
A novel method to decorate Au clusters onto graphene via a mild co-reduction process for ultrahigh catalytic activity†
Zhongqian
Song
ac,
Weiyan
Li
c,
Fushuang
Niu
b,
Yuanhong
Xu
b,
Li
Niu
ac,
Wenrong
Yang
ad,
Yao
Wang
b and
Jingquan
Liu
*ab
aShandong Provincial Key Laboratory of Detection Technology for Tumor Markers, College of Chemistry and Chemical Engineering, Linyi University, Linyi 276005, China
bCollege of Materials Science and Engineering Laboratory of Fiber Materials and Modern Textile, The Growing Base for State Key Laboratory, Qingdao University, Qingdao 266071, China. E-mail: jliu@qdu.edu.cn; Tel: +86 532 83780128
cEngineering Laboratory for Modern Analytical Techniques, c/o State Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, Jilin, China
dSchool of Life and Environmental Sciences, Deakin University, Geelong, VIC 3217, Australia
First published on 21st November 2016
Abstract
To achieve high catalytic activity and stability with low noble-metal loadings on special supports has triggered much research interest in the past few years. Herein, a mild co-reduction strategy was exploited to fabricate glutathione decorated Au clusters (with a size of ∼1.4 nm) on reduced graphene oxide (Au@HSG-rGO) with low Au loadings and high catalytic activity in an aqueous medium. The resultant Au@HSG-rGO complex exhibited 20.8 times higher catalytic activity than Au nanoparticle supported graphene for catalysis of the reduction of 4-nitrophenol (4-NP). The Au@HSG-rGO was packed in a filtering platform to afford a fixed-bed system, with which the catalytic conversion reached 96.03% for 0.2 mM 4-NP solution at a flow rate of 1 mL min−1. In addition, the poly(2-(dimethylamino) ethyl acrylate) modified Au@HSG-rGO (Au@HSG-rGO-PDMAEA) via π–π stacking interactions exhibited good recyclability and tunable catalytic activity and only showed slight loss of activity after recycling five times. The PDMAEA served as forest-like shelters to efficiently protect the Au@HSG clusters from aggregation and also endowed the system with enhanced stability and temperature-controlled catalytic activity. Meanwhile, the Au@HSG-rGO showed excellent electrocatalytic activity for the oxygen reduction reaction in alkaline electrolytes. This simple, economical and mild strategy could be generalized to the preparation of other metal cluster complexes for broad catalytic and analytical applications.
1. Introduction
Owing to its highly effective catalytic performance under mild reaction conditions, Au has attracted wide attention in many catalytic reactions such as hydrogenation of aldehydes, oxidation of alcohols and aldehydes, carbon–carbon bond formation and reduction of 4-nitrophenol.3–7 Dispersing catalysts with tens of metal atoms, mononuclear metal complexes or even single metal atoms on supports has become a burgeoning theme in the scientific community for obtaining cost-effective catalysts with maximum atom efficiency.8–14 However, during the catalytic reaction, the small-sized catalysts tend to aggregate to larger particles due to their high surface area and surface energy, which could easily result in the deactivation or loss of their catalytic activity.16 To address these inherent shortcomings, significant efforts should be devoted to developing feasible routes to anchor uniform and highly active Au catalysts on special supports.Some defective oxides (e.g., TiO2 and CeO2),18,19 graphene21,22,55 and C3N423 have been chosen as efficient supports to stabilize noble metal catalysts. Among all these available supports, graphene with a unique two dimensional structure of sp2 hybridized carbon has proved to be one of the most promising platforms owing to its high surface area, outstanding electronic mobility and good stability.24 The metal cluster catalyst anchored on graphene endows the system with excellent catalytic activity because of the enhanced charge transfer from the clusters to the graphene due to their synergistic effect.24 Meanwhile, the stability of the catalytic system could also be significantly improved via the interaction between the metal clusters and the defects of graphene.25 Therefore, it is expected that an efficient route could be designed to prepare a stable catalytic system by controlling the nucleation and growth processes on the surface of graphene.
Catalysts for the oxygen reduction reaction (ORR) are the basis for the further development of fuel cell technologies. Among the platinum-group metals, Au-based catalysts for the ORR have not attracted enough attention in electrocatalysis due to their poor catalytic performance. Nevertheless, the properties of materials could show a dramatic variation when the dimension of the Au catalyst is reduced to the nanoscale.26 In this respect, nanosized Au-based catalysts with diameters smaller than 10 nm have been actively pursued for CO oxidation27 and oxygen reduction.28,29 However, these catalysts frequently suffer from compact assembly and high Au loading for limited ORR catalytic activity, which can result in reduced efficiency. To overcome this obstacle, smaller sized nanostructured catalysts supported on active carbon, carbon nanotubes or graphene deserve more exploration to maximize their electroactive surface area and improve their catalytic activity. In addition, a high fraction of surface atoms with low coordination numbers can be tuned via manipulating the dimensions of the nanoparticles to obtain unusual catalytic activity.30,31 These studies have evaluated the relationships between the catalytic activity of Au-based catalysts and their size larger than 2 nm in diameter. More recently, Herzing et al.32 discovered that Au clusters containing only about 10 gold atoms (ca. 0.5 nm in diameter) exhibited high catalytic activity for CO oxidation, which suggests that sub-nanometer-sized Au clusters could envision more extensive catalytic applications in more versatile areas.
Herein, we reported a co-reduction strategy to fabricate glutathione protected Au clusters (Au@HSG) on reduced graphene oxide (rGO) in an aqueous medium, followed by the attachment of poly(2-(dimethylamino) ethyl acrylate) (PDMAEA) via π–π stacking interactions to obtain a temperature-tunable catalytically active system. The PDMAEA served as forest-like shelters to efficiently protect the Au@HSG clusters from aggregation and also endowed the system with temperature-controlled catalytic activity. The as-synthesized Au@HSG-rGO exhibited 20.8 times higher catalytic activity than Au nanoparticle supported graphene for selective reduction of 4-nitrophenol. The mild co-reduction strategy does not require rigorous conditions or toxic agents, and could be developed into an efficient and green approach for the fabrication of low noble-metal loading catalysts with high catalytic activity.
2. Experimental section
2.1 Materials
Glutathione (AR) was purchased from Shanghai Yuanye. 4-Nitrophenol (AR) was purchased from Tianjin Guangfu. HAuCl4 (AR) was purchased from Sigma-Aldrich. Natural graphite powders with an average size of 3 mm were supplied by Xiamen Knano Graphite Technology Corporation. Sulfuric acid (98%, AR), phosphorus(V) oxide (98%, AR), potassium permanganate (AR), and hydrogen peroxide (30%, AR) were purchased from Shanghai Jinlu Chemical. Ultrapure water (18.2 MΩ cm−1) was obtained with a Milli-Q ultrapure system (Qingdao, China).2.2 Preparation of Au@HSG-rGO
Graphene oxide was synthesized by a modified Hummers' method according to our previous work.33 2 mL of purified graphene oxide aqueous solution (6 mg mL−1) was mixed with a solution containing 7 mL deionized water and 50 mg glutathione. After sonication for 10 min, 1 mL of HAuCl4 aqueous solution (2 wt%) was added to this mixture in an ice-water bath. The resulting mixture was then continually sonicated for 20 min. The reaction mixture was transferred to a glass bottle and treated at 90 °C for 12 h. The as-synthesized product was collected by centrifugation and washed with deionized water three times. Finally, the Au@HSG-rGO aqueous suspension was obtained and stored at 4 °C for subsequent use.2.3 Synthesis of pyrene-terminated PDMAEA
Firstly, a pyrene-terminated RAFT agent was synthesized via our previously reported method.34 The pyrene-terminated RAFT agent (30 mg, 0.067 mmol), DMAEA (962.7 mg, 6.7 mmol) and AIBN (3.9 mg, 0.024 mmol) were dissolved in 5 mL dioxane to obtain a homogeneous solution. The resulting solution was sealed with rubber septa and deoxygenated with nitrogen for 30 min, and then incubated in a preheated oil bath at 75 °C. After 15 h, the solution was evaporated under vacuum to remove the solvent. The obtained polymer was precipitated in hexane three times to remove the unreacted monomer, yielding a viscous light yellow product and then dried under vacuum. 1H NMR (600 MHz, CDCl3, δ): 3.88–4.17 (m, OCH2CH2 of DMAEA), 2.06–2.49 (m, N(CH3)2 of DMAEA), 7.64–8.21 (m, 9H, CH of pyrene).2.4 Functionalization of Au@HSG-rGO with pyrene-terminated PDMAEA
The Au@HSG-rGO can be functionalized with the pyrene-terminated PDMAEA polymer via π–π stacking interactions. Typically, a 20 mL aqueous suspension of Au@HSG-rGO (20 mg) was mixed with the pyrene-terminated PDMAEA polymer (120 mg) solution and the resulting mixture was sonicated at room temperature for 30 min to give a homogeneous suspension. The suspension was then centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min to remove the excess tri-block copolymer and the process was repeated three times to afford the PDMAEA functionalized Au@HSG-rGO (Au@HSG-rGO-PDMAEA).
000 rpm for 15 min to remove the excess tri-block copolymer and the process was repeated three times to afford the PDMAEA functionalized Au@HSG-rGO (Au@HSG-rGO-PDMAEA).
2.5 Catalytic activity testing
The catalytic activity of the Au@HSG-rGO for the reduction of 4-NP was tested as follows. Typically, 1 mL of fresh NaBH4 aqueous solution (0.02 M) and 2 mL of 4-NP aqueous solution (0.2 mM) were homogenized in a quartzose vessel with shaking. Then, 25 μL of Au@HSG-rGO aqueous suspension (10 mg mL−1) was added into the quartzose vessel. UV-vis absorption spectra were recorded to monitor the changes during the catalytic reaction in the wavelength range of 250–550 nm. The bright yellow solution faded gradually to colorless during the catalytic reaction. The catalytic activity of the Au@HSG-rGO-PDMAEA for the reduction of 4-NP was tested using the same procedure. The recycling performance of Au@HSG-rGO and Au@HSG-rGO-PDMAEA was tested by separating the catalysts via centrifugation at 12000 rpm for 15 min and then they were directly reused under the same catalytic reaction conditions.
3. Results and discussion
The procedures for the fabrication of Au@HSG-rGO catalysts are schematically illustrated in Fig. 1. Homogenous GO aqueous solution was prepared by sonication and dialyzed in water for three days to remove impurities. Au@HSG-rGO was first synthesized via a thermal co-reduction method from HAuCl4 and GO suspension in the presence of GSH. GO was reduced into rGO and simultaneously Au@HSG clusters were formed and anchored onto the rGO surface. As demonstrated in Fig. 1, the dark brown mixture changed gradually into a dark suspension, which was collected by centrifugation and washed with pure water three times. Finally, dark Au@HSG-rGO powders were obtained by the freeze-drying process.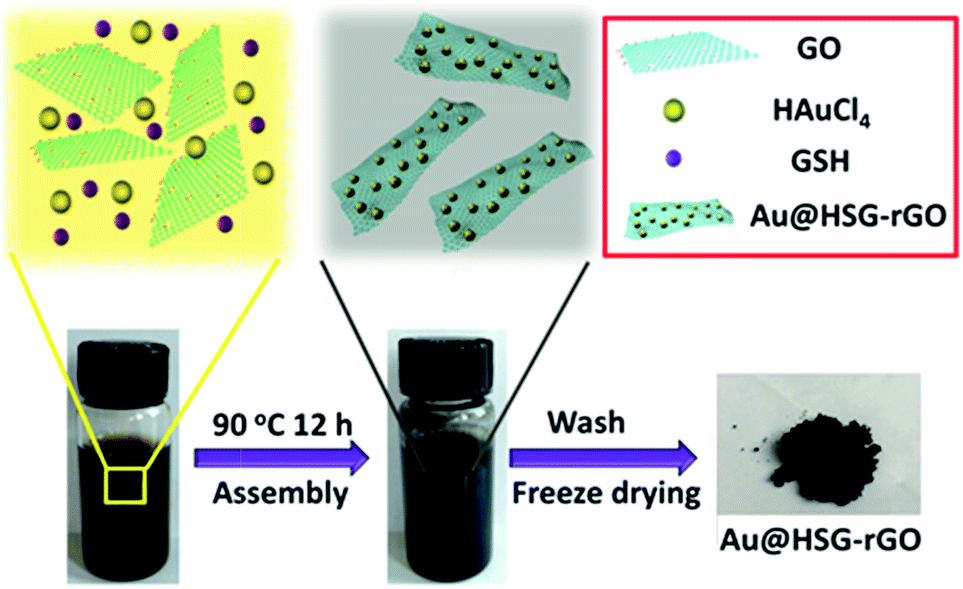 | ||
| Fig. 1 Schematic of the stepwise formation process of Au@HSG-rGO. | ||
The possible mechanism for the formation of Au@HSG-rGO was also proposed. Firstly, GO contains plenty of oxygen functional groups, such as –COOH, –OH and epoxy groups, which can facilitate the adsorption of metal ions onto the surface of GO via physisorption, electrostatic binding, or charge-transfer interactions, and then act as nucleation centres for metal anchoring.35,36 Secondly, GSH can facilitate the formation of Au@HSG clusters and reduction of GO into rGO via a co-reduction process. When GO is reduced into rGO by GSH, AuCl4− anions anchored on rGO are reduced to Au clusters, which are then protected by GSH. The reduction process is proposed as follows: GSH can generate charged particles, such as electrons and protons, which can react with oxygen-functional groups to transform GO into rGO. Since metal atoms tend to grow into three-dimensional structures on graphene surfaces, more interaction states and transmission channels could be generated between metal and graphene for metal binding to graphene37,38 Compared with the Au/graphene composites prepared via a hydrothermal process at high temperature,2 or reduced by tannic acid,39 ascorbic acid40 or trisodium citrate,41 the as-prepared Au@HSG-rGO clusters possess a smaller size and more uniform dispersity, which endow the metal catalyst with superiority. Hence, this mild method could undoubtedly be a new and effective technique for the preparation of metal/graphene composites with a small and well distributed size and excellent catalytic activity.
The representative transmission electron microscopy (TEM) images of Au@HSG-rGO are shown in Fig. 2a–d. The TEM images in Fig. 2a and b clearly show that Au@HSG clusters are homogenously dispersed on the surface of rGO sheets. It can be observed that the graphene sheets retain a lot of wrinkles, which can prevent re-stacking of graphene during the drying process and thus provide a high surface area.42 As shown in Fig. 2c and the inset, the high-resolution TEM (HRTEM) clearly displays uniform Au@HSG clusters with a narrow size distribution and an average size of 1.4 nm. The HRTEM image (Fig. 2d) shows well resolved lattice planes with average spacings of 0.235 nm and 0.216 nm, which correspond to the (111) and (200) lattice spacing of the Au clusters.43 The chemical composition of the as-synthesized Au@HSG-rGO was studied by energy dispersive X-ray spectrum (EDX, Fig. 2e). The appearance of C, N, O, S and Au elements from the EDX of Au@HSG-rGO indirectly indicates that the obtained catalysts are composed of the Au@HSG nanoclusters.
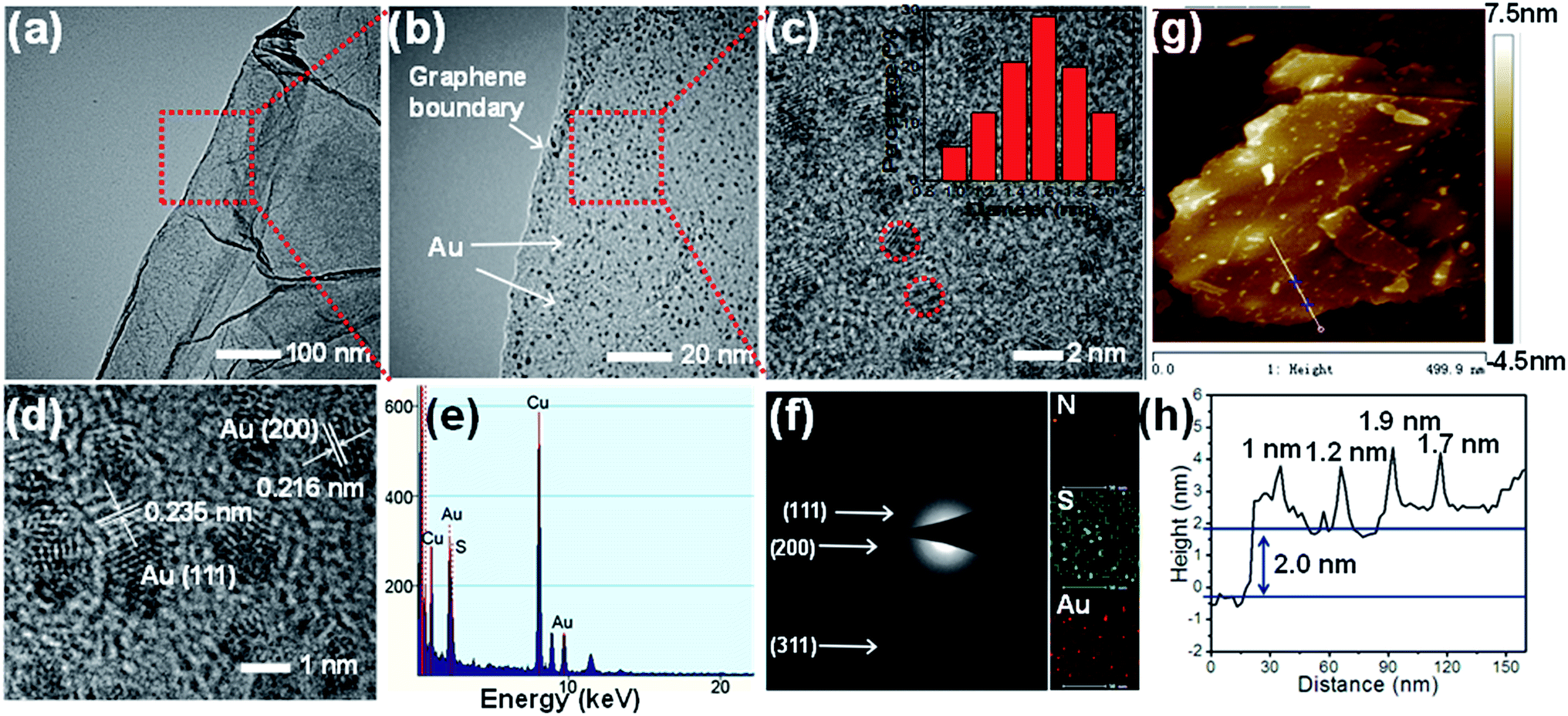 | ||
| Fig. 2 (a–d) HRTEM images of Au@HSG-rGO with different magnifications; the inset in (c) represents a size distribution diagram of Au@HSG clusters; (e) the EDX spectrum of Au@HSG-rGO; (f) SAED pattern of Au@HSG-rGO and the elemental mappings of N, S and Au; (g) AFM image and (h) height profile along the line in (g) of Au@HSG-rGO. | ||
The selected area electron diffraction (SAED) pattern (Fig. 2f) displays three d-spacings at 2.4, 2.1 and 1.3 Å, which should correspond to the (111), (200), and (331) Miller indices of fcc (face-centered cubic) gold crystals, respectively.44 The diffuse SAED pattern is due to the small diameter of the Au@HSG-rGO cluster. The EDX technique was also adopted to visualize the spatial distribution of the elements (inset of Fig. 2f). It can be seen that all elements (N, S and Au) are uniformly distributed on the whole surface of rGO. The UV-vis absorption spectra of GO, rGO and Au@HSG-rGO are shown in Fig. S1.† It can be seen that its maximum peak at 231 nm redshifted to 262 nm due to the restoration of electrons after the reduction of GO by GSH. No obvious peaks at around 520 nm for surface plasma resonance absorption were observed, indicating the small size of Au nanoclusters on rGO (Fig. S1†).
It can be observed from the SEM images (Fig. 3a and b) that the Au@HSG-rGO composite has a hierarchical hollow structure with a hollow size of several hundred nanometres. The wrinkles as indicated by the white arrow on the surface of Au@HSG-rGO should contribute to the high specific area, which can guarantee the high contact area and catalytic activity during the catalytic reaction. Owing to the small size of Au@HSG clusters, it is difficult to identify clusters located on the surface of rGO in the SEM images. To confirm the existence of Au@HSG clusters, the EDX technique was used to analyze the Au@HSG-rGO catalyst and the results shown in Fig. 3c evidence the existence of the N, S, and Au elements and a Au loading of 0.23%. As shown in the XRD spectra (Fig. 3d), no significant diffraction peaks indicative of the Au phase were observed due to the uniform dispersion, low content and extremely tiny Au cluster size.45
 | ||
| Fig. 3 (a and b) SEM images and (c) EDX spectrum of Au@HSG-rGO; (d) XRD pattern of GO, rGO, and Au@HSG-rGO. | ||
The Au@HSG-rGO was also investigated by the XPS technique, and the results are shown in Fig. 4a–e. The typical XPS spectra revealed the existence of C, N, O, S and Au elements (Fig. 4a), which confirmed that the graphene was decorated with Au@HSG nanoclusters. The C 1s spectrum in Fig. 4b exhibits five peaks of C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.8 eV), CN/C–S (285.5 eV), C–O (286.6 eV) and CO (288.5 eV), which confirm the successful decoration of Au@HSG nanoclusters. In the N 1s spectrum (Fig. 4c), the peaks located at 400.2 eV and 401.9 eV are assigned to C–N and N–H functionalities in GSH, respectively. The S 2p spectra of the Au@HSG-rGO can be deconvoluted into three peaks (Fig. 3d), which correspond to S–S/S–C bonds at 163.8 eV, S–H at 164.9 eV and sulfate of –SO3H (oxidized form –SH) at 169.3 eV. For the Au 4f spectra of free Au nanoparticles decorated on graphene (Fig. 4e, blue line), there are two distinct doublets: the Au 4f7/2 peak at 83.3 eV and the Au 4f5/2 peak at 86.8 eV. However, with regard to the Au@HSG-rGO, the Au 4f7/2 and Au 4f5/2 peaks at 84.4 and 88.1 eV are consistent with the Au(0) state,54 which has a 1.1 eV and 1.3 eV red shift compared with that of bulk Au. This shift in binding energy is a typical phenomenon of metal clusters (including Au) on a variety of supporting materials,46,47 and is generally attributed to the reduced core–hole screening in small metal particles.48 The electronic properties of the small sized metal clusters are superior to those of the corresponding bulk material, and this size-dependent alteration of the electronic structure gives rise to unusual catalytic properties.47
C (284.8 eV), CN/C–S (285.5 eV), C–O (286.6 eV) and CO (288.5 eV), which confirm the successful decoration of Au@HSG nanoclusters. In the N 1s spectrum (Fig. 4c), the peaks located at 400.2 eV and 401.9 eV are assigned to C–N and N–H functionalities in GSH, respectively. The S 2p spectra of the Au@HSG-rGO can be deconvoluted into three peaks (Fig. 3d), which correspond to S–S/S–C bonds at 163.8 eV, S–H at 164.9 eV and sulfate of –SO3H (oxidized form –SH) at 169.3 eV. For the Au 4f spectra of free Au nanoparticles decorated on graphene (Fig. 4e, blue line), there are two distinct doublets: the Au 4f7/2 peak at 83.3 eV and the Au 4f5/2 peak at 86.8 eV. However, with regard to the Au@HSG-rGO, the Au 4f7/2 and Au 4f5/2 peaks at 84.4 and 88.1 eV are consistent with the Au(0) state,54 which has a 1.1 eV and 1.3 eV red shift compared with that of bulk Au. This shift in binding energy is a typical phenomenon of metal clusters (including Au) on a variety of supporting materials,46,47 and is generally attributed to the reduced core–hole screening in small metal particles.48 The electronic properties of the small sized metal clusters are superior to those of the corresponding bulk material, and this size-dependent alteration of the electronic structure gives rise to unusual catalytic properties.47
 | ||
| Fig. 4 (a) XPS survey spectrum of Au@HSG-rGO and narrow scan spectra of C 1s (b), N 1s (c), and S 2p (d) of Au@HSG-rGO; (e) Au 4f spectra of Au@HSG-rGO and free Au nanoparticles; (f) Raman spectra of the GO, rGO and Au@HSG-rGO. | ||
The crystalline degree of GO, rGO and Au@HSG-rGO was investigated via Raman spectroscopy. The quality of graphene nanomaterials could be evaluated by the intensity ratio of the D to G bands (ID/IG).49 As shown in Fig. 4f, the two peaks at 1345 cm−1 and 1594 cm−1 in the spectrum of GO are assigned to the disorder-induced D band and the graphitic G band, respectively. The G band shifts to 1584 cm−1 after reduction by GSH (blue line) due to ordered in-plane stretching vibrations of sp2 sites, which demonstrates that the oxygenated groups are removed partly by GSH. The slight increase of the ID/IG ratio from 0.97 (GO) to 1.04 (Au@HSG-rGO) implies the structural changes after the reduction of GO, which are attributed to the marginal decrease of the sp3 domains and the creation of small graphitic domains.
The catalytic activity of the Au@HSG-rGO catalyst was measured via the catalytic reduction of 4-nitrophenol (4-NP) in the presence of NaBH4. This reaction is often chosen as a model to evaluate the catalytic activities of various metallic catalysts, such as Ag,20 Au,7 Ni,50 Pt,50 and Pd.51 The mechanism of catalytic reduction of 4-NP by NaBH4 on the Au@HSG-rGO is shown in Fig. 5a. The excellent catalytic performance of the Au@HSG-rGO may be attributed to the synergistic effect between the Au@HSG and the graphene sheets, which increases the electron transfer rate across the surface of the graphene sheets.52,54 The fast electron transfer should increase the electron concentration on the surface of graphene and accelerate efficient electron transfer from BH4− to 4-NP.
 | ||
| Fig. 5 (a) Schematic illustration of the mechanism of catalytic reduction of 4-NP to 4-AP by Au@HSG-rGO; (b) UV-vis absorbance spectra of the 4-NP reduction reaction in the absence and presence of Au@HSG-rGO. Inset shows the color change with the conversion of 4-NP to 4-AP; (c) ln(C/C0) versus reaction time for the reduction of 4-NP in the presence of rGO, free Au, Au/rGO and Au@HSG-rGO at room temperature. (d) Photographs showing the fixed bed system for the reduction of 4-NP; (e) UV-vis spectra recorded at different flow rates; (f) the conversion for the reduction of 4-NP at different flow rates. | ||
The kinetics of the reaction were monitored by UV-vis spectroscopy measured at room temperature. It can be seen from Fig. 5b that the peak at 401 nm for the absorption of 4-nitrophenolate ions decreases rapidly with increasing time. The concomitant peak at 300 nm also increases with increasing time because of the generation of 4-aminophenol (4-AP) during the reduction process. Meanwhile, as shown in the inset of Fig. 5b, the colour of the solution changes from yellow to colourless after the addition of the Au@HSG-rGO catalyst. The control test was performed under the same conditions using the Au nanoparticle supported graphene (Au/rGO) as the catalyst, which was prepared using ascorbic acid as the reductant. Although Au/graphene composites can be generated by the reduction of GO using ascorbic acid, the size of generated Au particles is much larger than that of Au@HSG (Fig. S4†).
Since the initial concentration of NaBH4 solution is much higher than that of 4-NP, the reaction rate can be assumed to be a constant during the reduction process. The kinetic equation can be written as ln(At/A0) = −kt, where A0 and At are the absorbance values of 4-NP at time 0 and t, respectively, and k is the apparent rate constant. So the catalytic rate can be evaluated via this pseudo-first-order kinetic equation. The plot of ln(At/A0) versus reaction time is shown in Fig. 5c, from which a linear relationship against the reaction time is derived. As shown in Fig. 5c, rGO has no significant catalytic ability for the reduction of 4-NP, whereas the Au/rGO and Au@HSG-rGO possessed good catalytic ability for the reduction of 4-NP. However, the Au@HSG-rGO catalysts possess much higher catalytic activity (k = 5.62 min−1) in the 4-NP reduction reaction than Au/rGO (k = 0.27 min−1). The activity difference between Au@HSG-rGO and Au/rGO can be ascribed to the smaller size and more active sites of the Au@HSG-rGO composite, suggesting that these Au@HSG nanoclusters have the potential to be an excellent candidate for catalytic reactions. Much more catalytic sites are accessible for 4-NP to be reduced to 4-AP compared to Au/rGO. In addition, the catalytic activity of free Au nanoparticles is lower than those supported on rGO, suggesting enhanced catalytic activities due to the charge transfer from Au nanoparticles to rGO.24 The Au@HSG-rGO shows better or comparable catalytic activity to the previously reported catalysts (Table 1). The turnover frequency (TOF) calculated by the moles of reactants consumed per mole of the catalyst per 1 s for the catalyst was also investigated to compare the catalyst efficiency. As shown in Table 1, the Au@HSG-rGO exhibits a higher TOF value than previously reported values even at relatively low concentration of NaBH4. The TOF of Au@HSG-rGO was found to be 1902.6 s−1. Meanwhile, to examine the scope of the Au@HSG-rGO catalytic system for the reduction of the nitro group, the other four nitro derivatives were also examined and the results are listed in Table S1.† It can be noted that the four nitro derivatives exhibit comparable conversion time and rate constants. The system exhibits the highest catalytic activities towards the transformations of o-nitrophenol (TOF = 11415.6 s−1). Thus, it can be concluded that the mild temperature and reductant are especially helpful for the in situ fabrication of efficient metal/graphene-based catalysts and will have a promising application in catalysis.
| Catalysts | C NaBH4 (M) | C 4-NP (mM) | k (min−1) | TOF (s−1) | Ref. |
|---|---|---|---|---|---|
| rGO/Fe3O4/Au | 0.2 | 5 | 0.686 | 11.9 | 1 |
| Au/graphene | 0.1 | 0.1 | 0.19 | 0.19 | 2 |
| Pd–CNT–rGO | 0.3 | 0.1 | 0.43 | 25.8 | 15 |
| Au/PMMA | 0.65 | 0.09 | 0.48 | NA | 17 |
| Au–Ag/GO | 0.88 | 1.54 | 8.56 | NA | 20 |
| Au-GO | 0.2 | 0.67 | 6.478 | 400 | 56 |
| Free Au | 0.02 | 0.2 | 0.16 | 50.8 | This work |
| Au@HSG-rGO | 0.02 | 0.2 | 5.62 | 1902.6 | This work |
| Au@HSG-rGO-PDMAEA | 0.02 | 0.2 | 1.217 | 393.6 | This work |
To improve the recyclability, a fixed-bed system was constructed by packing the Au@HSG-rGO catalysts into a filtering platform (Fig. 5d). The color of the aqueous mixture of 4-NP and NaBH4 changed from bright yellow to completely colorless after flowing through the catalytic system, which indicated the complete reduction of 4-NP. The flow rate can be controlled by changing the pressure on the top of the setup. The UV-vis spectra recorded at different flow rates are shown in Fig. 5e. It can be noted that the characteristic absorption peak of 4-NP decreases gradually with decreasing flow rate and 96.03% conversion was obtained when the flow rate of the aqueous mixture was controlled at 1 mL min−1 (Fig. 5e and f). The well designed fixed-bed system exhibited superior performance due to the great enhancement for the efficiency of the catalysts.
In addition, the TEM images of Au@HSG-rGO after recycling 5 times (Fig. 6a) clearly show that large Au nanoparticles are scattered on the wrinkled graphene sheets with an average size of ∼7 nm. The Au@HSG nanoclusters on the graphene sheets aggregate easily into large Au crystals during the catalytic process (Fig. 8a). The crystalline fingerprints of the aggregation-induced Au nanoparticles can be observed in Fig. 6c. In addition, the selected area electron diffraction (SAED) pattern of the Au@HSG-rGO recycled five times is shown in Fig. 6d. The diffraction rings from inner to outer are assigned to Au crystal faces (111), (200), (220), (311) and (222), respectively. However, numerous Au@HSG nanoclusters still exist on the surface of graphene sheets even after recycling 5 times (Fig. 6b). So the Au@HSG-rGO catalysts still exhibited better catalytic activity than Au particle supported graphene.
 | ||
| Fig. 6 (a–c) HRTEM images of Au@HSG-rGO at different magnifications after recycling five times; (d) the SAED pattern of Au@HSG-rGO after recycling five times. | ||
To minimize the aggregation of the Au@HSG nanoclusters, the pyrene-terminal PDMAEA (Fig. S5†) was chosen as the sheltering agent for modification of Au@HSG-rGO via π–π stacking interactions to endow the Au@HSG-rGO with good catalytic stability and recyclability. π–π stacking interactions have been frequently applied to attach polycyclic aromatic precursors onto the basal planes of graphene sheets to generate various graphene composites.33 To illustrate the π–π stacking interactions between graphene and pyrene groups of the PDMAEA polymer, fluorescence spectroscopy and UV-vis absorbance spectroscopy were performed and the results are shown in Fig. 7a and b. The peaks at 331 nm and 347 nm for pyrene groups in PDMAEA polymer decrease obviously after PDMAEA decoration. In addition, the emission fluorescence of the pyrene group at an excitation wavelength of λex = 345 nm is efficiently quenched after being attached onto graphene, showing the successful decoration of Au@HSG-rGO.
 | ||
| Fig. 7 (a) UV-vis absorption spectra and (b) fluorescence emission spectra of pyrene terminated PDMAEA and the supernatant after being functionalized with Au@HSG-rGO; (c) FT-IR spectra of PDMAEA, Au@HSG-rGO and Au@HSG-rGO-PDMAEA; (d) TGA curves of PDMAEA, rGO, Au@HSG-rGO and Au@HSG-rGO-PDMAEA. | ||
The FT-IR spectra of PDMAEA, Au@HSG-rGO and Au@HSG-rGO-PDMAEA were also collected. The peaks at 2968 cm−1 (C–H), 1731 cm−1 (CO), and 1166 cm−1 (C–N) were observed with the pure PDMAEA. Comparing the FT-IR spectra of Au@HSG-rGO and Au@HSG-rGO-PDMAEA in Fig. 7c, the peaks at 2772 cm−1 and 2948 cm−1 (C–H), and stronger peaks at 2922 cm−1 (C–H) and 1166 cm−1 (C–N) should have resulted from the attached PDMAEA chains. Thermogravimetric analysis (TGA) was employed to quantitatively determine the composition of the Au@HSG-rGO. As shown in Fig. 7d, the weight loss of rGO is more than 30.5 wt% at 300 °C due to the removal of oxygen functional groups. It can be observed that PDMAEA synthesized via RAFT polymerization has a three-step degradation process: the first degradation step at 244 °C, the second one at 332 °C and the third one at 497 °C. In addition, since the Au cannot be degraded in the heating process, the TGA curve of Au@HSG-rGO (Fig. 7d, green line) exhibits 3.2% more residues at 600 °C than that of pure rGO reduced by GSH, which is ascribed to the load of Au@HSG nanoclusters. After modification of Au@HSG-rGO with PDMAEA, the weight loss of the as-prepared Au@HSG-rGO-PDMAEA increased to 22.1 wt% at 600 °C.
Since the surface free energy of metal increases significantly with decreasing particle size,53 the smaller particles tend to aggregate into larger clusters (Fig. 8a). Hence, enhanced catalytic stability and temperature-controlled catalytic activity can be achieved by modifying Au@HSG-rGO with a pyrene-functionalized polymer PDMAEA, which served as a polymer shelter to minimize the aggregation of the Au@HSG nanoclusters (Fig. 8b). As depicted in Fig. 8c, the kinetics of the 4-NP reduction exhibited the characteristics of a pseudo-first-order reaction. Fig. S6† and 8c show the UV-vis absorbance spectra and plots of ln(At/A0) versus the reaction time for the reduction of 4-NP at different temperatures using Au@HSG-rGO-PDMAEA as the catalysts, where At and A0 are the absorption values at time t and 0, respectively. The Au@HSG-rGO-PDMAEA exhibits a negligible catalytic activity at the temperature of 0 and 20 °C with the rate constants of 0.006 and 0.01 min−1, respectively. The kinetic rate constants are calculated from the slope to be 0.214, 0.915, and 1.217 min−1 at 30 °C, 35 °C, and 50 °C, respectively (Fig. 8d). In the catalytic process for Au@HSG-rGO-PDMAEA catalysts, both the diffusion barrier and thermal activity are ascribed to the tunable catalytic activity. The catalytic activity shows an obvious turning point at 33 °C, which should be attributed to the phase transition of the surface bound PDMAEA from hydrophilic to hydrophobic. At the low temperature below 33 °C, the Au@HSG nanoclusters hide in the forest of stretching PDMAEA polymer chains (Fig. 8b left), causing worse catalytic activity for 4-NP reduction. However, the polymer chains tend to shrink and lead to the exposure of Au@HSG nanoclusters (Fig. 8b right) when the temperature is above 33 °C, resulting in effective enhanced catalytic activity.
 | ||
| Fig. 8 (a) Schematic diagram of the probable aggregation process of Au clusters on a graphene nanosheet; (b) schematic diagram of the temperature-tunable catalytic activity of Au@HSG-rGO-PDMAEA; (c) ln(At/A0) versus the time at different temperature; (d) kinetic rate constant k at different temperatures. (e) The conversion versus cycles of Au@HSG-rGO-PDMAEA and Au@HSG-rGO as a catalyst for the reduction of 4-NP. | ||
The decoration of PDMAEA can not only endow the Au@HSG-rGO with tunable catalytic activity via temperature manipulation, but also minimize the aggregation and coalescence of the Au@HSG nanoclusters. Even though the catalytic activity can be slightly weakened during repeated recycling, the reusability and stability can be obviously enhanced via the surface modification with PDMAEA (Fig. 8e and 9).
 | ||
| Fig. 9 (a) TEM image and (b) HR-TEM image of Au@HSG-rGO-PDMAEA after recycling five times. | ||
The Au@HSG-rGO also exhibited excellent electrocatalytic activity for the ORR in alkaline electrolytes. Cyclic voltammetry at a scan rate of 100 mV s−1 was first carried out to investigate the ORR activity of Au@HSG-rGO in a N2- and O2-saturated 0.1 M aqueous KOH electrolyte solution (Fig. 10a). It can be seen that Au@HSG-rGO showed a substantial reduction process in the presence of oxygen with the cathodic reduction peak at around −0.29 V and a high cathodic current density (−0.69 mA cm−2), whereas no obvious reduction peak was observed with N2-staturated 0.1 M aqueous KOH electrolyte solution, suggesting effective electrocatalytic activity of the Au@HSG-rGO in oxygen reduction. Subsequently, linear sweep voltammograms (LSVs) were recorded in an O2-saturated 0.1 M KOH electrolyte at a scan rate of 10 mV s−1 using a rotating disk electrode (RDE) at different rotation rates from 400 to 3025 rpm to evaluate the reaction kinetics of Au@HSG-rGO (Fig. 10b). The LSVs of Au@HSG-rGO show much better current density plateaus than those with larger Au nanoparticles31 and the current density increases with increasing rotation rates and the corresponding Koutecky–Levich plots (Fig. 10c) within the potential range of +0.30 to +0.54 V exhibit good linearity with a rather consistent slope, suggesting the first order reaction kinetics of the ORR with respect to the concentration of dissolved O2. In addition, it can be observed that the onset potentials of oxygen reduction are approximately −0.17 V, which is close to that obtained from cyclic voltammetric measurements as shown in Fig. 10b (−0.15 V). Compared with Pt/C (Fig. S7†), the Au@HSG-rGO exhibits a relatively low cathodic density and a more negative onset potential. However, the Au@HSG-rGO shows a comparable peak potential and onset potential to the reported references (Table S2†). Moreover, the accelerated durability test was also performed in O2-saturated 0.1 M KOH solution by applying a cyclic potential sweep between −0.8 and 0 V. As shown in Fig. 10d, the half-wave potential of the Au@HSG-rGO shows only an 8 mV negative shift and only a 0.022 mV cm−2 decrease for the current density is noticed after 1000 cycles.
 | ||
| Fig. 10 (a) CVs of Au@HSG-rGO in N2- and O2-staturated 0.1 M aqueous KOH electrolyte solution at a scan rate of 100 mV s−1. (b) LSVs of Au@HSG-rGO in O2-saturated 0.1 M KOH at a scan rate of 10 mV s−1 at different RDE rotation rates. (c) Koutecky–Levich plots at different electrode potentials of Au@HSG-rGO at different electrode potentials. (d) Accelerated durability test of Au@HSG-rGO performed for 1000 cycles in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 and a 2400 rpm rotation rate. | ||
4. Conclusions
In summary, a mild co-reductive method to immobilize Au@HSG nanoclusters onto graphene sheets for the fabrication of catalysts with high catalytic activity has been explored. Well-dispersed Au@HSG nanoclusters with an average size of 1.4 nm were self-assembled in situ on graphene sheets with the assistance of GSH using GO and HAuCl4 as precursors. The as-prepared Au@HSG-rGO catalysts exhibited much better catalytic activity for the reduction reaction of 4-NP than Au particles supported on graphene sheets prepared either by ascorbic acid or sodium citrate reduction, which can be attributed to their smaller size and uniform dispersity. The PDMAEA was chosen as a sheltering agent to avoid aggregation and coalescence of Au@HSG nanoclusters during the catalytic process. In addition, the catalytic activity of Au@HSG-rGO-PDMAEA can be adjusted by tuning the reaction temperature and meanwhile the reusability and stability can be obviously enhanced. The Au@HSG-rGO also possesses good electrocatalytic activity for the ORR in alkaline electrolytes. Therefore, the mild co-reduction methodology combined with the unique structure of graphene sheets would become a promising strategy for the design of various transition or noble metal nanocluster or even single atom decorated graphene catalysts for efficient catalytic applications.Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 21305133 and 21575071), Qingdao Innovation Leading Expert Program, Qingdao Basic & Applied Research Project (15-9-1-100-jch), Open Funds of the State Key Laboratory of Electroanalytical Chemistry (SKLEAC201601) and Science & Technology Fund Planning Project of Shandong Colleges and Universities (J16LA13).References
- Y. Wang, H. Li, J. Zhang, X. Yan and Z. Chen, Phys. Chem. Chem. Phys., 2016, 18, 615–623 RSC.
- J. Li, C.-Y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed.
- Z. Zhang, C. Liu, R. E. Kinder, X. Han, H. Qian and R. A. Widenhoefer, J. Am. Chem. Soc., 2006, 128, 9066–9073 CrossRef CAS PubMed.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- P. Luo, C. Li and G. Shi, Phys. Chem. Chem. Phys., 2012, 14, 7360–7366 RSC.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- X.-N. Li, Z. Yuan and S.-G. He, J. Am. Chem. Soc., 2014, 136, 3617–3623 CrossRef CAS PubMed.
- Z.-Y. Li, Z. Yuan, X.-N. Li, Y.-X. Zhao and S.-G. He, J. Am. Chem. Soc., 2014, 136, 14307–14313 CrossRef CAS PubMed.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed.
- Y. Liu, Y. Xu, Y. Tian, C. Chen, C. Wang and X. Jiang, Small, 2014, 10, 4505–4520 CrossRef CAS PubMed.
- Z. Zhang, T. Sun, C. Chen, F. Xiao, Z. Gong and S. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 21035–21040 CAS.
- R. Ouyang, J. X. Liu and W. X. Li, J. Am. Chem. Soc., 2012, 135, 1760–1771 CrossRef PubMed.
- K. Kuroda, T. Ishida and M. Haruta, J. Mol. Catal. A: Chem., 2009, 298, 7–11 CrossRef CAS.
- X. Q. Gong, A. Selloni, O. Dulub, P. Jacobson and U. Diebold, J. Am. Chem. Soc., 2008, 130, 370–381 CrossRef CAS PubMed.
- C. Zhai, M. Zhu, D. Bin, H. Wang, Y. Du, C. Wang and P. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 17753–17761 CAS.
- T. Wu, L. Zhang, J. Gao, Y. Liu, C. Gao and J. Yan, J. Mater. Chem. A, 2013, 1, 7384–7390 CAS.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed.
- C. Wang, H. Wang, C. Zhai, F. Ren, M. Zhu, P. Yang and Y. Du, J. Mater. Chem. A, 2015, 3, 4389–4398 CAS.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS PubMed.
- M. Zhou, A. Zhang, Z. Dai, Y. P. Feng and C. Zhang, J. Phys. Chem. C, 2010, 114, 16541–16546 CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- B. E. Hayden, D. Pletcher and J. P. Suchsland, Angew. Chem., Int. Ed., 2007, 46, 3530–3532 CrossRef CAS PubMed.
- J. Hernández, J. Sollagullón, E. Herrero, A. Antonio Aldaz and J. M. Feliu, J. Phys. Chem. C, 2007, 111, 14078–14083 Search PubMed.
- A. Morozan, S. Donck, V. Artero, E. Gravel and E. Doris, Nanoscale, 2015, 7, 17274–17277 RSC.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- W. Tang, H. Lin, A. Kleimanshwarsctein, G. D. Stucky and E. W. Mcfarland, J. Phys. Chem. C, 2008, 112, 10515–10519 CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- L. Cui, J. Liu, R. Wang, Z. Liu and W. Yang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4423–4432 CrossRef CAS.
- Z. Song, Y. Xu, W. Yang, L. Cui, J. Zhang and J. Liu, Eur. Polym. J., 2015, 69, 559–572 CrossRef CAS.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263–5266 CAS.
- W. Xu, X. Wang, Q. Zhou, B. Meng, J. Zhao, J. Qiu and Y. Gogotsi, J. Mater. Chem., 2012, 22, 14363–14368 RSC.
- X. Chen, G. Wu, J. Chen, X. Chen, Z. Xie and X. Wang, J. Am. Chem. Soc., 2011, 133, 3693–3695 CrossRef CAS PubMed.
- P. A. Khomyakov, G. Giovannetti, P. C. Rusu, G. Brocks, J. V. D. Brink and P. J. Kelly, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 026803 CrossRef.
- Y. Zhang, S. Liu, W. Lu, L. Wang, J. Tian and X. Sun, Catal. Sci. Technol., 2011, 1, 1142–1144 CAS.
- M. Iliut, C. Leordean, V. Canpean, C.-M. Teodorescu and S. Astilean, J. Mater. Chem. C, 2013, 1, 4094–4104 RSC.
- J. Huang, L. Zhang, B. Chen, N. Ji, F. Chen, Y. Zhang and Z. Zhang, Nanoscale, 2010, 2, 2733–2738 RSC.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. C, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- L. Shang, R. M. Dörlich, S. Brandholt, R. Schneider, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Nanoscale, 2011, 3, 2009–2014 RSC.
- J. Ftouni, M. Penhoat, A. Addad, E. Payen, C. Rolando and J.-S. Girardon, Nanoscale, 2012, 4, 4450–4454 RSC.
- F.-X. Zhu, W. Wang and H.-X. Li, J. Am. Chem. Soc., 2011, 133, 11632–11640 CrossRef CAS PubMed.
- L. Ono, D. Sudfeld and B. R. Cuenya, Surf. Sci., 2006, 600, 5041–5050 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS PubMed.
- A. Santra and D. Goodman, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 15, R31 CrossRef.
- Z. Ni, Y. Wang, T. Yu and Z. Shen, Nano Res., 2008, 1, 273–291 CrossRef CAS.
- S. K. Ghosh, M. Mandal, S. Kundu, S. Nath and T. Pal, Appl. Catal., A, 2004, 268, 61–66 CrossRef CAS.
- Z. Dong, X. Le, C. Dong, W. Zhang, X. Li and J. Ma, Appl. Catal., B, 2015, 162, 372–380 CrossRef CAS.
- X. Wen, P. Yu, Y. R. Toh, Y. C. Lee, K. Y. Huang, S. Huang, S. Shrestha, G. Conibeer and J. Tang, J. Mater. Chem. C, 2014, 2, 3826–3834 RSC.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- Y. Qin, J. Li, Y. Kong, X. Z. Li and H. G. Xue, J. Electrochem. Soc., 2013, 161, H172–H177 CrossRef.
- R. Yue, H. Wang, D. Bin, J. Xu, Y. Du, W. Lu and J. Guo, J. Mater. Chem. A, 2015, 3, 1077–1088 CAS.
- Y. Choi, H. S. Bae, E. Seo, S. Jang, H. P. Kang and B. S. Kim, J. Mater. Chem., 2011, 21, 15431–15436 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: S1, UV-vis absorbance spectra of GO, rGO and Au@HSG-rGO; S2, TEM image of GO synthesized via our modified Hummers' method; S3, time-dependent UV-vis absorption spectra of the 4-nitrophenol reduced by NaBH4 catalyzed by Au/rGO; S4, (a) TEM image, (b) SEM image and (c) EDX spectrum of Au/rGO; S5, 1H NMR spectrum of PDMAEA; S6, time-dependent UV-vis absorption spectra of the 4-nitrophenol reduced by NaBH4 catalyzed by Au@HSG-rGO-PDMAEA at 0, 20, 30, 35, and 50 °C. S7, LSVs of Au@HSG-rGO and Pt/C in O2-saturated 0.1 M KOH at a scan rate of 10 mV s−1 at a 1600 rpm rotation rate. Table S1, reduction of other relevant nitro derivatives catalyzed by Au@HSG-rGO and Au@HSG-rGO-PDMAEAa. Table S2, electrochemical parameters for the ORR estimated from CV and LSV curves in 0.1 M KOH solution. See DOI: 10.1039/c6ta08284j |
| This journal is © The Royal Society of Chemistry 2017 |