
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of poly(lauryl methacrylate)-b-poly(benzyl methacrylate) nano-objects synthesised by ATRP and their temperature-responsive dispersion properties†
Melody
Obeng
a,
Amir H.
Milani
 a,
Muhamad S.
Musa
a,
Zhengxing
Cui
a,
Lee A.
Fielding
*a,
Louise
Farrand
b,
Mark
Goulding
b and
Brian R.
Saunders
*a
a,
Muhamad S.
Musa
a,
Zhengxing
Cui
a,
Lee A.
Fielding
*a,
Louise
Farrand
b,
Mark
Goulding
b and
Brian R.
Saunders
*a
aSchool of Materials, The University of Manchester, Manchester, M13 9PL, UK. E-mail: brian.saunders@manchester.ac.uk
bMerck Chemicals Ltd, Chilworth Technical Centre, University Parkway, Southampton, SO16 7QD, UK
First published on 24th February 2017
Abstract
Self-assembling poly(lauryl methacrylate)-b-poly(benzyl methacrylate) (PLMAx–PBzMAy) diblock copolymers were synthesised for the first time using solution atom transfer radical polymerisation (ATRP). The PLMA degree of polymerisation (x) was fixed at 14 and the PBzMA degree of polymerisation (y) was varied from 34 to 74. Post-polymerisation transfer of this new series of diblock copolymers from chloroform into n-dodecane (a poor solvent for PBzMA) resulted in self-assembly of polymeric nano-objects. The morphologies for the latter (spheres, worms and vesicles) were controlled by y. The observed morphologies generally agreed with those reported for related PLMAx–PBzMAy diblock copolymers (x ≥ 16) prepared by polymerisation induced self-assembly (PISA) via reversible addition–fragmentation chain transfer (RAFT) polymerisation (Fielding et al., J. Am. Chem. Soc., 2014, 136, 5790). However, a number of differences were observed such as de-gelation behaviour and the phase boundary positions compared to those expected from Fielding et al. Variable-temperature dynamic light scattering studies for the PLMA14–PBzMA34 spheres revealed that the aggregation number was unaffected by a temperature increase over the range of 20–90 °C, which differed markedly from the behaviour observed for PLMA14–PBzMA64 worms. This difference is a new observation with mechanistic importance for the worm-to-sphere breakdown mechanism. We show that concentrated PLMA14–PBzMAy dispersions (20% w/w) in n-dodecane can be prepared using post-polymerisation transfer. The dispersion with a mixed spherical and worm-like copolymer phase exhibited reversible de-gelation when heated. Surprisingly, the dispersions containing only the worm phase remained as gels (which were white) at temperatures up to 90 °C. Our new ATRP approach for preparing temperature-responsive non-aqueous nano-object dispersions presented here decoupled chain growth and self-assembly and will apply to other copolymer dispersions.
Introduction
Self-assembly of amphiphilic diblock copolymers into dispersions of nano-objects is an important area of soft matter science which is generating increasingly sophisticated nanomaterials with useful properties.1–9 The use of a selective solvent for a diblock copolymer that is a good solvent for one of the blocks but a poor solvent for the other block triggers nano-object self-assembly.10 Depending on the packing parameter of the diblock copolymer a wide array of nano-objects can be obtained which include spheres, worms, and vesicles.5 Nano-objects are often prepared by means of polymerisation induced self-assembly (PISA) via reversible addition–fragmentation chain transfer (RAFT) polymerisation.10 Variations of RAFT polymerisation and alternative methods for preparing nano-objects are being actively pursued11,12 in order to expand architectural versatility and property performance. The majority of diblock copolymers prepared by PISA contain one block that has a glass transition temperature (Tg) higher than room temperature. This raises the question of the extent to which these nano-objects can be regarded as “living” in terms of their ability to reversibly disassemble. The latter question led us to investigate an alternative solution-based synthetic method to prepare non-aqueous dispersions of a self-assembling diblock copolymer. Here, we investigate poly(lauryl methacrylate)-b-poly(benzyl methacrylate) diblock copolymers (PLMA–PBzMA) prepared using atom transfer radical polymerisation (ATRP). The hypothesis for this study was that self-assembly of PLMA–PBzMA nano-objects would be independent of the copolymer synthesis method. We also aimed to explore the mechanism for temperature-responsive PLMA–PBzMA nano-object disassembly. The practical motivation for this study was to show that concentrated (gel-forming) dispersions of self-assembled nano-objects with tuneable morphologies and properties could be prepared using a polymerisation method that decoupled polymer chain growth from self-assembly.The self-assembly of amphiphilic copolymers into cylindrical (worm-like) polymer micelles using RAFT-based synthesis in aqueous13–17 and non-polar solvents18–22 is well established. Here, we focused on diblock copolymers that self-assembled into gel-forming nano-objects in a non-polar solvent (n-dodecane). Organogels have a range of potential applications which includes delivery,23,24 cosmetics25 and energy transfer.26 Fielding et al. synthesised a series of PLMAx–PBzMAy (x ≥ 16) diblock copolymers using RAFT-based PISA in n-dodecane.27 Systematic variation of the PLMA and PBzMA block lengths enabled spherical, worm-like and vesicle morphologies to be prepared. The worms formed temperature-responsive gels, which reversibly de-gelled at temperatures above 50 °C. The mechanism for the gel-to-fluid transition is believed to involve worm fragmentation into spheres.27 The subsequent fluid-to-gel formation upon cooling is relatively slow. Whilst sphere-fusion is believed to be involved the mechanistic details are not fully resolved. Derry et al. also studied PLMAx–PBzMAy (x ≥ 16) diblock copolymers and focussed on the effect of solvent on nano-object morphology.28 In the present study a new series of PLMAx–PBzMAy (x = 14) copolymers is studied and their self-assembly and dispersion properties investigated and compared to the previous work.
Whilst the PISA approach has achieved a considerable success10 there are some limitations which may constrain versatility. Firstly, the shell-forming block must be soluble in the selective solvent used and the latter must be a thermodynamically bad solvent for the core-forming block. Furthermore, the sulphur-based end groups may be undesirable for some industrial applications as they confer colour and a degree of toxicity.29 Whilst end-group modification methods are known,30 additional synthetic steps are required which would add to cost. Here, we use a potentially versatile solution based ATRP method to synthesise PLMA–PBzMA copolymers in a good solvent for both blocks. Because our new approach to PLMA–PBzMA nano-object preparation decouples copolymer synthesis from self-assembly, and should be apply to other copolymers, it offers new versatility for nano-object formation.
ATRP and other living polymerisation methods such as ring-opening polymerisation have been widely used to prepare a number of self-assembled copolymers.31–35 ATRP is a versatile method for the synthesis of diblock copolymers and enables high blocking efficiencies with targeted block lengths, well-defined architectures36 and is well-suited to the polymerisation of many vinyl monomers with diverse functionalities.37 However, there have been relatively few reports of the use of ATRP to prepare polymer worms. Banerjee et al. described using ATRP in xylene to prepare worm-like nanostructures from poly(chloromethyl styrene)-g-poly(benzyl methacrylate)38 with average lengths less than 100 nm. Worm-like nanostructures were also reported to self-assemble in bulk films of poly(methyl methacryate)-b-poly(azobenzene methacrylate).39 There have not been any reports using ATRP to prepare PLMA-based nano-objects to the best of our knowledge.
The approach used in the present study is depicted in Scheme 1a. A PLMA14–Br macroinitiator (abbreviated as L14) was prepared via ATRP of lauryl methacrylate (LMA), followed by solution ATRP of benzyl methacrylate (BzMA) to give PLMA14–PBzMAy (L14-By). The copolymer was dissolved in a low boiling point good solvent for both blocks (chloroform, Scheme 1b) and then added to a high boiling point selective solvent (n-dodecane), which is a good solvent for the PLMA block. The chloroform was then removed by evaporation. The PBzMA and PLMA blocks formed, respectively, the core and corona of the nano-objects. We studied the effect of PBzMA degree of polymerisation, i.e., y, on the morphology and response to temperature of the self-assembled L14-By nano-objects and gels.
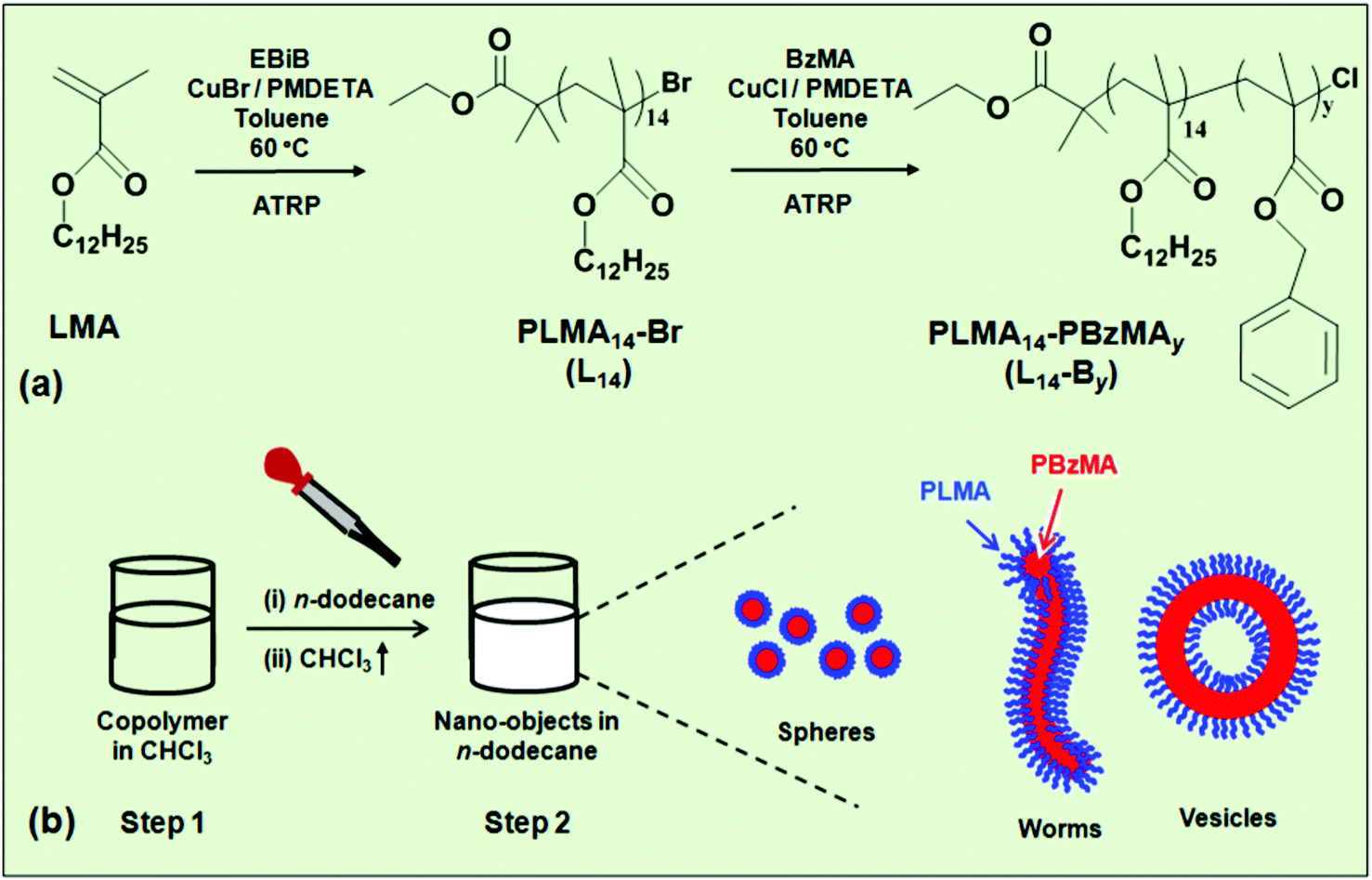 | ||
| Scheme 1 (a) Depiction of the synthesis of poly(lauryl methacrylate) macroinitiator (L14) via solution ATRP using α-bromoisobutyrate (EBiB) and the subsequent addition of benzyl methacrylate (BzMA) via halogen exchange to form L14-By diblock copolymers. (b) Self-assembly of nano-objects by post-polymerisation transfer which involved replacing a volatile good solvent (chloroform, step 1) with a selective solvent (n-dodecane, step 2). Spherical, worm-like or vesicle-like nano-objects assembled depending on y. | ||
The factors governing the self-assembly in aqueous dispersions of diblock copolymers are well understood.40 The two dominant free energy contributions are the elastic energy (entropic) of the corona and surface energy of the core. The elastic energy of stretched chains within the core also contributes to the total free energy. As the morphology changes from spheres, to worms and then to vesicles the stretching of the chains in the core decreases; however, crowding of the chains within the corona increases.41 The design rules governing nano-object morphology for diblock copolymers in non-polar solvents are still being established and the extent to which the nano-objects are at equilibrium is of considerable interest.10 In the present study we systematically vary the degree of polymerisation of the core-forming block (PBzMA) and probe temperature-triggered PLMA–PBzMA nano-object morphology and gelation changes in dilute and concentrated n-dodecane dispersions.
In the first part of the study the L14-By diblock copolymers are characterised by GPC and 1H NMR spectroscopy. Nano-object morphology and self-assembly behaviours are then explored using TEM and variable temperature 1H NMR spectroscopy and dynamic light scattering (DLS). Tube inversion studies and dynamic rheological data of concentrated dispersions are then investigated to study gel behaviour and it is shown that the mixed spherical and worm-like compositions are capable of forming temperature responsive physical gels. By comparing the temperature-triggered nano-object and gel-to-fluid transformations of dispersions of spheres and worms we find that the PBzMA degree of polymerisation affects reversibility. We also study the response of PLMA–PBzMA spheres to temperature for the first time and assess their importance for the temperature-triggered gel-to-fluid transitions. We show that the self-assembly of PLMA–PBzMA copolymers is generally independent of the synthesis method. However, the copolymers used in this study also show differences to previous work in terms of their morphologies and properties which are potentially important.
Experimental section
Materials
LMA (96%) was purified using a basic alumina column before use. BzMA, (96%) was passed through a 1/1 neutral/basic alumina column before use. Toluene (99.5%), ethyl α-bromoisobutyrate (EBiB, 98%), N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA, 99%), copper bromide (CuBr, 99.999%) and copper chloride (CuCl, ≥99.995%) were all purchased from Aldrich and used as received. Chloroform (ACS reagent grade), methanol (HPLC grade), tetrahydrofuran (HLPC grade) were all purchased from Fisher and used as received. Chloroform-d (99.8 atom% D), dodecane-d26 (98 atom% D) and n-dodecane (99+%) were purchased from Aldrich and Alfa Aesar, respectively, and were all used as received.Synthesis of PLMA–Br macroinitiator
The PLMA14–Br macroinitiator (L14) synthesis was adapted from a literature method.42 The ATRP homopolymerisation was terminated at 64% to retain a high percentage of bromo-terminated moieties for the subsequent diblock growth step.43 A Schlenk flask was charged with toluene (2 mL), LMA (10 mL, 34.1 mmol), EBiB (220.0 µL, 1.55 mmol) and PMDETA (323.0 µL, 1.55 mmol). The reagents were deoxygenated with Ar whilst stirring magnetically at room temperature and subjected to repeated freeze–pump–thaw cycles. CuBr (0.22 g, 1.55 mmol) was quickly added to the frozen mixture under Ar during the final cycle. The sealed reaction flask was immersed in a pre-heated oil bath at 60 °C for 150 min. The product was passed through a basic alumina column and precipitated from excess methanol before it was dried in a vacuum oven at 30 °C for 12 h.PLMA–PBzMA diblock copolymer synthesis
Table 1 shows the compositions and codes for the PLMA14–PBzMAy copolymers. L14-B34 represents the PLMA14–PBzMA34 copolymer. The following gives an example synthesis for L14-B34. The concentration ratio of monomer [M], initiator [I], Cu(I) [Cu(I)] and ligand [L] used was 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.8:0.8. Accordingly, a Schlenk flask was charged with the L14 (0.5 g, 0.133 mmol) that had been dissolved in toluene (3.63 mL), followed by BzMA (1.50 mL, 8.911 mmol) and PMDETA (23.0 µL, 0.11 mmol). The reagents were deoxygenated with Ar whilst stirring magnetically at room temperature before being subjected to repeated freeze–pump–thaw cycles. CuCl (11 mg, 0.11 mmol) was quickly added to the frozen mixture under Ar during the final cycle. The sealed reaction flask was immersed in an oil bath at 60 °C for 15 h. The resulting copolymer was passed through a 1/1 neutral/basic alumina column before being purified and dried as described for L14. The same method as described above was used to prepare L14-B46, L14-B64 and L14-B74. For those copolymers the ratios for [M]:[I]:[Cu(I)]:[L] were [M]:1:0.8:0.8 with [M] = 50, 67 and 80, respectively. The conversions for all copolymers were in the range of 92 to 96%.
:1:0.8:0.8. Accordingly, a Schlenk flask was charged with the L14 (0.5 g, 0.133 mmol) that had been dissolved in toluene (3.63 mL), followed by BzMA (1.50 mL, 8.911 mmol) and PMDETA (23.0 µL, 0.11 mmol). The reagents were deoxygenated with Ar whilst stirring magnetically at room temperature before being subjected to repeated freeze–pump–thaw cycles. CuCl (11 mg, 0.11 mmol) was quickly added to the frozen mixture under Ar during the final cycle. The sealed reaction flask was immersed in an oil bath at 60 °C for 15 h. The resulting copolymer was passed through a 1/1 neutral/basic alumina column before being purified and dried as described for L14. The same method as described above was used to prepare L14-B46, L14-B64 and L14-B74. For those copolymers the ratios for [M]:[I]:[Cu(I)]:[L] were [M]:1:0.8:0.8 with [M] = 50, 67 and 80, respectively. The conversions for all copolymers were in the range of 92 to 96%.
| Code | Compositiona | M n(NMR)/g mol−1 | M n(GPC)/g mol−1 | M w/Mn | d z /nm | Morphologyc |
|---|---|---|---|---|---|---|
| a Compositions determined from 1H NMR spectroscopy. b Apparent sphere-equivalent z-average diameter of the diblock copolymers measured at 20 °C. c Morphologies based on TEM data shown in Fig. 2. | ||||||
| L14 | PLMA14 | 3800 | 4900 | 1.31 | — | — |
| L14-B34 | PLMA14–PBzMA34 | 9700 | 13700 |
1.50 | 39 | Spheres |
| L14-B46 | PLMA14–PBzMA46 | 11800 |
15400 |
1.50 | — | Spheres and worms |
| L14-B64 | PLMA14–PBzMA64 | 15000 |
20900 |
1.58 | 480 | Worms |
| L14-B74 | PLMA14–PBzMA74 | 16700 |
22600 |
1.55 | — | Vesicles |
Physical measurements
Gel permeation chromatography (GPC) was performed using a Max Viscotek instrument that comprised two mixed B columns (average porosity of 500 Å). The eluent was THF and calibration was performed using linear polystyrene standards. The standards had molecular weights ranging of 1 × 103 to 2 × 106 g mol−1 and a Viscotek VE 3580 refractive index detector was used. 1H NMR spectroscopy was conducted using a Bruker AVI-400 MHz spectrometer and CDCl3 as the solvent. Variable-temperature 1H NMR spectroscopy was conducted using a Bruker Instrument (AVI-500 MHz spectrometer) using dodecane-d26. The copolymer dispersion was prepared by dissolving neat copolymer in chloroform (20 µL). The solution was then transferred into the appropriate volume of dodecane-d26 and chloroform evaporated with stirring at 40 °C for 48 h to give a dispersion (5.0% w/w). DLS studies were conducted using a Zetasizer Nano-ZS instrument (Malvern Instruments, UK) at a fixed scattering angle of 173°. The copolymer dispersions underwent 20 to 90 to 20 °C temperature cycles with an equilibration time of 5 min at each temperature. The z-average diameters (dz) correspond to sphere-equivalent values for the worm-containing systems. The dispersions used for DLS (0.1% w/w) used n-dodecane and were prepared using a similar method as described above. For transmission electron microscopy (TEM) samples diluted dispersions in n-dodecane (0.05% w/w) were used. The dispersion was deposited on a carbon-coated grid before being exposed to ruthenium tetra-oxide vapour for 10 min. TEM measurements were obtained using a Philips CM20 200 kV instrument. Number-average sizes were determined from TEM by counting 94 spheres, 27 worms and 32 vesicle shells. Dynamic rheology measurements were conducted using an AR-G2 rheometer equipped with aluminium cone geometry (2° and 20 mm diameter). Measurements were conducted using a strain of 1.0% and gap of 800–1200 µm. Variable-temperature studies were conducted at a frequency of 1 Hz with a ramping rate of 1 °C min−1 and an equilibration of 5 min for each step. The dispersions (20% w/w in n-dodecane) measured by rheology were prepared as described above using concentrated copolymer solutions in chloroform (∼45% w/w) and subsequent chloroform removal.Results and discussion
Macroinitiator and diblock copolymer characterisation
The macroinitiator was synthesised via solution ATRP of LMA in toluene (Scheme 1a). The 1H NMR spectrum (Fig. 1a) enabled calculation of the number-average degree of polymerisation (x) via end group analysis using the integrals for the methylene groups from PLMA (h), the methyl groups of EBiB (c) and the terminal methyl group of EBiB (a) (see eqn (S1), Fig. S1 and the details given in the ESI†). The calculated x value was 14.0, which was significantly lower than those reported previously for PLMA–PBzMA diblock copolymers prepared via RAFT-based PISA (x ≥ 16) by Fielding et al.27 and Derry et al.28 Consequently, the diblock PLMA14–PBzMAy copolymers reported here are a new series of copolymers. The GPC data (Fig. S2, ESI† and Table 1) gave an Mn of 4900 g mol−1 and a polydispersity of 1.31. The latter value is in the range reported for other PLMA homopolymers prepared by ATRP.42,44,45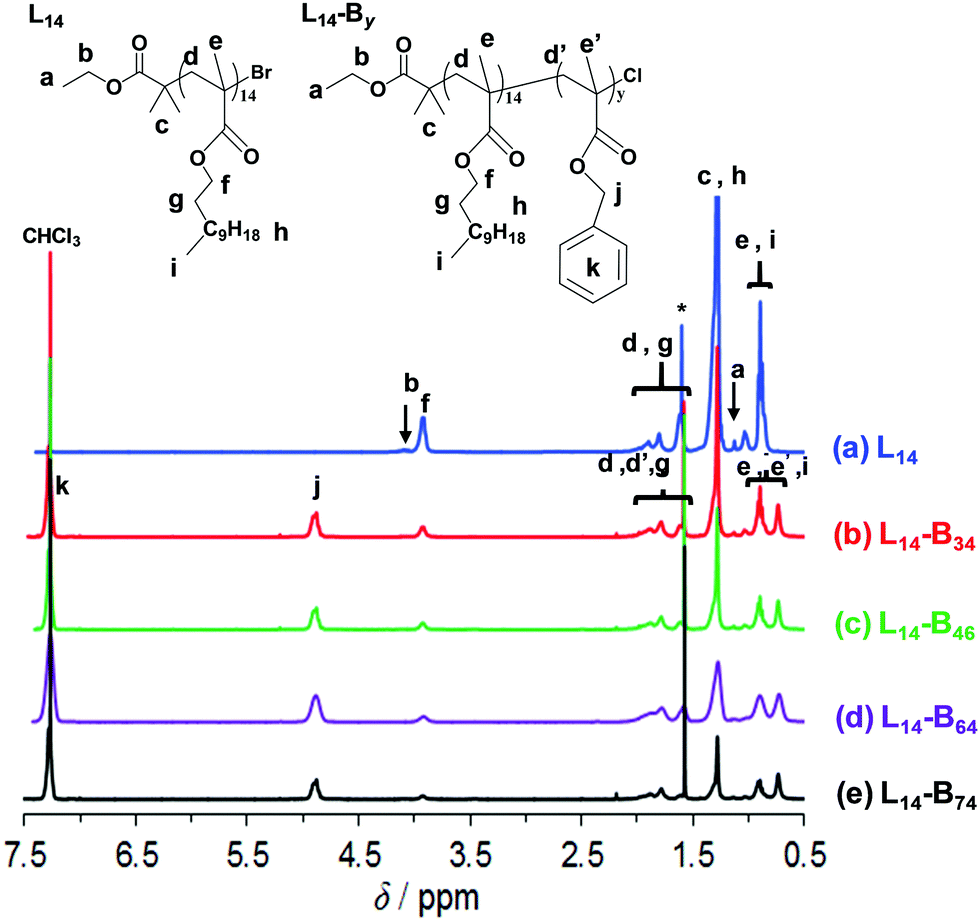 | ||
| Fig. 1 1H NMR spectra for the macroinitiator (L14) and the diblock copolymers. The identities are shown. The peak labelled with * was due to water. | ||
L14-By copolymer synthesis was conducted using L14 and Cu(I)Cl following the halogen exchange method46,47 (Scheme 1a). We found that the latter approach48,49 was essential to prepare the diblock copolymers and minimise their polydispersity. After PBzMA polymerisation the 1H NMR spectra (Fig. 1b–e) showed that signals from the BzMA methylene protons (4.7 ppm, j) and aromatic protons (7.3 ppm, k) were present, as expected.50 The relative intensity from the PLMA oxyethylene group (4.0 ppm, f) decreased after the growth of the PBzMA block. The number-average value for y was calculated using the integration values for the methylene PBzMA group (j) and the values for the methylene groups from PLMA (h) and the methyl groups from EBiB (c) using eqn (S2) (see ESI†). The y values can be seen from the L14-By compositions shown in Table 1. The GPC chromatograms indicated that the diblock copolymers were mostly monomodal (Fig. S2, ESI†) with polydispersities in the range of 1.50–1.58 (Table 1). It was challenging to gain control over BzMA chain growth and consequently a catalyst switch was used in this work (Scheme 1a). It is likely that a minor proportion of dead PLMA chains were present which subsequently contributed to the polydispersity of the copolymers.
Nano-object morphology
A key difference between the method used here and PISA19 is that our approach used copolymer synthesis under good solvency conditions. The formation of nano-objects involved the transfer of the diblock copolymer from a chloroform solution (which is a good solvent for both blocks) into the selective solvent n-dodecane (Scheme 1b) followed by chloroform removal (see Experimental section). 1H NMR analysis indicated a very low content of residual chloroform (less than 0.2 vol%) remained after this treatment. TEM data for L14-B34 deposited from n-dodecane showed spheres with low size polydispersity (Fig. 2a). The other three systems showed mixtures of spheres and worms (L14-B46, Fig. 2b), worms (L14-B64, Fig. 2c) and vesicles (L14-B74, Fig. 2d). Additional TEM images obtained using lower magnifications are shown in Fig. S3 (ESI†). Whilst there were occasional spheres present for L14-B64 and L14-B74 the overwhelming majority of the copolymer was present as worms or vesicles, respectively. The vesicles had a range of sizes with some less than 100 nm (Fig. S3d, ESI†). The number-average diameter of the spheres (L14-B34) was 20.5 ± 2.3 nm, whilst the worms (L14-B64) had an average cross-sectional diameter of 38.5 ± 3.4 nm. The vesicles (from L14-B74) had average wall thickness of 24.0 ± 1.6 nm. These morphologies are in accord with the reduction of strain for the PBzMA core chains afforded by the decrease in curvature associated with the transitions from spheres to worms and then vesicles.5,51 The TEM images show that the L14-By nano-objects prepared via ATRP had the same general morphologies as those reported for PLMAx–PBzMAy (x ≥ 16) diblock copolymers prepared by PISA.27 It follows that thermodynamics had a strong role in controlling PLMA–PBzMA nano-object morphology. However, there were also morphology differences which are described below. | ||
| Fig. 2 TEM images of diblock copolymer nano-objects deposited from n-dodecane dispersions. The samples were stained with ruthenium tetra-oxide. The scale bars in the main and inset figures represent 200 and 20 nm, respectively. | ||
To provide a better comparison of the morphologies of L14-By nano-objects prepared here by ATRP with the PLMAx–PBzMAy (x ≥ 16) nano-objects prepared using PISA,27 the morphological data from both studies were plotted on a common phase diagram (see Fig. S4, ESI†). In agreement with the results of Fielding et al.,27 as y increased the morphology changed from spheres, to worms and then vesicles. However, the positions of the nano-object phases identified from this study are not in the positions expected if the phase boundary lines from Fielding et al.27 are extrapolated to the PLMA degree of polymerisation value of 14.0. For example, L14-B64 existed as worms at a much higher y value than expected. Furthermore, L14-B34 existed as spheres in this work; whereas PLMA16–PBzMA37 (L16-B37) from Fielding et al.27 formed worms. One may question whether the x or x/y values are the more pertinent parameter for discussing the morphological differences. The x/y values for L14-B34 (this study) and L16-B37 (from ref. 27) were 0.41 and 0.43, respectively. These x/y values were within 5% of each other and are not considered distinguishable. Therefore, the differences in the morphologies for these two systems cannot be explained using x/y values. However, the x values were significantly different.
Clearly, a key factor affecting L14-By micelle formation and morphology is the preparation method. As mentioned above, in the present study the preformed copolymer was dissolved in a (low boiling point) good solvent for both blocks (chloroform, Scheme 1b) and then added to n-dodecane, which is a good solvent for the PLMA block. The chloroform was then removed by evaporation, which triggered micelle formation. The present approach is very different to the PISA method of Fielding et al.27 where the n-dodecane was used as the (selective) solvent for the polymerisation of BzMA using a PLMA macromolecular chain transfer agent. For the latter work PBzMA block growth triggered micelle formation. Consequently, the large differences in micelle formation conditions employed would have significantly contributed to the fact (noted above) that L14-B34 and L16-B37, respectively, existed as spheres in this study and worms when prepared by PISA.27
Whilst differences in copolymer polydispersities and possibly end group effects may have contributed to the L14-By morphologies and differences apparent from Fig. S4 (ESI†) it does appear that the phase boundary positions for these self-assembling copolymers depends on the manner in which the copolymers are synthesised. The presence of a mixed phase for L14-B46 (Fig. 2b) is a feature frequently reported for this family of self-assembling copolymers.28,52,53 Worm formation and growth is believed to involve fusion of cores which is a relatively slow process.54 It is suggested that driving force for core fusion was less pronounced for L14-B46 due to the relatively short chains which was resulted in the mixed phase.
Ruthenium tetra-oxide reacts preferentially with double bonds and enables contrast to be enhanced for TEM.55,56 Here, we used this species to stain the PBzMA blocks within the spheres (Fig. 2a), which enabled the number-average diameter (dTEM) of 20.5 nm to be used as a measure of the L14-B34 core diameter. The repeat unit length for BzMA is estimated as ∼0.25 nm and the fully stretched length for a PBzMA34 chain is calculated as ∼8 nm. Consequently, the diameter of a micelle core of fully stretched PBzMA chains would be ∼16 nm, which is close to dTEM. It follows that the PBzMA chains were highly stretched in the L14-B34 micelle cores. Furthermore, using the calculated molecular weight of the PBzMA34 block (5984 g mol−1) an aggregation number (Nagg) of 530 can be estimated (details are given in the ESI†). This estimation assumed that solvation of the core did not occur at room temperature which is consistent with 1H NMR data (below) and has previously been shown for related systems.22 From the average surface area of the L14-B34 spheres and the Nagg value an average separation of the PLMA chains at the surface of ∼1.6 nm was calculated (see ESI† for the details). The latter value can be compared to the diameter of a cross-sectional slice of a PLMA cylinder which can be shown to be ∼6 nm. This geometric analysis suggests that the PLMA chains were sterically crowded in the micelle corona. It follows that both blocks of the copolymer that formed the spheres were in somewhat entropically unfavourable environments. It is likely that the micelles were preferred because of the strong (enthalpic) incompatibility between PBzMA and n-dodecane at room temperature.
Temperature-triggered morphology transitions
The effect of temperature on the local environments of the PBzMA and PLMA blocks was probed using variable-temperature 1H NMR spectroscopy for the L14-B34 (spheres) and L14-B64 (worms) dispersed in dodecane-d26 (Fig. 3). With increasing temperature, both the methylene signal from PBzMA at 4.9 ppm (j) and the PLMA oxyethylene group (f) increased in intensity (Fig. 3a and b). The latter trend is consistent with related data reported for PLMA16–PBzMA37 prepared by PISA.27 The increases in the signal intensities apparent from scrutiny of Fig. 3a and b indicate that solvation increased for both blocks with temperatures greater than or equal to 40 °C. Interestingly, relatively enhanced solvation occurred for the PBzMA block at higher temperatures as can be seen from Fig. 3c which shows the ratio of the integrals (Aj/Af) for the PBzMA and PLMA signals. The onset temperatures for enhanced PBzMA solvation were about 55 and 70 °C, respectively, for both L14-B34 and L14-B64. This difference may indicate that the shorter PBzMA34 chains were more easily solvated. Upon cooling the signals corresponding to PBzMA groups were not detectable (Fig. 3a and b) which shows that the solvation changes were fully reversible for both the spheres and worms. | ||
| Fig. 3 Variable-temperature 1 H NMR spectra for dilute dispersions of (a) L14-B34 and (b) L14-B64 in dodecane-d26. The temperature was increased from 25 to 115 °C before cooling to 25 °C. (c) Signal integral ratios of j to f for the dispersions containing L14-B34 spheres (red circles) and L14-B64 worms (black squares). The changes in Aj/Af relate to solvation changes. | ||
The response of the dispersions of spheres (L14-B34) and worms (L14-B64) to elevated temperatures was also studied using variable temperature DLS. The dz and PDI data are shown in Fig. 4 and the size distributions appear in Fig. S5 (ESI†). Whilst DLS data were published for PLMA16–PBzMA37 worms prepared by PISA,27 variable dz data for spheres have not been reported. Considering the spheres first (Fig. 4a) the dz range of 37–39 nm at 20 °C is larger than the TEM-derived number-average core diameter of 20.5 nm. A sphere consisting of a 20.5 nm core and corona of PLMA14 chains with a fully extended backbone would have a calculated total diameter of 27 nm. If the LMA chains were forced to protrude outwards due to lateral crowding (as discussed above) an extra 6 nm could be added, resulting in a total diameter of ∼33 nm. Taking into account chain polydispersity and the fact that the dz values are strongly weighted by the largest spheres which scatter more light it is reasonable to attribute the dz range at 20 °C to spherical L14-B34 micelles.
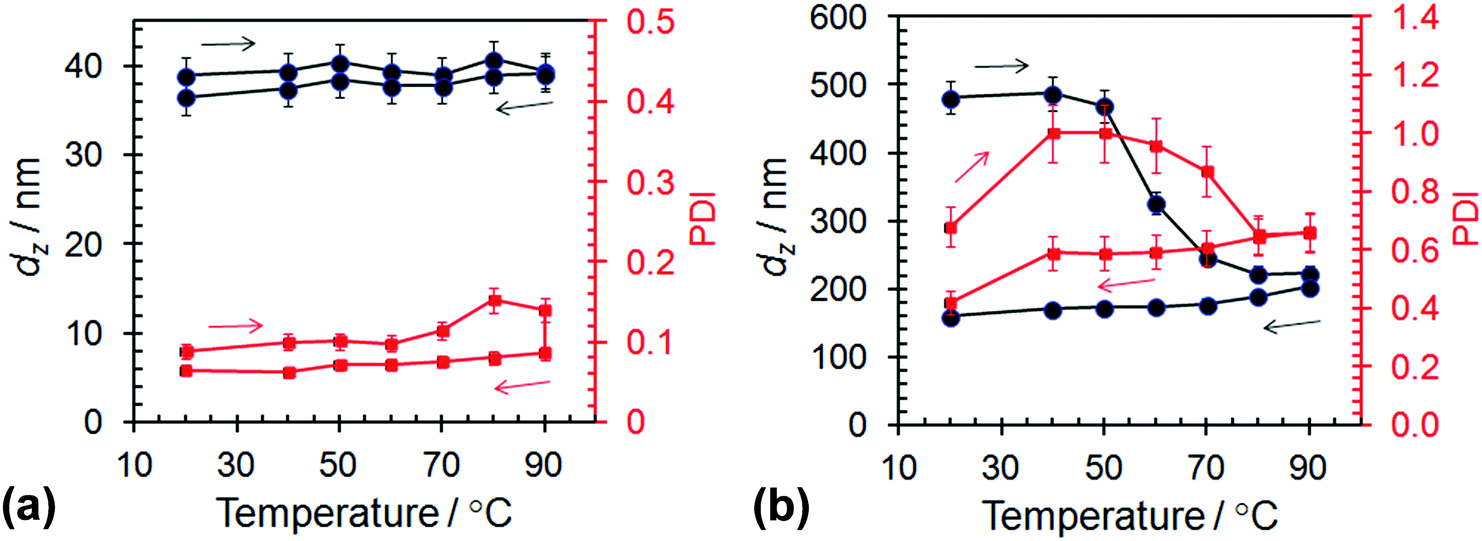 | ||
| Fig. 4 Variable-temperature DLS data for dispersions of (a) L14-B34 spheres and (b) L14-B64 worms. The z-average diameter is represented by black circles, while the polydispersity index is represented by red squares. The arrows indicate the directions of temperature ramps. The initial and final temperatures were 20 °C. | ||
The variable-temperature DLS data for the spheres (Fig. 4a) shows that dz did not change significantly over the temperature range of 20–90 °C. The polydispersity of the spheres decreased slightly due to heating. The invariance of dz with temperature is noteworthy because the core PBzMA chains became increasingly solvated with increasing temperature (as discussed above in connection with Fig. 3c) and must, therefore, have occupied an increasingly larger (average) volume. An increase in diameter of 2 nm for the spheres (from 39 nm at 20 °C to 41 nm at 80 °C during the temperature increase) would be too small to detect using DLS. However, this diameter increase would equate to a 16% increase in volume. Therefore, 1H NMR spectroscopy was more sensitive to small solvation changes than DLS. Because the dz values were not significantly affected by heating there was no evidence of the spheres dissociating into smaller species and a significant decrease of Nagg. The data support the view that the spheres were the lowest energy state for these systems.
By contrast to the L14-B34 spheres, the L14-B64 worms demonstrated irreversible changes for dz when heated (Fig. 4b). The irreversible nature of the morphology changes is supported by the DLS size distributions measured at each temperature (Fig. S5b, ESI†). For the L14-B64 dispersion at 20 °C, dz was 480 nm and decreased to 223 nm upon heating to 90 °C. However, upon subsequent cooling to 20 °C a dz of only 159 nm was measured. Whilst this trend agrees with that reported for L16-B37 worms prepared using PISA27 the dz values for our L14-B64 worms were a factor of ∼3 larger and the onset of the dz decrease occurred at a much lower temperature (50 °C here cf. 70 °C27). Moreover, the dz values for our L14-B64 worms decreased at temperatures well below that where solvation of the PBzMA core was detectable by 1H NMR (∼84 °C from Fig. 3c). This behaviour differs greatly to that observed for the spheres and indicates that the worms were relatively unstable and fragmented before the majority of the PBzMA had been solvated. A process akin to fracture can be envisaged whereby the worms cleaved into smaller segments, moving toward spheres.27 The latter conclusion is generally supported by the PDI data which decreased due to heating. Whilst the DLS size distributions and PDI values for the worms must be considered with caution due to the inherent broad distribution of lengths associated with self-assembled polymer worms,10 it appears that the temperature-triggered fragmentation process may have also involved some relatively large species (Fig. S5b, ESI†). More study on this process in the future is warranted.
As the temperature subsequently decreased the formation of worms was favoured. However, worm formation was relatively slow because worms grow by core–core fusion.54,57 These differences in temperature-triggered nano-object behaviours for the L14-B34 spheres (Fig. 4a) and the L14-B64 worms (Fig. 4b) are likely to have contributions from the relatively low dispersion concentration used for the present DLS measurements and the PBzMA degree of polymerisation. In a study on a related poly(stearyl-methacrylate-b-phenylpropyl methacrylate) system Pei et al.58 observed good worm-to-sphere reversibility using DLS of samples taken from concentrated dispersions. For the present system, the increased y value for the L14-B64 worms resulted in less favourable PBzMA solvation at elevated temperatures (Fig. 3c) and consequently kinetic trapping due to a lower mobility core may also have retarded core fusion.
Temperature-triggered gelation
We next investigated the properties of concentrated L14-By dispersions (20% w/w) prepared using our post-polymerisation transfer method (Scheme 1b). The L14-B34, L14-B46 and L14-B64 dispersions became more opaque with increasing y value (Fig. 5a). The use of solution ATRP afforded gels that were white, which contrasts to the pink colour normally associated with RAFT copolymers. This lack of colour is an inherent advantage for the present ATRP approach because it would facilitate inclusion of chromophores to prepare coloured or fluorescent responsive gels59 without the complication of end group removal. Tube inversion tests for the diblock copolymers in n-dodecane showed that L14-B46 and L14-B64 formed self-supporting physical gels (Fig. 5a). By contrast, our L14-B34 (spheres) and L14-B74 (vesicles) were free-flowing liquids. | ||
| Fig. 5 (a) Concentrated dispersions of L14-By diblock copolymers in n-dodecane (20% w/w) at room temperature. Dispersions of L14-B34 and L14-B74 were free-flowing liquids. The vesicles in L14-B74 showed evidence of sedimentation. L14-B46 and L14-B64 formed self-supporting gels with the former being less turbid. (b) L14-B46 changed from a gel to a fluid when heated to 90 °C which subsequently re-gelled when cooled. | ||
Only the L14-B46 dispersion showed reversible gel formation behaviour. Heating this gel to 90 °C for 4 h resulted in a free-flowing viscous fluid that could reform into a physical gel upon cooling to 20 °C over a 24 h period (Fig. 5b). Surprisingly, the gel formed from L14-B64 did not show reversible gel formation when tested by tube inversion even after 16 h of heating at 90 °C. It is likely that the relatively high PBzMA degree of polymerisation of 64 for L14-B64 was responsible for the lack of reversibility. The longer PBzMA64 chains had stronger intra-core attractive interactions and weaker solvation (Fig. 3c). This conclusion implies that the reversibility of temperature-triggered gelation for L14-By dispersions is tuneable using y.
To gain insight into the gelation behaviour dynamic rheology was used to probe concentrated L14-B46 and L14-B64 dispersions at 20 °C. Fig. 6 shows frequency-sweep data and it can be seen that the G′ (storage modulus) and tan δ values (=G″/G′, where G″ is the loss modulus) for both physical gels were frequency-dependent. These data imply that the gels were soft and viscoelastic. The physical gel formed from L14-B64 had a greater G′ value (1450 Pa) compared to the gel formed from L14-B46 (1180 Pa) at 1 Hz indicating the former gel had about 20% more elastically-effective chains. The latter conclusion is from rubber elasticity theory wherein the modulus is proportional to the number-density of elastically effective chains.60 For the present gels the details of the network are not fully understood. Nevertheless, the trend of the G′ values above is expected because of the higher proportion of anisotropic worms which are undoubtedly the load bearing elements of these networks. The fact that the gels had tan δ values (Fig. 6b) that were significantly larger than zero (the value for an ideal elastic solid) shows that viscous dissipation mechanisms were operative. We speculate that these involved breaking and reforming of worms and worm-to-worm contacts.
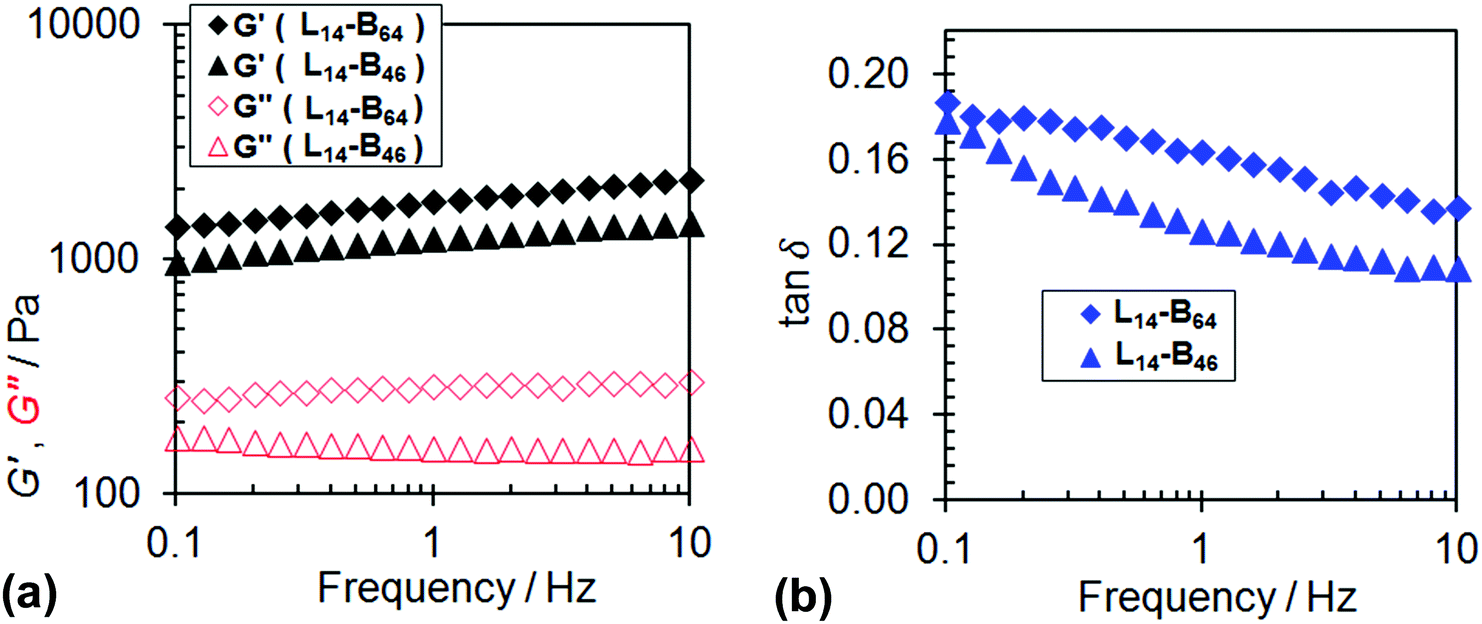 | ||
| Fig. 6 (a) Dynamic frequency-sweep rheology data for the physical gels formed from L14-B64 and L14-B46 dispersed in n-dodecane (20% w/w). The G′ (storage modulus) and G″ (loss modulus) values are indicated by closed and open data points, respectively. (b) Variation of tan δ (=G″/G′) with frequency for L14-B64 and L14-B46. All data were measured at 20 °C. | ||
Variable-temperature rheological data were measured to probe the temperature-triggered changes in the gel networks for L14-B46 and L14-B64 (Fig. 7). The critical gelation temperature (CGT) describes the temperature at which the worm-to-sphere transition occurs to a sufficient extent whereby G′ becomes equal to G″ and tan δ = 1.0. Following Fielding et al. the CGT values during the first heating cycle could be estimated as 55 °C for L14-B46. However, close inspection of the data for the variation of tan δ with temperature (shown in Fig. S6, ESI†) reveals that the tan δ values fluctuated with temperature and became less than 1.0 at higher temperatures. This behaviour is a combination of a weak gel-to-fluid transition, possible structural rearrangement, and the difficulty of measuring small G′ and G″ values. It is noted that the tan δ values for L14-B64 remained less than 1.0 at all temperatures (Fig. S6b, ESI†) and this system did not exhibit a CGT. Of greater significance than the CGT value is the onset of the pronounced G′ decreases for both systems (Fig. 7). These values occurred at ∼45 and 50 °C, respectively for L14-B46 and L14-B64, respectively. These values can be considered as “softening temperatures (TSoft)”. The latter values reside within the temperature range where solvation of the PBzMA blocks was first detectable from the 1H NMR spectroscopy data (≥40 °C, Fig. 3a and b). The beginning of temperature-triggered solvation of the PBzMA cores strongly decreased the load distribution abilities of the worm-based networks.
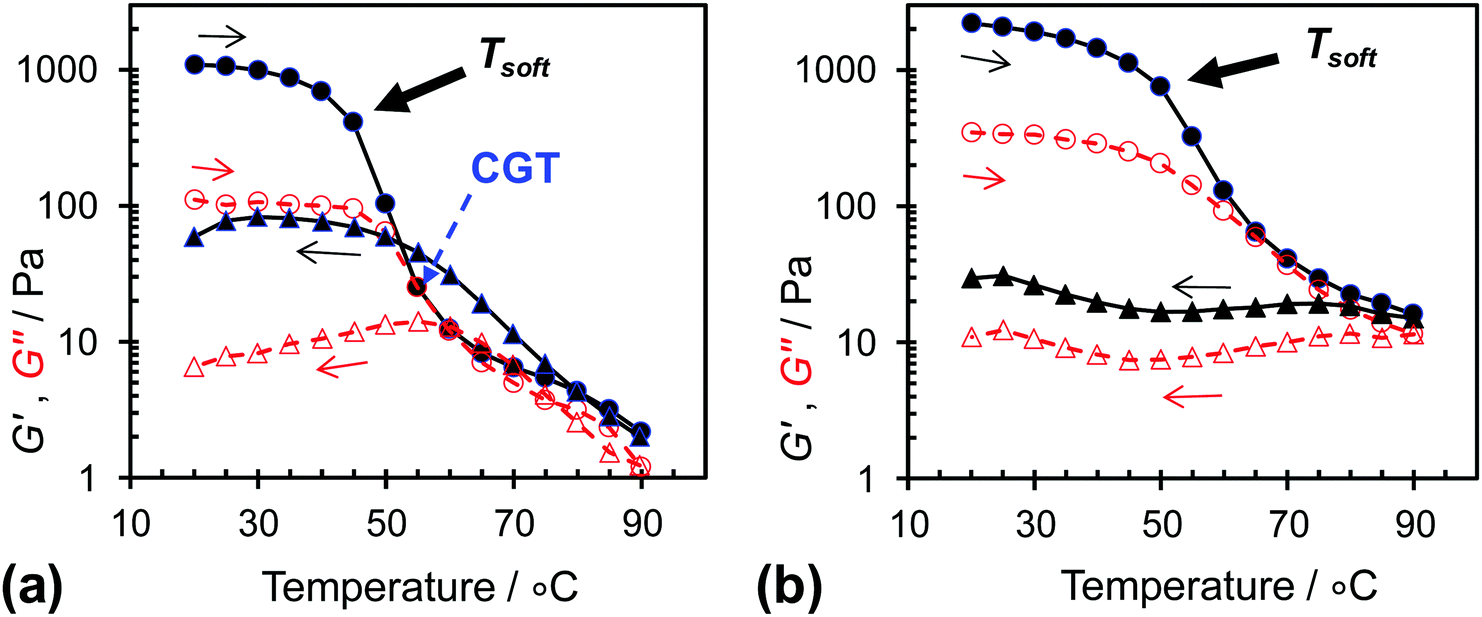 | ||
| Fig. 7 Variable temperature dynamic rheological data for (a) L14-B46 and (b) L14-B64 dispersed in n-dodecane (20% w/w). The G′ (storage modulus) and G″ (loss modulus) values are indicated by closed data points and open data points, respectively. Tsoft and CGT are the gel softening temperature and critical gelation temperatures, respectively (see text). | ||
It is noteworthy that temperature-triggered reversibility was not observed for the L14-B64 gels because the G′ values remained higher than G″ (and tan δ < 1.0) after the samples were held at 90 °C and then cooled (Fig. 7b and Fig. S6b, ESI†). This behaviour is consistent with the DLS data (Fig. 4b) and tube inversion study (Fig. 5) as discussed above. However, the lack of gel reversibility contrasts to the reversible gel formation reported for RAFT-based PLMA–PBzMA worm gels.27 We attribute the lack of reversibility for the L14-B64 gels to the relatively long timescales involved in the reformation of worms which probably involved core fusion. These results lead to the suggestion that PLMAx–PBzMAy copolymers with higher BzMA contents (such as L14-B64) are more kinetically frozen systems. This claim is supported by the delayed and weaker solvation of the PBzMA chains evident from Fig. 3c. Therefore, the mechanical properties of these concentrated worm-based dispersions are tuneable in terms of their reversibility and G′ variation with temperature through control of y.
Conclusions
The present study is not the first to investigate PLMAx–PBzMAy nano-objects. However, it is the first to successfully use ATRP combined with post-polymerisation transfer to prepare PLMAx–PBzMAy spheres, worms, and vesicles at high concentrations (20% w/w). Uniquely, our approach decoupled copolymer synthesis and assembly for this family of copolymers. Furthermore, we examined the temperature-responsive behaviour of the spheres for the first time. Variable-temperature 1H NMR spectra for L14-B34 and L14-B64 showed that reversible solvation of the PBzMA occurred with increasing temperature. Temperature-triggered worm breakdown occurred for L14-B64 as judged by DLS and was poorly reversible. By contrast DLS data also showed that the sphere diameter was not affected by temperature over the range of 20–90 °C and they were considered to be equilibrium systems. Concentrated dispersion studies revealed that only the mixed phase system (L14-B46) showed reversible temperature-triggered gelation behaviour as measured by tube inversion, whilst the pure L14-B64 worms softened (but did not de-gel) at 90 °C. It was proposed that as y increased the morphological transition changed from being largely thermodynamically controlled to having a greater kinetic contribution which is consistent with the core-fusion mechanism. The extent of “livingness” observed appears to depend on the PBzMA block length as well as the nano-object concentration. The ability to use ATRP and post-polymerisation transfer to prepare temperature-responsive, colour-free, gel-forming non-aqueous dispersions should also apply to other diblock copolymer/solvent combinations and may bring forward the application of worm-based systems for next generation lubricants, gels and displays. Whilst this work demonstrates that PISA via RAFT dispersion polymerisation is not a prerequisite for preparing self-assembling PLMA–PBzMA nano-objects the present approach has some disadvantages. These include the requirement for the removal of a toxic catalyst and chlorinated solvent. Also, the copolymers prepared here were less well defined. Nevertheless, the present study adds to the toolbox for constructing future self-assembling temperature-responsive non-aqueous nano-object dispersions.Acknowledgements
We would gratefully acknowledge funding for this research through an EPSRC iCASE grant (Voucher 12220937).References
- N. P. Truong, M. R. Whittaker, A. Anastasaki, D. M. Haddleton, J. F. Quinn and T. P. Davis, Polym. Chem., 2016, 7, 430–440 RSC.
- L. Bes, S. Angot, A. Limer and D. M. Haddleton, Macromolecules, 2003, 36, 2493–2499 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- Y. C. Chen, K. Zhang, X. J. Wang, F. W. Zhang, J. H. Zhu, J. W. Mays, K. L. Wooley and D. J. Pochan, Macromolecules, 2015, 48, 5621–5631 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- F. L. Baines, N. C. Billingham and S. P. Armes, Macromolecules, 1996, 29, 3416–3420 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- C. J. Mable, K. L. Thompson, M. J. Derry, O. O. Mykhaylyk, B. P. Binks and S. P. Armes, Macromolecules, 2016, 49, 7897–7907 CrossRef CAS PubMed.
- N. J. W. Penfold, Y. Ning, P. Verstraete, J. Smets and S. P. Armes, Chem. Sci., 2016, 7, 6894–6904 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- J. Tan, C. Huang, D. Liu, X. Zhang, Y. Bai and L. Zhang, ACS Macro Lett., 2016, 5, 894–899 CrossRef CAS.
- L. Xiao, Y. Chen and K. Zhang, Macromolecules, 2016, 49, 4452–4461 CrossRef CAS.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jerome, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- S. Boisse, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M. H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- J. Rosselgong, A. Blanazs, P. Chambon, M. Williams, M. Semsarilar, J. Madsen, G. Battaglia and S. P. Armes, ACS Macro Lett., 2012, 1, 1041–1045 CrossRef CAS.
- L. Sun, A. Pitto-Barry, A. W. Thomas, M. Inam, K. Doncom, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2016, 7, 2337–2341 RSC.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 128–139 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Polym. Chem., 2014, 5, 3466–3475 RSC.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- V. K. Singh, K. Pal, D. K. Pradhan and K. Pramanik, J. Appl. Polym. Sci., 2013, 130, 1503–1515 CrossRef CAS.
- S. Sahoo, N. Kumar, C. Bhattacharya, S. S. Sagiri, K. Jain, K. Pal, S. S. Ray and B. Nayak, Des. Monomers Polym., 2011, 14, 95–108 CrossRef CAS.
- M. E. Morales, V. Gallardo, B. Clares, M. B. Garcia and M. A. Ruiz, J. Cosmet. Sci., 2009, 60, 627–636 CAS.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS PubMed.
- Z. C. Li, Y. Z. Liang, G. Q. Chen and F. M. Li, Macromol. Rapid Commun., 2000, 21, 375–380 CrossRef CAS.
- W. L. Zhang, J. L. He, Z. Liu, P. H. Ni and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1079–1091 CrossRef CAS.
- N. Karanikolopoulos, M. Zamurovic, M. Pitsikalis and N. Hadjichristidis, Biomacromolecules, 2010, 11, 430–438 CrossRef CAS PubMed.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- J. Wang, P. Gao, L. Ye, A. Y. Zhang and Z. G. Feng, Polym. Chem., 2011, 2, 931–940 RSC.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- S. Banerjee, T. Maji and T. K. Mandal, Colloid Polym. Sci., 2014, 292, 2217–2226 CAS.
- H. Yu, A. Shishido, T. Iyoda and T. Ikeda, Macromol. Rapid Commun., 2007, 28, 927–931 CrossRef CAS.
- E. B. Zhulina, M. Adam, I. LaRue, S. S. Sheiko and M. Rubinstein, Macromolecules, 2005, 38, 5330–5351 CrossRef CAS.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Langmuir, 2006, 22, 9409–9417 CrossRef CAS PubMed.
- V. Raghunadh, D. Baskaran and S. Sivaram, Polymer, 2004, 45, 3149–3155 CrossRef CAS.
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Chem., 1986, A23, 839–852 CrossRef CAS.
- D. P. Chatterjee and B. M. Mandal, Macromolecules, 2006, 39, 9192–9200 CrossRef CAS.
- W. J. Xu, X. L. Zhu, Z. P. Cheng and J. Y. Chen, J. Appl. Polym. Sci., 2003, 90, 1117–1125 CrossRef CAS.
- C. H. Peng, J. Kong, F. Seeliger and K. Matyjaszewski, Macromolecules, 2011, 44, 7546–7557 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- K. Matyjaszewski, D. A. Shipp, J. L. Wang, T. Grimaud and T. E. Patten, Macromolecules, 1998, 31, 6836–6840 CrossRef CAS.
- S. Munirasu, J. Ruhe and R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2848–2861 CrossRef CAS.
- Q. B. Li, Y. Y. Bao, H. Wang, F. F. Du, Q. Li, B. K. Jin and R. K. Bai, Polym. Chem., 2013, 4, 2891–2897 RSC.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 1525–1568 RSC.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- L. Wang, Y. Wang, H. Miao and D. Chen, Soft Matter, 2016, 12, 4891–4895 RSC.
- J. S. Trent, J. I. Scheinbeim and P. R. Couchman, Macromolecules, 1983, 16, 589–598 CrossRef CAS.
- P. P. Soo, B. Huang, Y.-I. Jang, Y.-M. Chiang, D. R. Sadoway and A. M. Mayes, J. Electrochem. Soc., 1999, 146, 32–37 CrossRef CAS.
- M. Williams, N. J. W. Penfold, J. R. Lovett, N. J. Warren, C. W. I. Douglas, N. Doroshenko, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2016, 7, 3864–3873 RSC.
- Y. Pei, O. R. Sugita, L. Thurairajah and A. B. Lowe, RSC Adv., 2015, 5, 17636–17646 RSC.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- L. R. G. Treloar, The physics of rubber elasticity, 3rd edn, Clarendon, Oxford, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm02656g |
| This journal is © The Royal Society of Chemistry 2017 |