
Exfoliated MoS2 nanosheets confined in 3-D hierarchical carbon nanotube@graphene architecture with superior sodium-ion storage†
Tuhin Subhra
Sahu
a,
Qianqian
Li
b,
Jinsong
Wu
b,
Vinayak P.
Dravid
*b and
Sagar
Mitra
*a
aDepartment of Energy Science and Engineering, Indian Institute of Technology Bombay, Powai, Mumbai 4000 76, India. E-mail: sagar.mitra@iitb.ac.in; Tel: +91 22 2576 7849
bDepartment of Materials Science and Engineering, NUANCE Center, Northwestern University, Evanston, Illinois 60208, USA. E-mail: v-dravid@northwestern.edu
First published on 9th November 2016
Abstract
Sodium-ion batteries (SIBs) have undergone extensive research efforts as compatible successors of Li-ion batteries (LIBs) for grid-scale energy storage owing to the abundance of sodium resources. However, the poor cycling stability and low rate capability of existing anodes has prevented the practical application of SIBs. To mitigate the situation we have created a 3D heterostructure electrode based on alternative layers of 2D (MoS2–graphene) and 1D (CNTs) materials via a hydrothermal route that is fundamentally different from the usual composites. For comparison, composites were prepared using the same experimental conditions with either rGO or MWCNTs. While discharging at 100 mA g−1 and 500 mA g−1, the MoS2–MWCNT@rGO could deliver a high discharge capacity of 664 mA h g−1 and 551 mA h g−1, and retained 100% and 98.4% capacity after 80 and 250 discharge–charge cycles, respectively. At 2 A g−1, it can yield an initial discharge capacity of 375 mA h g−1, maintaining 81.3% and 67% capacity after 250 and 500 cycles, respectively. The excellent performance of the MoS2–MWCNT@rGO hybrid is mainly attributed to the robust MWCNT@rGO framework with improved 3D electrical conductivity, additional porosity and excellent buffering capability. Furthermore, an in situ TEM technique was employed to explore the sodiation mechanism of the MoS2 nanosheets.
Rechargeable sodium-ion (Na-ion) batteries have recently drawn attention as potential alternatives to lithium-ion batteries owing to their low cost, inexhaustibility and widespread terrestrial reserves. In addition, sodium provides a very large negative redox potential of −2.71 V vs. SHE and almost similar ion insertion chemistry as that of lithium. Therefore, a Na-ion battery could be a very promising candidate for future large scale electrical energy storage (EES) applications like solar energy and wind farms.1–3 However, the large ionic radius (1.02 Å vs. Li 0.76 Å) and higher ionization potential of the Na-ion restricts the selectivity and structural variability of the host materials, particularly for anodes, where large voltage polarization and poor cyclic performance are still some of the major concerns.4 Unlike lithium, sodium cannot intercalate into graphite layers, due to the smaller channel size of graphite compared to the large sodium ionic radii.5 Therefore, a variety of other carbonaceous materials have been investigated for sodium-anode applications. These include hard carbon,6 bio-inspired carbon,7 and hollow carbon nanowires.8 Recently, Wen et al. reported expanded graphite that can reversibly intercalate sodium-ions with good capacity retention.5 However, Na-electroplating onto the carbon surface at a lower potential and poor rate performance are the main problems that need to be addressed.9 Moreover, the specific capacities of the carbonaceous materials against sodium are limited and the maximum achieved is 300 mA h g−1, which is insufficient for large scale applications. In this aspect, layered transitional metal oxides or di-chalcogenides could serve as better alternatives to graphite.10–18 They can provide sufficient gallery space to accommodate large solvated sodium-ions and are also capable of storing sodium-ions in various combined ways, like intercalation–deintercalation/conversion or first conversion and then alloying–dealloying. Specific capacities corresponding to any one of these individual mechanisms can be achieved just by tuning the potential window during their reaction with sodium.19–21 In addition, they are environmentally benign and also commercially viable. Unlike conversion/alloying systems where the active materials suffer from severe volume change (up to 420%) during sodiation/de-sodiation,22 intercalation/conversion systems are more preferable since they can give more reversible and stable capacities. Molybdenum sulphide (MoS2) possesses a typical layered structure, providing a large interlayer spacing (0.615 nm vs. 0.335 nm for graphite) along the c-axis, which could eventually store Na-ions. However, owing to their large surface energy, associated with weak van der Waals attraction, these two dimensional (2-D) layers always have a tendency to restack.23 More importantly, they have an intrinsically poor electrical conductivity, especially along the c-direction. Therefore, the synthesis of a few-layered structure with enlarged interlayer spacing, and grafting onto a conductive matrix like CNT or rGO, could partly overcome the above-mentioned limitations. Here, such conductive matrixes would have much improved electrical conductivity, increased porosity for better transport of Na-ions and enhanced mechanical strength.24,25 Adaptation of a suitable conductive scaffold with large surface area and high porosity is very crucial to utilize the maximum capacity of MoS2. Although, Zhang et al. demonstrated that vertically grown MoS2 on a MWCNT matrix can retain 89.3% (at 50 mA g−1) and ∼84.8% (at 200 mA g−1) of their initial capacities after the 100th and 80th cycles, respectively.26 However, during a long run at high current density, there is always a possibility of the detachment of MoS2 nanosheets from the CNT surface that would result in awful cycling performance. In addition, a one-dimensional (1D) conductive network of MWCNTs inherently limits the ion and electron transfer pathway to merely along the long axis of the CNTs, which results in low rate performance.27 Such problems can be circumvented by tailoring reduced graphene oxide (rGO) nanosheets as conformal building blocks for the layered sulphides. The high surface area of mechanically resilient rGO sheets can hold the MoS2 nanoplates, even after cracking. However, rGO layers are very prone to irreversible agglomeration due to their strong π–π interactions, leading to a substantially reduced surface area for the access of electrolytes,28 and breakdown of the connected channels for fast ion/electron transport. Although the in situ growth of MoS2 nanoplates onto rGO surfaces can prevent the stacking of rGO sheets to some extent, it also lowers the overall conductivity of the resulting graphene-based composite, especially cross-plane conductivity. Therefore, incorporation of 1D MWCNTs into the 2D rGO interlayers could preserve the high surface area of rGO, and can provide a unique three dimensional (3D) conductive network with extraordinary in-plane and cross-plane electron conductivity. A previous study of in situ transmission electron microscopy (TEM) by Gao et al.29 only reveals the phase transformation of MoS2 nanosheets due to Na-intercalation, but the structural evolution of Na-inserted MoS2 beyond intercalation remains unclear.
Herein, we report a facile polyallylamine hydrochloride (PAH) assisted hydrothermal route for preparing the binary composites of MoS2 with either MWCNTs (referred as MoS–C) or rGO (referred as MoS–G) and a ternary composite with both MWCNTs and rGO framework (termed as MoS–GC hereafter). Since the negatively charged surface of MWCNTs or GO could inhibit the approach of MoO42− precursors, the whole synthetic strategy was driven by the idea of modifying the surfaces of carboxy-functionalized MWCNTs or GO by the adsorption of positively charged PAH molecules. As such, negatively charged MoO42− precursors could approach the MWCNTs or GO surfaces via electrostatic interaction and thus MoS2 could nucleate (represented in Scheme 1). Owing to the unique molecular structure of PAH, a long carbon backbone with abundant –NH3+ groups not only promotes the nucleation but can serve as a spacer between adjacent MoS2 nanosheets. By controlling the charge for each component, we can further synthesize composites with a unique three dimensional (3D) architecture from MoS2, MWCNTs and rGO, i.e. by anchoring the PAH-modified MWCNTs onto the negatively charged GO surface before incorporation of the MoO42− precursors, since the MWCNTs@GO skeleton can provide large reactive interfacial areas that allow easy incorporation of Mo-precursors. Here, PAH modification of MWCNTs can prevent agglomeration for GO nanosheets in the MoS–GC composite. This unique feature not only facilitates the easy access of electrolytes and fast electron transport but also accommodates the large volume change in the active material during sodiation/de-sodiation. There are very few reports on the syntheses of such ternary composites. Meanwhile, compared to the simple mixing of individual components,30 the in situ growth of MoS2 onto the MWCNTs, rGO or their hybrid framework can provide a better attachment to the conductive matrixes, and thus significantly improve the interfacial electron transfer.31 The function of the graphene is relatively clear in MoS2–graphene composites, i.e. to provide a highly efficient pathway for electrons and Na-ions to diffuse, and to provide a stronghold/support for the active materials. MoS2 is the most effective component to host Na-ions during sodiation. We applied in situ TEM to explore the structural changes of MoS2 nanosheets during sodiation. It revealed that MoS2 reacts with Na-ions in a two-step manner. A Na-intercalated phase has been observed in the first step, while Na2S and Mo nanocrystals are identified as the final products at the end of a full discharge due to the conversion reaction.
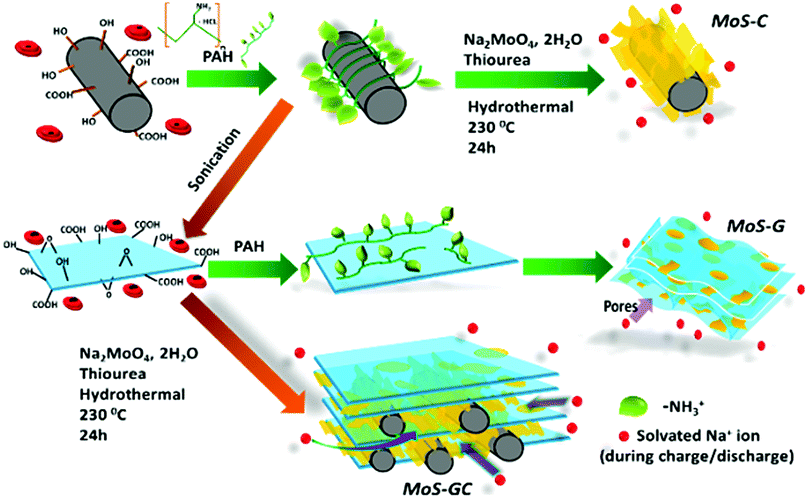 | ||
| Scheme 1 Schematic illustration showing the PAH-assisted hydrothermal preparation of MoS–C, MoS–G and MoS–GC. | ||
Fig. 1A represents the X-ray diffraction patterns of the MoS–C, MoS–G and MoS–GC hybrid composites. For both MoS–C and MoS–G, a sharp diffraction peak at 2θ = 8.97° with d(002) = 0.99 nm (assigned as 1#) is observed due to the formation of significantly expanded MoS2 layers caused by the probable insertion of PAH molecules in between MoS2 layers during their growth. This low 2θ diffraction peak was not found for MoS–C1, a MoS2–MWCNT composite prepared by the same hydrothermal route without PAH modification of the MWCNTs. Another broad peak at 2θ = 17.31° (marked as 2#), corresponding to d-spacing 0.51 nm can be attributed to the distance between MoS2 and the carbon layer.32 Whereas the XRD patterns for MoS–GC and MoS–C1 demonstrate the typical 2H-MoS2 phase (JCPDS card no. 37-1492). The lower intensity (002) peak in MoS–GC compared to MoS–C1 indicates the growth of MoS2 layers along the c-direction was inhibited by the incorporation of graphene.33 Moreover, the (002) diffraction peak at 2θ = 13.4° in MoS–GC implies an exfoliated nature for MoS2, with interlayer spacing of 0.69 nm. A broad peak appearing at 2θ = 25.69°, associated with the (002) plane of hexagonal rGO or MWCNTs, disappeared only in MoS–G. This indicates that restacking of graphene sheets can be prevented significantly with incorporation of MoS2. Raman spectroscopy was employed to confirm further the crystalline phase of MoS2 and the existence of MWCNTs or rGO (Fig. 1B). The two characteristic peaks at 373.9 cm−1 (E12g, the vibration of opposite S atoms with respect to the Mo atom) and 398.1 cm−1 (A1g, the out-of-plane vibrations of S atoms only) are in good agreement with the typical Raman modes of MoS2 and the D-band (the disorder induced phonon mode, 1352 cm−1) and G-band (the graphite band, found at 1572 cm−1 for MWCNTs and 1583 cm−1 for rGO) are either due to MWCNT or rGO. Moreover, the less intense MoS2 peaks with a frequency difference of 24.2 cm−1 (vs. 28 cm−1 for pristine MoS2) clearly confirmed the semi-crystalline, few-layered nature of the MoS2 nanosheets in the corresponding composites.34–36 X-ray Photoelectron Spectroscopy (XPS) was used to investigate the surface electronic state and the chemical composition of the MoS–GC nanocomposite. The XPS profile in Fig. 1C confirms the expected presence of the elements Mo, S, C and O in MoS–GC. The high-resolution XPS spectrum in Fig. 1D reveals two strong peaks at around 233.05 eV and 229.9 eV, which can be attributed to the Mo 3d3/2 and Mo 3d5/2 orbitals of Mo4+ in MoS2 crystals, respectively, while the peak located at 227.1 eV can be indexed as S 2s.37,38 CHN analysis of MoS–C, MoS–G and MoS–GC confirm these composites contain 16.52, 12.01 and 13.23% carbon, respectively (Fig. S1†).
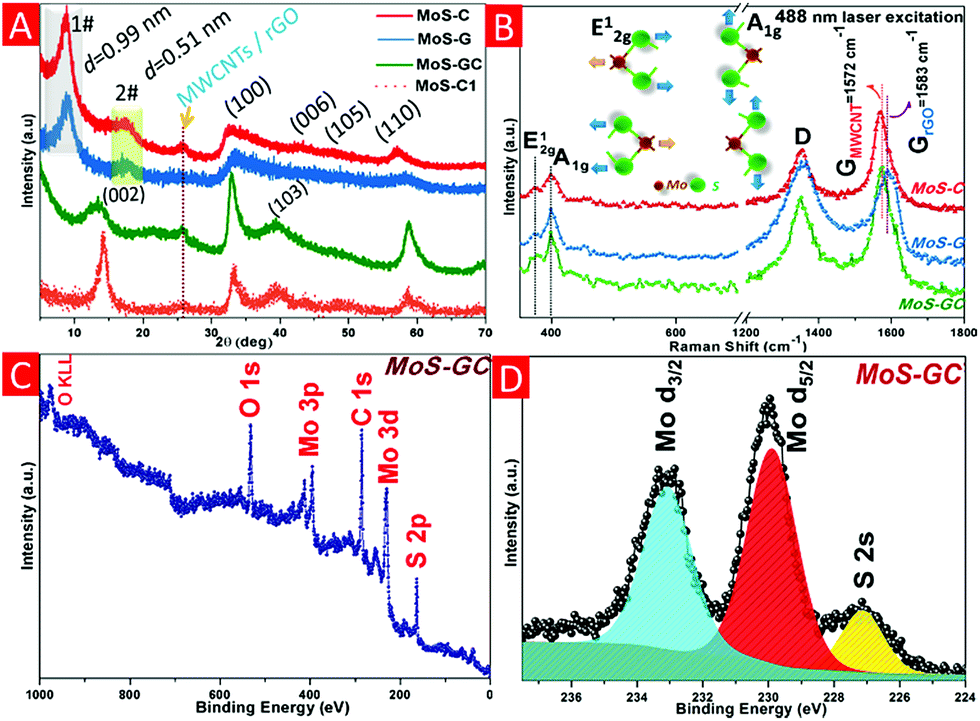 | ||
| Fig. 1 Crystal structure analysis of MoS–C, MoS–G, MoS–GC and MoS–C1. (A) X-ray diffraction (XRD) patterns. (B) Raman spectra of all three composites (inset: atomic displacements of the two Raman-active modes in the unit cell of the bulk MoS2 crystal). (C) XPS survey of the MoS–GC hybrid. (D) Mo 3d region XPS spectrum of MoS–GC. | ||
The morphology and microstructure of MoS–C, MoS–G and MoS–GC were analysed by field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM). Fig. 2A and B display the SEM images of MoS–C, providing a general view of the morphology over a large area. The close-up view of MoS–C in Fig. 2C depicts uniform coating of MoS2 nanosheets onto the MWCNTs. However, without PAH modification of the MWCNTs, there was no such vertical growth of MoS2 on the MWCNT surface (Fig. S2†). This implies that PAH molecules, a cationic polyelectrolyte, can uniformly wrap the negatively charged MWCNT surface in an aqueous suspension,39 which could facilitate the nucleation of MoS2 on the CNT surface. A similar phenomenon is observed for GO,40 since it also can behave as an anionic polyelectrolyte due to the abundant oxygen functionalities on its surface. Fig. 2D presents the crumpled, quasi-3D structure of MoS–G composite. Whereas in Fig. 2E there is evidence for the presence of several interconnected pores that are formed by the self-assembly of reduced GO during the hydrothermal process. These pores that are several nanometres in diameter might be beneficial for Na-ion diffusion throughout the hybrid-electrode during charge–discharge cycles. As indicated in Fig. 2F, MoS2 nanosheets with lateral lengths of 100–200 nm were attached to the rGO network. SEM images (Fig. 2G–I) show basic views of MoS–GC composites at three different places. In Fig. 2G, the SEM micrograph was taken from a typical cross-section of the MoS–GC composite, which possesses an almost layer-by-layer (LBL) assembled structure comprising MoS2, MWCNTs and rGO nanosheets. This robust 3D texture may be responsible for providing the multi-dimensional electron transport pathway and shortening channels for electrolyte access, Na+-ion diffusion, and could remarkably increase the volumetric energy density of the composite-electrode.41 However, it is essential to remove excess PAH from the MWCNT/GO suspension by washing, otherwise it will lead to bundle formation of the MWCNTs or GO in an aqueous solution. Therefore, after completion of the reaction, although there is growth of MoS2 on the MWCNTs or the rGO surface, PAH will lead to agglomerated structures (Fig. S3†). Repeated washing of the PAH-modified MWCNTs or GO sheets could cause the complete detachment of PAH molecules from their surface. Therefore, Fourier transform infrared (FTIR) spectroscopy of PAH-modified MWCNTs (after washing twice) was conducted to confirm the presence of PAH. As exhibited in Fig. S4,† the FTIR peaks at ∼2918 cm−1 and 2848 cm−1 can be attributed to the stretching vibrations of terminal –CH3 groups and –CH2 groups, most common for long chain alkanes, and a broad peak at 1640 cm−1 originates from the scissoring in-plane bending mode of the primary amine (–NH2) of the PAH molecules, while a wide band at ∼3449 cm−1 could be assigned to the N–H stretching mode of –NH2 that might couple with the carboxylic O–H stretching. This implies that the MWCNTs surface was well modified with adsorbed PAH molecules, even after repeated washing. All other IR peaks remain almost unchanged for carboxylated-MWCNTs after PAH-functionalization. To further investigate their morphological features, all three composites were characterized by transmission electron microscopy (TEM) and high resolution TEM (HRTEM). A low magnified TEM image of MoS–C, Fig. 3A, shows that the outer surface of the MWCNT was uniformly decorated with vertically aligned MoS2 nanosheets. As the magnified TEM image (Fig. 3B) reveals, most of the MoS2 nanosheets have an average thickness of ∼3 nm, indicating their few layered structure. Furthermore, the end of most of the lattice fringes turned into single layer MoS2. There is also single layer MoS2, as marked by the yellow circles. Whereas for the MoS–G composite, both Fig. 3D and E demonstrate that the MoS2 nanosheets with lateral size ranging between 100 nm and 200 nm are confined homogeneously throughout the rGO network. Here, PAH-modified GO sheets serve as a solid support for the nucleation and growth of MoS2 into a layered structure, due to the interaction between the positively charged PAH molecules adsorbed onto the GO surface and the negatively charged MoO42− precursors. Such features would help to alleviate the large volume change in MoS2 during sodiation and de-sodiation. Moreover, the presence of interconnected pores (shown by the green circles), with a size of several nanometres are in good agreement with the SEM observations (Fig. 2E). The TEM image in Fig. 3G reveals that cross-linked MWCNTs are firmly attached to the rGO network even after ultrasonication and hydrothermal treatments, suggesting strong interaction between the PAH-modified MWCNTs and GO nanosheets. Fig. 3H displays a typical sandwich structure of the MoS–GC composite. Furthermore, the transparent appearances of MoS2 nanosheets in all three composites suggest that these are fairly thin, probably composed of few-layered sheets. That can be proved further by HRTEM analysis. The HRTEM image of MoS–C in Fig. 3C reveals that the coaxially grown MoS2 coating layers typically consist of one to three walls parallel to the basal planes of the carbon nanotube matrixed with an interlayer spacing ranging between 0.92 nm and 1.25 nm. For MoS–G (Fig. 3F), the single layer (marked by the yellow circle) and the mostly few layered MoS2 nanosheets are found to be grown on the rGO surface. The average d-spacing is ∼0.93 nm. Therefore, it is noteworthy that positively charged PAH molecules adsorbed onto either the MWCNT or GO surfaces not only provide nucleation sites but also function as spacers, leading to extreme exfoliation of the as-formed MoS2 nanosheets. However, for MoS–GC, the interlayer spacing of the MoS2 nanosheets (∼0.69 nm) is smaller compared to those of MoS–C and MoS–G (Fig. 3I). This might be due to a lower availability of PAH molecules, as PAH was being used only to modify the MWCNT surface so that it could be anchored homogeneously onto the GO surface. More importantly, all of the HRTEM results are in accordance with the XRD analysis. It is very interesting to observe from HRTEM studies, that the MoS2 nanosheets grown on MWCNTs, rGO or MWCNTs@rGO, have a number of cracks on their basal surface (highlighted with red circles). These defect-rich structures can enormously increase the exposure of active edge sites,42 which can lower the diffusion path for solvated Na-ions to access the internal surface areas of the MoS2 nanosheets. Nitrogen adsorption–desorption measurements were employed to unveil the texture and porous nature of MoS–GC, MoS–G and MoS–C composites. As indicated in Fig. S6,† all the three composites exhibit type-IV isotherms with a distinct hysteresis loop at a relative pressure P/P0 ranging from 0.4 to 1.0. The Brunauer–Emmett–Teller (BET) surface area of MoS–GC (Fig. S6a†) is calculated to be approximately 217 m2 g−1, which is much larger than the MoS–G (∼133 m2 g−1, see Fig. S6b†) and the MoS–C (∼96 m2 g−1, Fig. S6c†). Interestingly, despite a lower degree of exfoliation of MoS2, the MoS–GC composite is showing a large specific surface area compared to other composites with slightly more exfoliated MoS2. This could be attributed to the high surface area of CNT/rGO matrixes compared to individual rGO or CNTs.43 Such high surface area is desirable for the enhancement of the power/energy density of a nano-composite. The pore size distributions of the composites were derived from the Barrett–Joyner–Halenda (BJH) method, and presented in the inset of Fig. S6.† For all three composites, pores with the maximum volume (∼0.6–1.2 cm3 g−1) are concentrated in the diameter range of 3.0–15 nm, displaying their mesoporous nature. Macropores (dia. approximately 176 nm) with pore volume ∼0.5 cm3 g−1 in MoS–GC could be attributed to the distance between MWCNTs trapped in the interlayer of the rGO nanosheets, also visible from FESEM and TEM analysis.
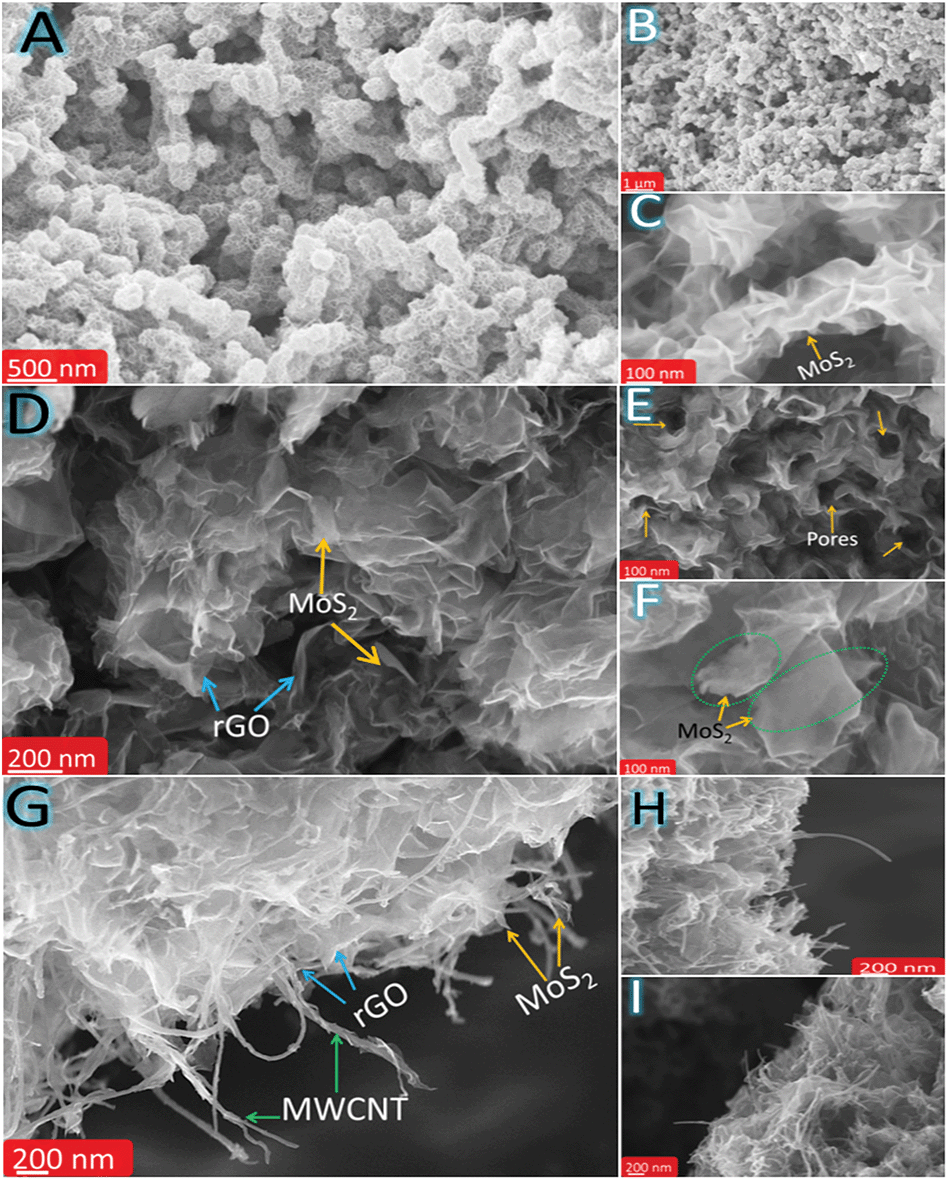 | ||
| Fig. 2 FESEM micrographs of MoS–C, MoS–G and MoS–GC nano-hybrids. (A and B) MoS–C at two different magnifications, (C) a magnified FESEM image of MoS–C showing the vertical growth of MoS2 nanosheets on the MWCNT surface. (D) Crumpled structure of MoS–G, (E) interconnected nano-pores (marked with yellow arrows) in MoS–G could favour the Na+-ion diffusion. (F) MoS2 nanosheets (100–200 nm, shown in dotted green circles) are perfectly held by rGO sheets, (G–I) different cross sections of 3-D MoS–GC displaying the highly porous texture. | ||
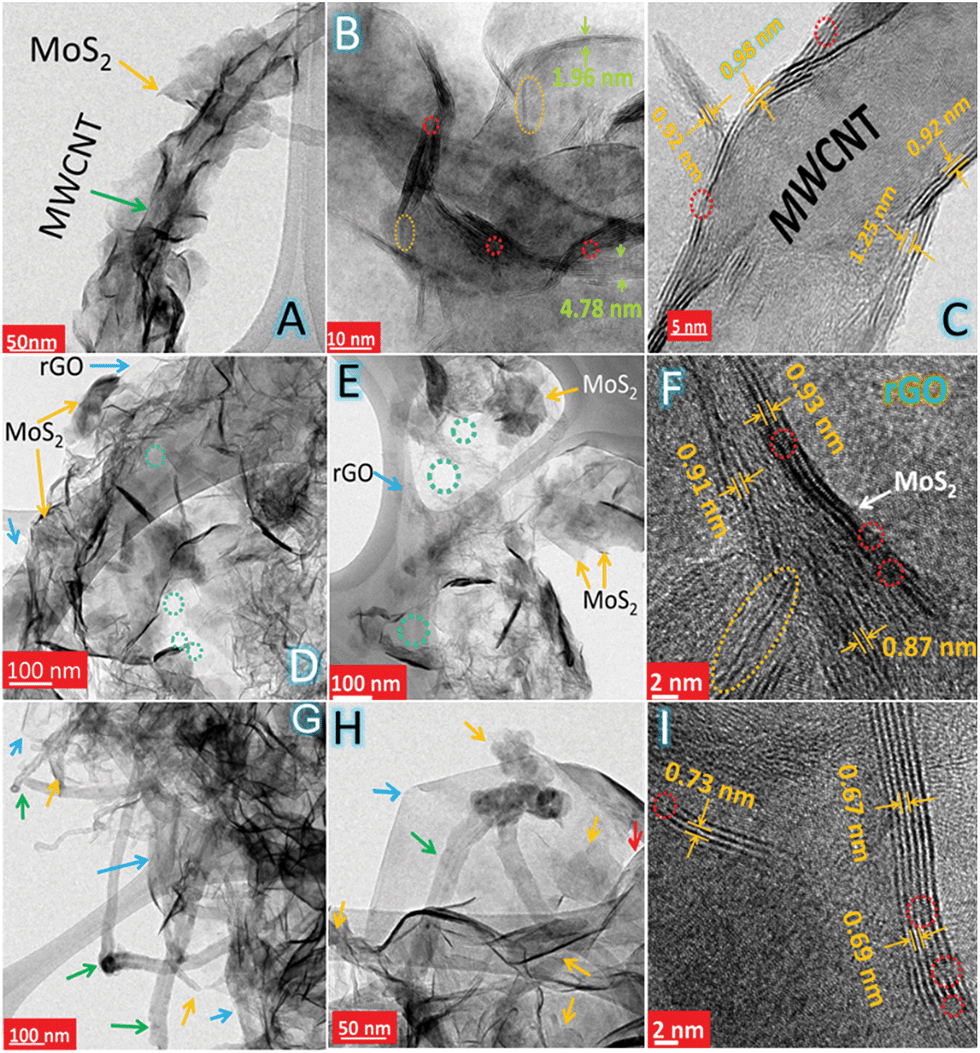 | ||
| Fig. 3 TEM images for (A and B) MoS–C, (D and E) MoS–G and (G and H) MoS–GC are showing the homogeneity of the corresponding hybrids (arrow colour code: yellow, MoS2; green, MWCNT; blue, rGO. Circle colour code: yellow, single layer MoS2; green, nanopores). HRTEM images for (C) MoS–C, (F) MoS–G and (I) MoS–GC are displaying their defect-rich (dotted red circles), few-layered structures with expanded interlayer spacing. | ||
The electrochemical performances of MoS–C, MoS–G and MoS–GC electrodes were evaluated using 2032 type coin cells with Na-metal as a counter/reference electrode. ESI Fig. S7† depicts the cyclic voltammetry (CV) curves of the MoS–GC electrode at a scan rate 0.1 mV s−1 with a potential window ranging between 2.6 V and 0.01 V. During the first cathodic sweep, a sharp peak located at ∼0.81 V and a broad peak around 0.54 V are attributed to the insertion of Na+ ions into MoS2 interlayers (MoS2 + xNa+ + xe−1 → NaxMoS2, x < 2) and the electrochemical conversion of MoS2 into ultra-small Mo nanoparticles [NaxMoS2 + (4 − x)Na+ + (4 − x)e−1 → 2Na2S + Mo]. Another peak around 0.01 V is originated from Na storage on the hetero-interfaces; while the anodic peak at ∼1.8 V should be derived from the reverse reaction between metallic (Mo) nano-grains with Na2S (Mo + 2Na2S → 4Na+ + MoS2 + 4e−1) as mentioned elsewhere.44 From the second cycle onwards, the CV curves overlap well, which suggests a highly reversible and stable sodiation–desodiation process. Fig. 4A–C demonstrates the galvanostatic discharge–charge voltage profiles for the MoS–GC, MoS–G and MoS–C electrodes at 100 mA g−1 in the voltage range 2.6–0.01 V. Each of the three composites show three apparent plateaus at (1.0–0.75 V), (0.75–0.4 V) and (0.4–0.01 V) corresponding to the stepwise intercalation and conversion reaction in the first discharge process, which further shifted to (1.5–1.0 V) and (0.75–0.5 V) from the second discharge onwards. Therefore, the above voltage profiles are well consistent with the reduction–oxidation peaks in the CV of the MoS–G electrode (Fig. S7†). Fig. 4D displays the cyclic stability of the composite electrodes at 100 mA g−1 within the same potential range. For MoS–GC, the initial discharge and charge capacities calculated based on the total active mass of the hybrid are found to be 1026 mA h g−1 and 760 mA h g−1, respectively, giving a relatively high Coulombic efficiency of 74% (vs. 67% of the MoS–G and 63% of MoS–C). The irreversible capacity loss could be attributed to the formation of SEI films on the electrode surfaces due to electrolytic decomposition, which are the common for nanostructured anode materials. During the 2nd cycle, the discharge capacity decreases to 684 mA h g−1 with a corresponding charge capacity 660 mA h g−1, leading to the much higher Coulombic efficiency 96.5%. The value further increases to >99% from the 3rd cycle onwards and maintains this up to the 80th charge–discharge cycle. While MoS–G and MoS–C exhibit 97–98% Coulombic efficiency during their sodiation–desodiation at 100 mA g−1, we also notice that, during 1st anodic run, both the MoS–GC and MoS–G displayed much higher charge capacities (760 mA h g−1 for MoS–GC and 695 mA h g−1 for MoS–G) compared to their nominal value of the total sum (670 × 0.88 + 149 × 0.13 = 602.27 mA h g−1 for MoS–GC and similarly 611.08 mA h g−1 for MoS–G; details are given in Fig. S8†). Such enhanced capacity might be attributed to the synergistic effects of the hetero-interface that exists between the MoS2 nanoplates and rGO sheets. Xie et al. have shown experimentally and theoretically that MoS2/rGO composites with more hetero-interfacial areas can store Na+ ions beyond their theoretical limits.45 Additionally, the high surface area of those nanostructured anodes could also provide numerous reaction sites that allow surface-dominated charge–discharge reactions with Na+ leading to some additional capacity.46 Now, considering the specific capacities, MoS–GC electrodes initially deliver an average capacity 640 mA h g−1, which then increased to 687 mA h g−1 after 80 cycles. Whereas, at the end of the 80th charge–discharge cycle, MoS–G and MoS–C retained 98% (≈582 mA h g−1) and 93% (≈484 mA h g−1) of their 3rd cycle capacities, respectively.
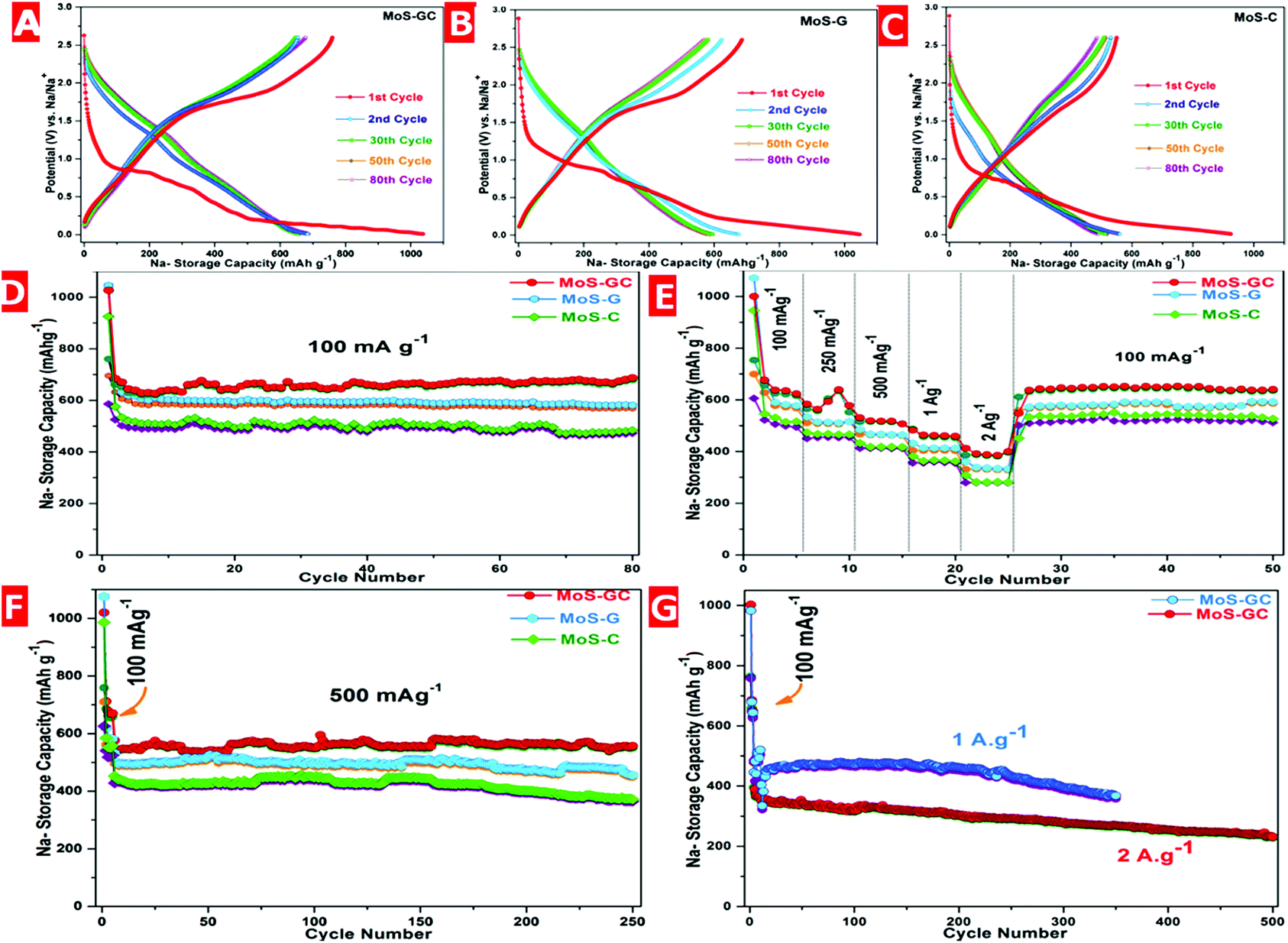 | ||
| Fig. 4 Electrochemical performances of the nano-composites for high-performance sodium storage. Galvanostatic discharge–charge profiles for selected cycles of (A) MoS–GC, (B) MoS–G and (C) MoS–C at a current density of 100 mA g−1. Comparative studies of (D) cycling performances, (E) rate capabilities and (F) long cycling performances of MoS–GC, MoS–G and MoS–C electrodes at different current densities. (G) Ultra-long cycling stability of MoS–GC electrodes at significantly high current rates. | ||
Fig. 4E shows a comparative study of the rate performances of the MoS–C, MoS–G and MoS–GC composites. As expected, the MoS–GC electrode exhibits much higher average capacities of 648 mA h g−1 (100 mA g−1), 583 (250 mA g−1), 535 (500 mA g−1), 482 (1 A g−1) and 381 (2 A g−1) mA h g−1 than those of 588, 512, 501, 413 and 332 mA h g−1 for MoS–G, and 510, 485, 418, 364 and 279 mA h g−1 for MoS–C at the same respective current densities applied for MoS–GC.
Fig. 4F presents the relatively long-term cycling performance of the MoS–C, MoS–G and MoS–GC electrodes measured at 500 mA g−1. As can be seen, the MoS–GC electrode delivers an initial discharge capacity of 551 mA h g−1 and maintains 542 mA h g−1 (∼98.4% capacity retention) even after 250 deep charge/discharge cycles, with a capacity-decay of only 0.01% per cycle. In contrast, MoS–G and MoS–C show average discharge capacities of 493 mA h g−1 and 387 mA h g−1 over 250 cycles with capacity retentions of 92.3% and 81.5%, respectively.
Inspired by their excellent cycling and rate performances, the MoS–GC electrodes were further charged–discharged at significantly higher current densities i.e. 1 A g−1 and 2 A g−1. As indicated by Fig. 4G, when discharged at 1 A g−1, MoS–GC initially exhibits a reversible capacity of 464 mA h g−1 and retains a capacity of 368 mA h g−1 (∼79.3% retention) at the end of 350 cycles. In addition, the hybrid electrode delivers an initial discharge capacity of 375 mA h g−1 at 2 A g−1. After 250th and 500th cycles, discharge capacities of 305 mA h g−1 (∼81%) and 251 mA h g−1 (∼67%) are still obtainable, indicative of the exceptional high-rate cycling stability of MoS–GC. During both studies the average Coulombic efficiencies were almost 100%. To explore the interfacial charge transfer kinetics in the as-prepared nanocomposites, electrochemical impedance spectroscopy (EIS) experiments were adopted at their open circuit voltage (OCV). The Nyquist plots for the MoS–GC, MoS–G and MoS–C electrodes were displayed in Fig. S9.† The analysis of the impedance spectra is based on the equivalent circuit presented in the inset of Fig. S9†, where the symbol R0 represents the ohmic resistance, which is almost constant (∼5.62 Ω) for all three composites, contributed to by the combination of the current collectors, electrode, separator and mainly electrolyte, and is indicated by the intercept at high frequency. Rct and Qdl, standing for the charge transfer resistance and Na+ crossing the SEI layer, are represented by a semicircle with large diameter at medium frequency and W, the Warburg impedance, is shown by a straight line in the low frequency region and is attributed to the diffusion of solvated Na+ ions through the bulk of the electrode. Among all the three composites, the MoS–GC electrode exhibits a lower Rct value (∼369 Ω) compared to MoS–G (∼432 Ω) and MoS–C (∼489 Ω). This lower charge transfer resistance in the MoS–GC electrode is dedicated to the low diffusion resistance to Na+ as well as easy electrolyte penetration, owing to the high electro-active surface area of MoS–GC and the presence of interconnected open channels among CNTs and graphene layers.
Ex situ TEM analysis of the cycled electrodes was performed to understand the superior electrochemical performance of the MoS–GC electrodes. Fig. S10a and b† for MoS–GC and Fig. S10c† for MoS–G showed 25–50 nm long few-layered MoS2 nanosheets (marked by the green circles) still anchored onto the MWCNT@rGO or rGO network after 10th discharge–charge cycles at 100 mA g−1. This indicates the excellent buffering capability of the rGO or MWCNT@rGO framework that helps to maintain the structural integrity of the electrode by alleviating volume changes in the active material. A HRTEM image of MoS–G (Fig. S10d†) reveals the presence of well crystallized, fine-sized Mo nanocrystals as marked by yellow circles. As depicted by Fig. S10f and g† for MoS–C, a thin layer (approximately 5–8 nm thick) of SEI is found to be deposited on the outer surface of the MWCNTs, into which single or few-layered ultra-small MoS2 particles seem to be trapped. Interestingly, despite such SEI formation, the MoS–C electrode is still able to retain 93% of its initial capacity (compared to 100% of MoS–GC and 98% in MoS–G) after 80 cycles at 100 mA g−1, it is probably thanks to fluoroethylene carbonate (FEC), used as an electrolyte additive, which leads to the formation of a stable SEI and passivation layer that allow reversible Na insertion–extraction.47
The EIS technique is further used to explain different electrochemical behaviours and nature of SEI formation among the three composite-electrodes upon cycling (Fig. S11a†). According to the quantitative analysis by the fitting model (Fig. S11b†), R0 (8.12 Ω) did not change significantly after 10 cycles, indicating a stable electrolytic medium. The high SEI-resistance (Rsl) in MoS–GC (22.63 Ω vs. 12.41 Ω in MoS–G and 16.61 Ω for MoS–C) is probably caused by more SEI deposition on MoS–GC electrode owing to its large surface area. Nevertheless, the minimum Rct (39.17 Ω) and inclined Warburg line clearly indicates the ultra-fast Na diffusion throughout the composite electrode. This is purely attributed to the presence of interconnected open channels in MoS–GC. In contrast, high Rct value (121.3 Ω) in MoS–C, reflecting relatively slow charge transfer kinetics through the SEI layer deposited on MWCNT surface.
Therefore, these superior electrochemical performances of the MoS–GC composite compared to MoS–C and MoS–G might be the consequences of the following facts: firstly, the impregnation of MWCNTs into the interlayer galleries of GO can provide enough diffusion paths on the surface of graphene, facilitating the electrolyte/ions diffusion and migration in the electrode during the rapid discharge/charge process. Additionally, the unique hierarchical porous texture combined with high surface area of MoS–GC can also favour the above mentioned ionic diffusion. In contrast, the ultralong-1D network of MWCNT restricts ion and electron conduction in MoS–C. Secondly, such a sandwich-type architecture as MoS–GC is equipped with an impressive substantial buffering capability to mitigate the pulverization and further enhance the structural stability of the electrode material. Thirdly, the half-closed electrochemical chamber in MoS–GC, could provide a unique 3D conductive network for the transport of electrons, thus reducing the internal resistance of the electrode, and improving the cross-plane electrical conductivity more effectively compared to single MWCNTs or rGO. Last but not least, both the rGO and its CNT hybrid can provide a large enough electro-active surface area for stable SEI formation as well as for the electrochemical reaction, and enormous nanopores for the easy access of electrolyte/ions during the course of discharge–charge (schematically illustrated in Fig. S12†). For MoS–C, even though the stable amorphous SEI layer allows the Na+ ions to diffuse through, it is well accepted that solid state diffusion is less favourable compared to the diffusion in liquid phase.
In our previous report,20 we tried to explain the sodiation of MoS2via ex situ XRD, ex situ TEM and in situ EIS techniques. However, recently the in situ TEM study has proven itself the most convenient way to monitor the structural changes of active materials during electrochemical processes.22,48,49 Hence, we have used in situ TEM to investigate the sodiation mechanism of the MoS2 nanosheets and identified a two-step reaction with Na-ions. In the first step of sodiation, the Na+ intercalated into the MoS2 layers and a crystalline phase termed as NaxMoS2 was formed, which is an ordering structure of MoS2 with an expanded lattice. In the second step, with more and more Na+ inserted into the structure and when the amount of Na+ reached a critical value, a conversion reaction happened with the formation of Na2S nanocrystals and Mo nanocrystals as the reaction products. Pristine MoS2 has a hexagonal structure with a = 3.16 Å and c = 12.29 Å. The orientation of the pristine MoS2 nanosheets before sodiation in the current experiment can be identified along the [−1−21] zone axis (Fig. 5A). Small super-lattice diffraction spots appeared after about 4 minutes' sodiation, as shown in Fig. 5B. The appearance of those extra diffraction spots is an indication of the formation of an ordered structure with the intercalation of Na-ions into the MoS2 lattice, termed as NaxMoS2. The detailed structure of the ordered phase is under investigation. There is a rotation of the crystals observed in diffraction patterns from Fig. 5A to D, during the sodiation. Such a rotation was caused by the mechanic deformation brought by the contact and pressing the Na-probe to the tested samples. Along with further sodiation, diffused scattering features appeared in the electron diffraction patterns (Fig. 5C and D). Such diffused scattering is caused by short-range ordering of Na+ in the intercalated structure NayMoS2. In the second stage of the reaction, the diffraction spots corresponding to the NayMoS2 phase disappeared gradually during the sodiation process as shown in Fig. 5E to F. Meanwhile, the intensity of the Na2S diffraction rings became stronger and stronger indicating the formation of Na2S nanocrystals. There is also a diffused ring of Mo that can be observed after about 22 minutes' sodiation reaction, which implies the formation of ultrafine Mo nanocrystals in this stage. Therefore, in the second stage of the sodiation, the reaction is identified as the conversion reaction: NayMoS2 + (4 − y)Na+ + (4 − y)e−1 = 2Na2S + Mo. The conversion reaction provides much larger capacity as compared to the intercalation reaction alone.
 | ||
| Fig. 5 In situ TEM of the sodiation of MoS2 nanosheets: the study of the structural evolution using electron diffraction. (A) The diffraction pattern of a pristine MoS2 nanosheet showing that the MoS2 nanosheet is oriented along the [−1−21] direction before sodiation. (B) Electron diffraction pattern taken after 4 minutes' sodiation shows many small super-lattice spots marked by red arrowheads. This is an indication of the formation of an ordered crystalline phase labeled as NaxMoS2. (C and D) From 7 to 15 minutes' sodiation, features due to diffused scattering can be seen in the patterns. Such diffused scattering is caused by short-range ordering of Na+ in the Na-intercalated structure NayMoS2 as more and more Na+ are inserted (y > x). In the second stage of the reaction from (E) to (F), the diffraction rings of Na2S gradually appear, as indicated by thin arrows, and a diffused ring of Mo, as indicated by the thick arrow, while the diffraction spots and diffused patterns of the Na-intercalated phase disappeared. It is a clear indication that in the second stage a conversion reaction occurs with the reaction products of Na2S and Mo nanoparticles. | ||
In summary, we have developed a novel and scalable cationic-polyelectrolyte assisted hydrothermal method to grow ultrathin MoS2 nanosheets on a MWCNT or rGO surface. Here, the long PAH molecules not only function as a nucleation centre but also prevent the aggregation of MoS2 nanosheets leading to an extremely exfoliated, defect-rich and few-layered (3–5 layers) MoS2 nanostructure. Furthermore, a unique 3D MWCNT@GO skeleton was introduced to serve as a conformal building block for the in situ construction of MoS2 nanosheets. The synergistic effect of the MWCNTs and rGO sheets in such a ternary composite can provide a better 3D conductive network and enormous pores for the easy access of both ions and electrolytes. As a consequence, this 3D architecture manifests an outstanding long-life for cycling at low rates as well as high rates; it can deliver a specific capacity of 664 mA h g−1 at 100 mA g−1 and no capacity fading after the 80th cycle. At 500 mA g−1, this electrode displays 551 mA h g−1 and retains 98.4% capacity at the end of 250 cycles. Even at high current densities of 1 and 2 A g−1, this electrode exhibits 468 and 378 mA h g−1 with excellent ultra-long cycling stability. The improved cycling and rate performances of the ternary composite over binary composites are purely attributed to its robust texture and high electro-active surface area, and hence it is very promising as an SIB anode as well as for other relevant applications.
Acknowledgements
The authors acknowledge the financial support provided by Solar Energy Research Institute for India and the United States (SERIIUS) funded by the U.S. Department of Energy (Office of Science, Office of Basic Energy Sciences, and Energy Efficiency and Renewable Energy, Solar Energy Technology Program, under Subcontract DEAC36-08GO28308 to the National Renewable Energy Laboratory). The instrumental supports were provided by the National Centre for Solar Photovoltaic Research and Education (NCPRE), funded by MNRE-Govt. of India. The In Situ TEM experiment was partly supported as part of the Center for Electrochemical Energy Science, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award # DEAC02-06CH11357, and the Initiative for Sustainability and Energy at Northwestern (ISEN). The TEM work was performed in the EPIC facility (NUANCE Center-Northwestern University), which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) resource (NSF NNCI-1542205); MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN.Notes and references
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- S. W. Kim, D. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dhabi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033–4043 CAS.
- A. D. Stevens and R. J. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef.
- J. Ding, H. Wang, Z. Li, A. Kohandehghan, K. Cui, Z. Xu, B. Zahiri, X. Tan, E. M. Lotfabad, B. C. Olsen and D. Mitlin, ACS Nano, 2013, 7, 11004–11015 CrossRef CAS PubMed.
- Y. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed.
- A. D. Stevens and R. J. Dahn, J. Electrochem. Soc., 2001, 148, 803–811 CrossRef.
- S. Hariharan, K. Saravanan and P. Balaya, Electrochem. Commun., 2013, 31, 5–9 CrossRef CAS.
- D. Su, H. J. Ahn and G. Wang, Chem. Commum., 2013, 49, 3131–3133 RSC.
- J. W. Jong, W. H. Ryu, S. Yu, C. Kim, S. H. Cho and I. D. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 26758–26768 Search PubMed.
- S. H. Choi and Y. C. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 24694–24702 CAS.
- Y. Zhang, P. Zhu, L. Huang, J. Xie, S. Zhang, G. Cao and X. Zhao, Adv. Funct. Mater., 2015, 25, 481–489 CrossRef CAS.
- W. Sun, X. Rui, D. Yang, Z. Sun, B. Li, W. Zhang, Y. Zong, S. Madhavi, S. Dou and Q. Yan, ACS Nano, 2015, 9, 11371–11381 CrossRef CAS PubMed.
- K. Zhang, Z. Hu, X. Liu, Z. Tao and J. Chen, Adv. Mater., 2015, 27, 3305–3309 CrossRef CAS PubMed.
- Y. Liu, Y. Zhao, L. Jiao and J. Chen, J. Mater. Chem. A, 2014, 2, 13109–13115 CAS.
- Y. Liu, H. Kang, L. Jiao, C. Chen, K. Kao, Y. Wang and H. Yuan, Nanoscale, 2015, 7, 1325–1332 RSC.
- G. S. Bang, K. W. Nam, J. Y. Kim, J. Shin, J. W. Choi and S. Y. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 7084–7089 CAS.
- T. S. Sahu and S. Mitra, Sci. Rep., 2015, 5, 12571 CrossRef CAS PubMed.
- P. K. Dutta, U. K. Sen and S. Mitra, RSC Adv., 2014, 4, 43155–43159 RSC.
- W. J. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett., 2012, 12, 5897–5902 CrossRef PubMed.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS PubMed.
- D. Y. W. Yu, P. V. Prikhodchenko, C. W. Mason, S. K. Batabyal, J. Gun, S. Sladkevich, A. G. Medvedev and O. Lev, Nat. Commun., 2013, 4, 2922 Search PubMed.
- S. Zhang, X. Yu, H. Yu, Y. Chen, P. Gao, C. Li and C. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 21880–21885 CAS.
- R. Chen, T. Zhao, J. Lu, F. Wu, L. Li, J. Chen, G. Tan, Y. Ye and K. Amine, Nano Lett., 2013, 13, 4642–4649 CrossRef CAS PubMed.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- P. Gao, L. Wang, Y. Zhang, Y. Huang and K. Liu, ACS Nano, 2015, 9, 11296–11301 CrossRef CAS PubMed.
- L. Zhang, W. Fan and T. Liu, RSC Adv., 2015, 5, 43130–43140 RSC.
- J. Zhou, H. Song, L. Ma and X. Chen, RSC Adv., 2011, 1, 782–791 RSC.
- Z. Wang, T. Chen, W. Chen, K. Chang, L. Ma, G. Huang, D. Chen and J. Y. Lee, J. Mater. Chem. A, 2013, 1, 2202–2210 CAS.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS PubMed.
- Y. Shi, Y. Wang, J. I. Wong, A. Y. S. Tan, C. L. Hsu, L. J. Li, Y. C. Lu and H. Y. Yang, Sci. Rep., 2013, 3, 2169–2177 Search PubMed.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695–2700 CrossRef CAS PubMed.
- A. C. Gomez, M. Barkelid, A. M. Goossens, V. E. Calado, H. S. j. Van der Zant and G. A. Steele, Nano Lett., 2012, 12, 3187–3192 CrossRef PubMed.
- Y. N. Liu, R. Ghosh, D. Wu, A. Ismach, R. Rouff and K. J. Lai, Nano Lett., 2014, 14, 4682–4686 CrossRef CAS PubMed.
- I. Song, C. Park, M. Hong, J. Baik, H. J. Shin and H. C. Choi, Angew. Chem., Int. Ed., 2014, 126, 1290–1293 CrossRef.
- F. T. Lopez, A. M. Gomez, S. M. Vega-Diaz, M. L. Garcia-Betancourt, N. P. Lopez, A. L. Elias, H. Muamatsu, R. Cruz-Silva, S. Tsuruoka, Y. A. Kim, T. Hayahsi, K. Kanoko, M. Endo and M. Terrones, ACS Nano, 2013, 7, 10788–10798 CrossRef PubMed.
- S. Park, D. A. Dikin, S. B. T. Nguyen and R. S. Rouff, J. Phys. Chem. C, 2009, 113, 15801–15804 CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- J. Xie, H. hang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- S. Y. Yang, K. H. Chang, H. W. Tien, Y. F. Lee, S. M. Li, Y. S. Wang, J. Y. Wang, C. C. M. Ma and C. C. Hu, J. Mater. Chem., 2011, 21, 2374 RSC.
- J. Wang, C. Luo, T. Gao, A. Langrock, A. C. Mignerey and C. Wang, Small, 2015, 11, 473–481 CrossRef CAS PubMed.
- X. Xie, Z. Ao, D. Su, J. Zhang and G. Wang, Adv. Funct. Mater., 2015, 25, 1393–1403 CrossRef CAS.
- Y. Y. Hu, Z. Liu, K. W. Nam, O. J. Borkiewicz, J. Cheng, X. Hua, M. T. Dunstan, X. Yu, K. M. Wiaderek, L. S. Du, K. W. Chapman, P. J. Chupas, X. Q. Yang and C. P. Grey, Nat. Mater., 2013, 12, 1130–1136 CrossRef CAS PubMed.
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168 CAS.
- J. Y. Huang, L. Zhong, C. M. Wang, J. P. Sullivan, W. Xu, L. Q. Zhang, S. X. Mao, N. S. Hudak, X. H. Liu, A. Subramanian, H. Fan, L. Qi, A. Kushima and J. Li, Science, 2010, 330, 1515–1520 CrossRef CAS PubMed.
- J. Wan, F. Shen, W. Luo, L. Zhou, J. Dai, X. Han, W. Bao, Y. Xu, J. Panagiotopoulos, X. Fan, D. Urban, A. Nie, R. S. Yasser and L. Hu, Chem. Mater., 2016, 28, 6528–6535 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, CHN analysis, FTIR spectra, Brunauer–Emmett–Teller (BET) analysis, cyclic voltammetry, impedance spectroscopy results before and after cycling, and ex situ Transmission Electron Microscopy (TEM) analysis. See DOI: 10.1039/c6ta07390e |
| This journal is © The Royal Society of Chemistry 2017 |