin, H. Su, X. Jing, F. Sun and G. Zhu, Chem. Commun., 2011, 47, 6389–6391 RSC.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- T. Ben, K. Shi, Y. Cui, C. Pei, Y. Zuo, H. Guo, D. Zhang, J. Xu, F. Deng, Z. Tian and S. Qiu, J. Mater. Chem., 2011, 21, 18208–18214 RSC.
- M. Rose, A. Notzon, M. Heitbaum, G. Nickerl, S. Paasch, E. Brunner, F. Glorius and S. Kaskel, Chem. Commun., 2011, 47, 4814–4816 RSC.
- H. C. Cho, H. S. Lee, J. Chun, S. M. Lee, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 917–919 RSC.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., 2009, 121, 9621–9624 CrossRef.
- O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934–937 RSC.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- T. Muller and S. Brase, RSC Adv., 2014, 4, 6886–6907 RSC.
- T. Muller and S. Bräse, Angew. Chem., Int. Ed., 2011, 50, 11844–11845 CrossRef CAS PubMed.
- P. Rekha, U. Sahoo and P. Mohanty, RSC Adv., 2014, 4, 34860–34863 RSC.
- J. R. Holst, E. Stöckel, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8531–8538 CrossRef CAS.
- P. Pandey, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Mater. Chem., 2011, 21, 1700–1703 RSC.
- O. Plietzsch, C. I. Schilling, T. Grab, S. L. Grage, A. S. Ulrich, A. Comotti, P. Sozzani, T. Muller and S. Brase, New J. Chem., 2011, 35, 1577–1581 RSC.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- S. N. Riduan and Y. Zhang, Chem. Soc. Rev., 2013, 42, 9055–9070 RSC.
- A. S. Shaplov, D. O. Ponkratov and Y. S. Vygodskii, Polym. Sci., Ser. B, 2016, 58, 73–142 CrossRef CAS.
- A. Dani, E. Groppo, C. Barolo, J. G. Vitillo and S. Bordiga, J. Mater. Chem. A, 2015, 3, 8508–8518 CAS.
- J. Cui, W. Zhu, N. Gao, J. Li, H. Yang, Y. Jiang, P. Seidel, B. J. Ravoo and G. Li, Angew. Chem., Int. Ed., 2014, 53, 3844–3848 CrossRef CAS PubMed.
- Q. Zhao, P. Zhang, M. Antonietti and J. Yuan, J. Am. Chem. Soc., 2012, 134, 11852–11855 CrossRef CAS PubMed.
- F. Yan and J. Texter, Angew. Chem., Int. Ed., 2007, 46, 2440–2443 CrossRef CAS PubMed.
- H. Zhao, Y. Wang and R. Wang, Chem. Commun., 2014, 50, 10871–10874 RSC.
- K. Thiel, R. Zehbe, J. Roeser, P. Strauch, S. Enthaler and A. Thomas, Polym. Chem., 2013, 4, 1848–1856 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- O. Mahmoodi Nosrat, I. Nikokar, M. Farhadi and A. Ghavidast, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 715 Search PubMed.
- H. Debus, Justus Liebigs Ann. Chem., 1858, 107, 199–208 CrossRef.
- B. Radziszewski, Ber. Dtsch. Chem. Ges., 1882, 15, 1493–1496 CrossRef.
- A. J. Arduengo, T. K. Prakasha, F. P. Gentry and H. E. Simmons, WO2002040454 A1, 2002.
- D. Esposito, S. Kirchhecker and M. Antonietti, Chem.–Eur. J., 2013, 19, 15097–15100 CrossRef CAS PubMed.
- J.-P. Lindner, Macromolecules, 2016, 49, 2046–2053 CrossRef CAS.
- K.-S. Krannig, D. Esposito and M. Antonietti, Macromolecules, 2014, 47, 2350–2353 CrossRef CAS.
- A. R. Hajipour and Z. Khorsandi, Catal. Commun., 2016, 77, 1–4 CrossRef CAS.
- W. Yu, Z. Wen-Li, C. Hao, Z. Li-Zhuo, Z. Qiu-Ju, Z. Huan, W. Yuan-Jiang, G. Wen-Hui, F. Yue, Z. Sheng-Liang and W. Hui, Asian J. Chem., 2015, 27, 3107–3110 CrossRef.
- M. Lee, U. H. Choi, D. Salas-de la Cruz, A. Mittal, K. I. Winey, R. H. Colby and H. W. Gibson, Adv. Funct. Mater., 2011, 21, 708–717 CrossRef CAS.
- N. Matsumi, K. Sugai, M. Miyake and H. Ohno, Macromolecules, 2006, 39, 6924–6927 CrossRef CAS.
- S. Zulfiqar, M. I. Sarwar and D. Mecerreyes, Polym. Chem., 2015, 6, 6435–6451 RSC.
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- Y. Zhou, J. Liu, M. Xiao, Y. Meng and L. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 5547–5555 CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Polym. Int., 2013, 62, 345–352 CrossRef CAS.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, San Diego, 3rd edn, 1990 Search PubMed.
- R. L. Frost and J. T. Kloprogge, J. Mol. Struct., 2000, 526, 131–141 CrossRef CAS.
- L. Yu, J. Clifford, T. T. Pham, E. Almaraz, F. Perry, G. A. Caputo and T. D. Vaden, J. Phys. Chem. B, 2013, 117, 7057–7064 CrossRef CAS PubMed.
- R. Holomb, A. Martinelli, I. Albinsson, J. C. Lassègues, P. Johansson and P. Jacobsson, J. Raman Spectrosc., 2008, 39, 793–805 CrossRef CAS.
- H. Zine, M. H. Baron and A. Piart-Goypiron, Spectrochim. Acta, Part A, 1995, 51, 457–470 CrossRef.
- N. N. Sa'adun, R. Subramaniam and R. Kasi, Sci. World J., 2014, 2014, 7 Search PubMed.
- Y. Kaneko, M. Shoiriki and T. Mizumo, J. Mater. Chem., 2012, 22, 14475–14478 RSC.
- M. L. Pinto, L. Mafra, J. M. Guil, J. Pires and J. Rocha, Chem. Mater., 2011, 23, 1387–1395 CrossRef CAS.
- M. M. Cecchini, J. Steinkoenig, S. Reale, L. Barner, J. Yuan, A. S. Goldmann, F. De Angelis and C. Barner-Kowollik, Chem. Sci., 2016, 7, 4912–4921 RSC.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880–2885 CrossRef CAS.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed.
- F. E. Hahn, L. Wittenbecher, D. Le Van and R. Fröhlich, Angew. Chem., Int. Ed., 2000, 39, 541–544 CrossRef CAS.
- A. Wilke, J. Yuan, M. Antonietti and J. Weber, ACS Macro Lett., 2012, 1, 1028–1031 CrossRef CAS.
- S. Soll, Q. Zhao, J. Weber and J. Yuan, Chem. Mater., 2013, 25, 3003–3010 CrossRef CAS.
- H. Cheng, P. Wang, J. Luo, J. Fransaer, D. E. De Vos and Z.-H. Luo, Ind. Eng. Chem. Res., 2015, 54, 3107–3115 CrossRef CAS.
- T. O. Magalhaes, A. S. Aquino, F. D. Vecchia, F. L. Bernard, M. Seferin, S. C. Menezes, R. Ligabue and S. Einloft, RSC Adv., 2014, 4, 18164–18170 RSC.
- X. Wang, Y. Zhou, Z. Guo, G. Chen, J. Li, Y. Shi, Y. Liu and J. Wang, Chem. Sci., 2015, 6, 6916–6924 RSC.
- E. I. Privalova, E. Karjalainen, M. Nurmi, P. Mäki-Arvela, K. Eränen, H. Tenhu, D. Y. Murzin and J.-P. Mikkola, ChemSusChem, 2013, 6, 1500–1509 CrossRef CAS PubMed.
- G. Gurau, H. Rodríguez, S. P. Kelley, P. Janiczek, R. S. Kalb and R. D. Rogers, Angew. Chem., Int. Ed., 2011, 50, 12024–12026 CrossRef CAS PubMed.
- J. Blath, N. Deubler, T. Hirth and T. Schiestel, Chem. Eng. J., 2012, 181–182, 152–158 CrossRef CAS.
- J. Tang, H. Tang, W. Sun, M. Radosz and Y. Shen, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5477–5489 CrossRef CAS.
- M. Saleh, S. B. Baek, H. M. Lee and K. S. Kim, J. Phys. Chem. C, 2015, 119, 5395–5402 CAS.
- L.-B. Sun, A.-G. Li, X.-D. Liu, X.-Q. Liu, D. Feng, W. Lu, D. Yuan and H.-C. Zhou, J. Mater. Chem. A, 2015, 3, 3252–3256 CAS.
- C. F. Martin, E. Stockel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475–5483 RSC.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 CAS.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed.
- T. Panda, P. Pachfule, Y. Chen, J. Jiang and R. Banerjee, Chem. Commun., 2011, 47, 2011–2013 RSC.
- D. P. Bezerra, R. S. Oliveira, R. S. Vieira, C. L. Cavalcante and D. C. S. Azevedo, Adsorption, 2011, 17, 235–246 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- A. Masala, F. Grifasi, C. Atzori, J. G. Vitillo, L. Mino, F. Bonino, M. R. Chierotti and S. Bordiga, J. Phys. Chem. C, 2016, 120, 12068–12074 CAS.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 CAS.
- J. Tang, W. Sun, H. Tang, M. Radosz and Y. Shen, Macromolecules, 2005, 38, 2037–2039 CrossRef CAS.
- J. Tang, H. Tang, W. Sun, H. Plancher, M. Radosz and Y. Shen, Chem. Commun., 2005, 3325–3327, 10.1039/B501940K.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- M. Fevre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS PubMed.
- H. Zhou, W.-Z. Zhang, Y.-M. Wang, J.-P. Qu and X.-B. Lu, Macromolecules, 2009, 42, 5419–5421 CrossRef CAS.
- L. Yang and H. Wang, ChemSusChem, 2014, 7, 962–998 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- M. Fèvre, P. Coupillaud, K. Miqueu, J.-M. Sotiropoulos, J. Vignolle and D. Taton, J. Org. Chem., 2012, 77, 10135–10144 CrossRef PubMed.
- J. Pinaud, J. Vignolle, Y. Gnanou and D. Taton, Macromolecules, 2011, 44, 1900–1908 CrossRef CAS.
- G. W. Nyce, J. A. Lamboy, E. F. Connor, R. M. Waymouth and J. L. Hedrick, Org. Lett., 2002, 4, 3587–3590 CrossRef CAS PubMed.
- M. Hans, L. Delaude, J. Rodriguez and Y. Coquerel, J. Org. Chem., 2014, 79, 2758–2764 CrossRef CAS PubMed.
- R. Lambert, P. Coupillaud, A.-L. Wirotius, J. Vignolle and D. Taton, Macromol. Rapid Commun., 2016, 37, 1143–1149 CrossRef CAS PubMed.
- J. Oomens and J. D. Steill, J. Phys. Chem. A, 2008, 112, 3281–3283 CrossRef CAS PubMed.
- P. M. MacQueen, R. A. Bach, C. T. P. MacLean and S. L. MacQuarrie, J. Phys. Chem. C, 2014, 118, 5239–5242 CAS.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Material syntheses, description of the techniques, HR-MS spectra and MSn fragmentation, additional nitrogen and carbon dioxide adsorption isotherms, thermogravimetric analyses data, SEM images and micro-calorimetry data. See DOI: 10.1039/c6ta08574a |
| This journal is © The Royal Society of Chemistry 2017 |