
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect demonstration of lipid phosphorylation in the lipid bilayer of the biomimetic bicontinuous cubic phase using the confined enzyme lipid A phosphoethanolamine transferase†
Leonie
van ’t Hag
 abc,
Anandhi
Anandan
d,
Shane A.
Seabrook
c,
Sally L.
Gras
abe,
Calum J.
Drummond
cf,
Alice
Vrielink
*d and
Charlotte E.
Conn
*f
abc,
Anandhi
Anandan
d,
Shane A.
Seabrook
c,
Sally L.
Gras
abe,
Calum J.
Drummond
cf,
Alice
Vrielink
*d and
Charlotte E.
Conn
*f
aDepartment of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, Victoria 3010, Australia
bBio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, Victoria 3010, Australia
cCSIRO Manufacturing, Clayton, Victoria 3168, Australia
dSchool of Chemistry and Biochemistry, University of Western Australia, Crawley, Western Australia 6009, Australia. E-mail: alice.vrielink@uwa.edu.au
eThe ARC Dairy Innovation Hub, The University of Melbourne, Parkville, Victoria 3010, Australia
fSchool of Science, College of Science, Engineering and Health, RMIT University, Melbourne, Victoria 3001, Australia. E-mail: charlotte.conn@rmit.edu.au
First published on 19th January 2017
Abstract
Retention of amphiphilic protein activity within the lipid bilayer membrane of the nanostructured biomimetic bicontinuous cubic phase is crucial for applications utilizing these hybrid protein–lipid self-assembly materials, such as in meso membrane protein crystallization and drug delivery. Previous work, mainly on soluble and membrane-associated enzymes, has shown that enzyme activity may be modified when immobilized, including membrane bound enzymes. The effect on activity may be even greater for amphiphilic enzymes with a large hydrophilic domain, such as the Neisserial enzyme lipid A phosphoethanolamine transferase (EptA). Encapsulation within the biomimetic but non-endogenous lipid bilayer membrane environment may modify the enzyme conformation, while confinement of the large hydrophilic domain with the nanoscale water channels of a continuous lipid bilayer structure may prevent full function of this enzyme. Herein we show that NmEptA remains active despite encapsulation within a nanostructured bicontinuous cubic phase. Full transfer of the phosphoethanolamine (PEA) group from a 1,2-dioleoyl-glycero-phosphoethanolamine (DOPE) doped lipid to monoolein (MO), which makes up the bicontinuous cubic phase, is shown. The reaction was found to be non-specific to the alkyl chain identity. The observed rate of enzyme activity is similar to other membrane bound enzymes, with complete transfer of the PEA group occurring in vitro, under the conditions studied, over a 24 hour timescale.
Introduction
Lipidic self-assembly materials, consisting of surfactant bilayer structures that mimic the cell membrane environment, can be used to encapsulate amphiphilic proteins and peptides.1 The inverse bicontinuous cubic phases (QII) are particularly suited for this purpose, due to their continuous three-dimensional bilayer structure and amphiphilic nature, which can incorporate both the hydrophobic and hydrophilic domains of proteins.2,3 For this reason, the cubic phase nanostructure may assist in the retention of protein activity following the encapsulation of soluble, peripheral and integral membrane proteins.1 Additionally, the high surface area to volume ratio of these structures allows incorporation of relatively large amounts of protein or peptide. The resulting self-assembling hybrid protein–lipid materials are increasingly used in applications such as in meso crystallization of membrane proteins,4 as drug delivery vehicles for amphiphilic and hydrophobic therapeutics and imaging agents,5–9 as biosensors10 and biofuel cells.11The relationship between the lipid nanomaterial and the encapsulated protein must be understood in order to fully develop and apply hybrid protein–lipid self-assembly materials. Retention of the bilayer membrane based cubic nanostructure is essential to the use of these materials in select applications, such as in meso crystallization and drug delivery. A number of recent studies have therefore investigated changes to the lipidic nanostructure caused by the incorporation of amphiphilic proteins and peptides within the bicontinuous cubic phases.1,12–17 Hydrophobic mismatch, hydrophilic domain size and charge were found to be the most important factors affecting the nanostructure. In addition, retention of the activity of the protein within the lipidic material is essential if the protein is to be used in its functionally active form.
While spectroscopic techniques that investigate the conformation of the proteins have been used as an indirect measure for protein activity,13,15,18,19 direct measurements of the activity of amphiphilic proteins within the bicontinuous cubic phase remain challenging. The enzyme activity of soluble oxidases in the nanoscale water channels of the cubic phase has been reported, for example for glucose oxidase,10,20,21 ceruloplasmin,20 lactate oxidase,21 urease,21 creatinine deiminase,21 cholesterol oxidase,10 horseradish peroxidase22,23 and laccase.11,24 Increased stability and activity was found for the mostly hydrophilic membrane bound enzyme fructose dehydrogenase when immobilized within the cubic phase compared to when present in buffer solution.25 Ion transport across the lipid membrane by the purely hydrophobic proteins and peptides OmpF,16 EcClC,26 and a synthetic four-helix bundle27 has also been reported. Ligand binding assays were recently used to show that the dopamine D2L receptor retained a conformation that preserves the ligand binding region when encapsulated in some bicontinuous cubic phases.28 However, to our knowledge the activity of a large amphiphilic protein, which has amphiphilic donor and receiver substrates, when encapsulated in the bicontinuous cubic phase has not yet been reported. These systems differ in that the hydrophilic domain must be confined within the nanoscale aqueous channel network of the cubic phase. Retention of protein activity is particularly important for these proteins, such as the large, non-rigid, hydrophilic domains from G-Protein Coupled Receptors (GPCRs) that are commonly removed or modified for successful crystal growth using in meso crystallization.29 These proteins are also of critical importance, as 60% of all pharmaceutical compounds currently on the market target GPCRs.30 The size of the hydrophilic domain is therefore also of great interest for applications of the bicontinuous cubic phase.
In this study the activity of the amphiphilic enzyme lipid A phosphoethanolamine transferase from Neisseria meningitidis (NmEptA) within the nanostructured bicontinuous cubic phase of monoolein (MO) is investigated. MO is the most commonly used bicontinuous cubic phase forming lipid31 and is readily available at low cost. It forms a robust diamond cubic QDII phase at room temperature and in excess water, Fig. 1(A), with a lattice parameter of ∼104 Å. The typical lipid bilayer thickness within the MO cubic phase is ∼32 Å, with water channels of ∼51 Å in diameter.32,33 Branched chain lipids such as phytantriol have a significantly higher lateral bilayer pressure profile, which in a recent study was found to be unfavorable for the encapsulation of transmembrane peptides and proteins.34,35
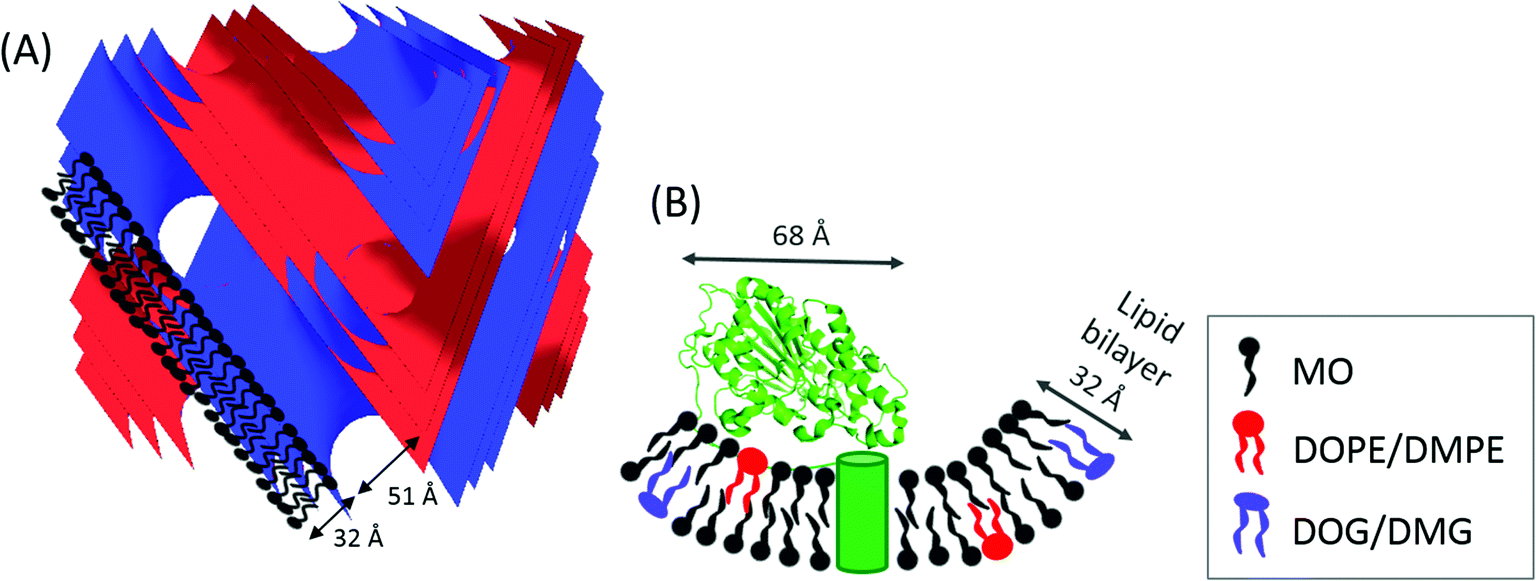 | ||
| Fig. 1 (A) Schematic representation of self-assembled diamond cubic QDII phase, with typical dimensions for lipid bilayer thickness and water channel diameter indicated for monoolein (MO). The blue and red surfaces indicate the QDII phase structure adopted by the amphiphile. (B) Lipid A PEA transferase protein structure (PDB ID: 4KAV and information about the location of the transmembrane domain from A. Anandan, G. L. Evans, K. Condic-Jurkic, M. L. O'Mara, C. M. John, N. J. Phillips, G. A. Jarvis, S. S. Wills, K. A. Stubs, I. Moraes, I. Moraes, C. M. Kahler and A. Vrielink, PNAS, 2017, in press) and the corresponding dimensions determined from the crystal structure.36 The transmembrane α-helices are indicated by the green cylinder in a lipid bilayer formed by MO doped with DOPE/DMPE, and with the formation of DOG/DMG. | ||
Lipid A phosphoethanolamine transferase (EptA) (62.2 kDa) from the Gram-negative bacteria Neisseria meningitides and N. gonorrhoeae is a cytoplasmic membrane localized enzyme consisting of five transmembrane α-helices and a large hydrophilic domain ∼45 × 45 × 68 Å in size, Fig. 1(B). EptA modifies the lipid A headgroups of lipopolysaccharides (bacterial endotoxin molecules), with phosphoethanolamine (PEA).37 This modification of the bacterial endotoxin prevents the binding of negatively charged antibiotics, such as polymyxin and naturally occurring cationic antimicrobial peptides (CAMPs), such as defensins, to the cell membrane. Additionally, this modification confers resistance to both the innate immune clearance mechanisms during bacterial colonization and the treatment of invasive infections with polymyxin.38 An improved understanding of the activity and catalytic mechanism of EptA can lead to the development of novel treatments for Gram-negative bacterial infections such as gonorrhea. A crystal structure of the soluble domain of NmEptA showed components of the enzyme active site, Fig. 1(B), but suggested that the full substrate binding site would require the transmembrane domain,36 thus emphasizing the need to characterize the full length amphiphilic protein. A crystal structure of the full length NmEptA protein has since been obtained from detergent micelles by Anandan et al. (PNAS, in press). This structure shows the enzyme in a compact conformation with a single molecule of n-dodecyl β-D-maltoside (DDM) bound to the proposed substrate binding site, leading to the hypothesis that this structure mimics the phosphatidylethanolamine substrate bound conformation. Molecular dynamics simulations and intrinsic fluorescence studies also indicate that the protein undergoes conformational changes, possibly to accommodate the binding of the two different types of lipid substrates: the small donor substrate, phosphatidylethanolamine, and the larger receiver substrate, lipid A containing lipooligosaccharide. This conformational flexibility may alter the total length of the enzyme from that observed in the crystal structure.
In vivo NmEptA transfers the PEA group from PEA containing lipids such as 1,2-dimystroyl-glycero-phosphoethanolamine (DMPE) in the inner- and outer membrane of the bacteria and attaches this molecule to lipid A as described above. In this study, the activity of NmEptA in the nanostructured cubic phase of MO was studied in the presence of the substrate DMPE, or 1,2-dioleoyl-glycero-phosphoethanolamine (DOPE). DMPE and DOPE were compared to investigate the specificity of the enzyme to phospholipids with saturated alkyl chains (DMPE), as commonly found in Gram-negative bacteria, or with unsaturated alkyl chains (DOPE), which are more miscible with the unsaturated alkyl chains of MO.39 The chemical structures and molecular weights of the lipids used are shown in Table 1. In the first part of the paper, the conversion of DMPE to 1,2-dimyristoyl-glycerol (DMG) and of DOPE to 1,2-dioleoyl-glycerol (DOG) by NmEptA in the bulk cubic phase of MO was investigated using reversed phase – high performance liquid chromatography (RP-HPLC) and electrospray ionization – time-of-flight (ESI-TOF) mass spectroscopy (MS). We also used high-throughput techniques, including high-throughput sample preparation and high-throughput synchrotron small-angle X-ray scattering (SAXS), to investigate the additional effect of NmEptA protein concentration and the concentration of either DMPE or DOPE.
| Lipid | Chemical structure | M w (g mol−1) | M w + 18 (g mol−1) | Phase + lp(A) |
|---|---|---|---|---|
| Monoolein (MO) |

|
356.5 | 374.5 | QDII 104 |
| Monoolein phosphoethanolamine (MOPE) |

|
479.7 | ||
| Dioleoyl-glycerol (DOG) |

|
621.0 | 639.0 | Fl |
| Dioleoyl-phosphoethanolamine (DOPE) |

|
744.0 | HII 74 | |
| Dipalmitoyl-glycerol (DMG) |

|
512.8 | 530.8 | Lα 42 |
| Dipalmitoyl-phosphoethanolamine (DMPE) |

|
635.9 | Lα 51 |
Experimental section
Materials
MO, Hepes, NaCl, n-dodecyl β-D-maltoside (DDM) were purchased from Sigma-Aldrich (St Louis, MO, USA). DOPE, DMPE, DOG and DMG were obtained from Avanti polar lipids (Alabaster, AL, USA). Ethanol and chloroform were bought from Merck & Co, Inc. (Whitehouse Station, NJ, USA). Methanol was purchased from Fischer Scientific Pty Ltd (Scoresby, VIC, Australia). SD-2 96-well crystallization plates were purchased from IDEX Corp (Rohnert Park, CA, USA).NmEptA expression and purification
Recombinant NmEptA was expressed and purified according to the procedures detailed in Anandan et al. (PNAS, in press). Briefly, recombinant C terminal His6-tagged NmEptA was expressed in E. coli strain (BL21(DE3) pLysS). Cells were harvested by centrifugation (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 290 × g for 1 hour at 4 °C) and lysed in 50 mM Na phosphate pH 7.5 containing 300 mM NaCl and 1 mM PMSF using an Emulsiflex C5 high-pressure homogenizer (Avestin, Mannheim, Germany). The membranes were isolated by ultracentrifugation of the lysate at 185511 × g for an hour at 4 °C. NmEptA was solubilized from the membranes by incubating the isolated membranes with 1% w/v DDM in 50 mM Na phosphate pH 7.5 containing 300 mM NaCl and 1 mM PMSF for 4 hours. The solubilized NmEptA was separated from the membrane debris by ultracentrifugation at 185511 × g for an hour at 4 °C.
290 × g for 1 hour at 4 °C) and lysed in 50 mM Na phosphate pH 7.5 containing 300 mM NaCl and 1 mM PMSF using an Emulsiflex C5 high-pressure homogenizer (Avestin, Mannheim, Germany). The membranes were isolated by ultracentrifugation of the lysate at 185511 × g for an hour at 4 °C. NmEptA was solubilized from the membranes by incubating the isolated membranes with 1% w/v DDM in 50 mM Na phosphate pH 7.5 containing 300 mM NaCl and 1 mM PMSF for 4 hours. The solubilized NmEptA was separated from the membrane debris by ultracentrifugation at 185511 × g for an hour at 4 °C.
Solubilized NmEptA was purified by metal chelating chromatography and size exclusion chromatography. The supernatant from ultracentrifugation was applied to a HisTrap crude extract chelating affinity column (GE Healthcare, Buckinghamshire, UK) equilibrated with binding buffer (50 mM sodium phosphate pH 7.5, 300 mM sodium chloride, 20 mM imidazole and 0.023% w/v DDM (3 × CMC)) using an ÅKTApurifier FPLC system (GE Healthcare). Bound NmEptA was eluted using the elution buffer (50 mM sodium phosphate pH 7.5, 300 mM sodium chloride, 500 mM imidazole, 0.023% w/v DDM). The eluted protein was buffer exchanged into 50 mM sodium phosphate pH 7.5, 150 mM sodium chloride and 0.023% w/v DDM using a PD-10 desalting column (GE Healthcare) and applied to a size exclusion chromatography column (Superdex 200 10/30, GE Healthcare) equilibrated with 50 mM Hepes pH 7.0, 100 mM NaCl and 0.023% w/v DDM. Fractions containing NmEptA eluted from the size exclusion chromatography column were pooled and concentrated using a 100 kDa molecular weight cutoff centrifugal filter unit (Vivaspin 20 MWCO 100000, GE Healthcare) to 18 mg mL−1, as determined by the absorbance at 280 nm using a calculated molar extinction coefficient for NmEptA (73980 M−1 cm−1).
Sample preparation for NmEptA assay
MO with 10% w/w DOPE was first co-dissolved in ethanol, and MO with 10% w/w DMPE in methanol/chloroform/water (65:35:8 v/v). The use of different solvents for sample preparation with DOPE and DMPE was necessary due to the limited solubility of DMPE in ethanol. A significant amount of chloroform had to be used to solubilize DMPE but chloroform is not compatible with subsequent sample treatment and analysis procedures. Most of the respective solvent was evaporated under a stream of N2 and the lipid mixture was then freeze dried. The cubic phase was prepared in coupled Hamilton syringes (Hamilton, Reno, NV, USA) at 40% w/w hydration,18 starting with the lipid mixture in one syringe and NmEptA at 18 mg mL−1 in buffer/DDM or just buffer/DDM solution in the second syringe. A volume of 25 μL of cubic phase was then transferred into a microfuge tube and centrifuged for 2 minutes at 14500 rpm (Eppendorf, Ontario, Canada) to obtain a smooth surface. An additional 25–50 μL of buffer solution was added to ensure the samples remained hydrated. After the appropriate reaction time, the excess water was removed and the MO/DOPE/NmEptA samples were dissolved in 500 μL of 70% v/v ethanol and the MO/DMPE/NmEptA samples were dissolved in 100 μL methanol/chloroform/ water (65:35:8 v/v). The MO/DMPE/NmEptA samples were then diluted 500 times in 70% v/v ethanol to reduce the amount of chloroform in the sample. The NmEptA protein was removed from the samples using a Vivaspin 500 centrifuge tube with 10000 MWCO from Satorius AG (Goettingen, Germany). The resulting lipid solutions at 30 mg mL−1 for MO/DOPE and 0.3 mg mL−1 for MO/DMPE in 70% v/v ethanol were used for composition analysis.
Lipid composition analysis NmEptA assay
The lipid composition in the NmEptA assay samples was determined after 1, 12 and 24 hours for separate samples using mass spectroscopy (MS) and reversed-phase – high performance liquid chromatography (RP-HPLC). MS was performed using an Agilent 6200 Series ESI-TOF spectrometer from Agilent Technologies (Santa Clara, CA, USA). RP-HPLC was carried out using an Agilent 1200 series HPLC system equipped with a quaternary pump (G1311A) and diode array (DAD)/multiple wavelength detector (G1365B). Lipid separation was performed using a reversed phase ProteCol C18 HQ203 column (SGE, Ringwood, VIC, Australia) with 200 Å pore size, 100 mm × 2.1 mm. The linear gradient was 70–100% methanol (30–0% water) in 30 minutes and 100% methanol from 30 to 45 minutes, with a flow rate of 0.25 mL min−1. Data of a 2,2,2-trifluoroethanol sample was subtracted from the spectra to correct for the baseline. Fractions for ESI-TOF analysis were collected every one minute, in a Corning 96-well plate (Corning, NY, USA).Plate setup for high-throughput NmEptA assay
Samples for the high-throughput NmEptA assay using small-angle X-ray scattering (SAXS) analysis were prepared in 96-well SD-2 crystallization plates.40 MO, DOPE and DOG stock solutions were prepared in ethanol at concentrations of 400 mg mL−1, 50 mg mL−1 and 50 mg mL−1 respectively. A DMG solution was prepared at 50 mg mL−1 in ethanol/methanol/chloroform (70:15:15 v/v). DMPE was dissolved at 25 mg mL−1 in methanol/chloroform (50:50 v/v) for plates without NmEptA and at 50 mg mL−1 in ethanol for plates with NmEptA. A volume of (diluted) lipid solutions was dispensed with the Mosquito robot (TTP Labtech, Cambridge, MA, USA) into the top and bottom subwells of the plates to obtain the required amount of each lipid. In every well 0.21 mg MO with up to 0.03 mg additional DOPE, DOG, DMPE, or DMG was deposited. The largest volume of lipid stock solution was always dispensed last. DMPE was poorly soluble and dispensed as an emulsion. Plates were dried under vacuum for 5–7 days to remove solvent. A volume of 20 μL of water or buffer (50 mM Hepes (pH 7.0), 100 mM NaCl) was dispensed into the reservoirs using the Phoenix robot (Arts Robins Instruments, Sunnyvale, CA, USA). A volume of 0.56 μL of water, buffer, buffer/DDM or NmEptA solution was dispensed on top of the dried lipid films using the Mosquito robot, resulting in 70–73% w/w (excess) hydration in the wells. Samples with NmEptA were measured after 2, 12 and 20 hours equilibration and SAXS patterns were taken at different positions in the well for each time point (top left, top right and bottom within the 1.5 mm diameter well, respectively) to minimize the effect of radiation damage.
Small-angle X-ray scattering (SAXS) measurements
Structural analysis of the cubic phase samples in SD-2 crystallization plates and sandwich plates with Kapton tape was performed at the SAXS/WAXS beamline at the Australian Synchrotron.41 Data acquisition was performed at 25 °C using a custom-designed, temperature-controlled plate holder. An X-ray wavelength of 1.0332 Å (12.0 keV) was used with a typical flux of 1013 photons s−1, 0.2 s exposure time and beam dimensions of ∼200 μm × 300 μm. Recording SAXS patterns for all wells in one SD-2 crystallization plate (128 wells) took ∼20 minutes, which is significantly faster than the time scales of the enzymatic reaction, which was observed to occur over a time frame of 12–24 hours but not before two hours. A Pilatus 1 M detector was used to record two-dimensional diffraction images. 1-D scattering profiles were obtained by azimuthally averaging the 2-D SAXS images. In the 1-D scattering profiles many orders of diffraction were observed, which allowed unambiguous identification of the bicontinuous cubic, inverse hexagonal and lamellar phases. The IDL-based AXCESS software package and the high-throughput analysis program RapidPhaseIdent were used for data analysis.42,43Results and discussion
NmEptA assay in the bulk bicontinuous cubic phase of MO with DOPE and DMPE
The bulk bicontinuous cubic phase was formed by the lipid MO with the second lipid (DOPE or DMPE) doped within the lipid bilayer at a concentration of 10% w/w. The amphiphilic NmEptA protein was then incorporated within the nanostructured cubic phase, with the transmembrane α-helices embedded in the lipid bilayer and the hydrophilic domain confined within the nanoscale water channels, as shown in Fig. 1. The substrate binding site of the enzyme is expected to be at the interface between the hydrophobic and hydrophilic domains, which allows the PEA moiety to be captured from lipids such as DOPE and DMPE found within the lipid bilayer. We first studied the transfer of the PEA moiety from DOPE and DMPE by NmEptA in the bulk bicontinuous cubic phase of MO using lipid composition analysis by MS and RP-HPLC. RP-HPLC was used to quantify the lipids present, with identification of the lipid fractions confirmed by MS. Mixtures of pure MO, DOPE, and DOG (49:25:26 w/w) and MO, DMPE, and DMG (47:27:26 w/w) in 70% v/v ethanol (in the absence of NmEptA) were initially analyzed to develop these MS and RP-HPLC methodologies.
Three lipid species can be clearly distinguished in the ESI-TOF spectra shown for the pure lipid mixtures in Fig. 2(A) and 4(A). Peaks for DOPE at m/z = 744 and DMPE at m/z = 636 dominate the spectra, because of the presence of a zwitterion in the PEA moiety: such charged species are analyzed more readily than non-charged species in ESI-TOF mass spectroscopy. The m/z values observed correlate well with the molecular weight of these lipids, Table 1. In contrast, peaks for the uncharged DOG and DMG at m/z = 639 and m/z = 531 were of much lower intensity, even though they were present at similar concentration in the samples. The m/z values observed were ∼18 mass units higher than the molecular weight of the lipids, Table 1. We suggest that these are the analyte adducts of DOG and DMG with ammonium (NH4+), as is commonly observed in positive ion ESI,†44 as applied here. For MO, peaks with m/z = 357 and m/z = 374 were observed for both lipid mixtures. This again correlates to the molecular weight of MO, as well as its ammonium adduct, Table 1. These data with the pure lipid mixtures confirm that ESI-TOF MS analysis can be used to identify the lipids present in these types of samples.
 | ||
| Fig. 2 Masses detected in the NmEptA assay with DOPE by ESI-TOF MS: (A) MO, DOPE, DOG, (B) MO, DOPE, buffer/DDM after 24 hours and MO, DOPE and NmEptA samples after (C) 1 hour, (D) 12 hours and (E) 24 hours. | ||
Note that RP-HPLC analysis was only performed for MO, DOPE, DOG samples and not for MO, DMPE, DMG samples due to the limited solubility of DMPE in ethanol and methanol. The large amount of chloroform needed for solubilization of DMPE prevented loading of sufficient lipid in 70% v/v ethanol or methanol in water onto the column for accurate separation and detection. The MO, DOPE, and DOG lipids were detected with the diode array detector (DAD) signal at 203 nm, similar to previous reports that phospholipids are detected readily at ∼206 nm.45 RP-HPLC fractions were collected every minute for lipid identification by ESI-TOF mass spectroscopy.
MO was found to elute after ∼22 minutes, which corresponds to ∼92% methanol, Fig. 3(A). The MO peak was found to have a small shoulder at ∼21 minutes, which is probably due to the presence of the isomeric 2-monoolein in the sample with >90% 1-monoolein. DOPE and DOG eluted after ∼36 and ∼38 minutes, respectively, which corresponds to 100% methanol, reflecting the different affinity of these two compounds with the reversed-phase C18-column. The ESI-TOF MS data for the relevant fractions is shown in ESI,† Fig. S1.1. The sequence of elution of MO, DOPE, and DOG is as expected, due to the increased hydrophobicity of these molecules. The relative areas of the peaks for MO, DOPE and DOG (65:14:21) were found to represent the relative amounts of the different lipids in the sample (65:15:20 mol mol−1) well, confirming this method can be used for quantification of the relative amount of each of the components present.
 | ||
| Fig. 3 RP-HPLC data with diode array detector (DAD) signal at 203 nm of NmEptA assay with DOPE. (A) MO, DOPE, DOG. (B–D) MO, DOPE, NmEptA (red solid lines) and buffer/DDM (dashed grey lines) after (B) 1 hour, (C) 12 hours, (D) 24 hours. (A–C) Were measured on the same HPLC system and (D) was analyzed on a different HPLC instrument. The slight differences in peak position and shape between (A–C) compared to (D) are ascribed to difference in HPLC systems. | ||
In the presence of 18 mg mL−1NmEptA ∼10% of the DOPE present was dephosphorylated to DOG after 1 hour (peak areas MO:DOPE:DOG 92:7:1), as shown by the MS and RP-HPLC data in Fig. 2 and 3 (B–E and B–D respectively) confirming that NmEptA is bioactive in the nanostructured cubic phase. After a further period of 12 hours approximately half of the DOPE molecules were converted to DOG (peak areas MO:DOPE:DOG 92:5:3) and after 24 hours ∼90% of the DOPE was converted to DOG (peak areas MO:DOPE:DOG 95:0.5:4.5) illustrative of the time dependence of conversion. The data also confirm that the dephosphorylation activity of this enzyme is not specific to saturated phospholipids that are native in the cell membrane of Gram-negative bacteria and the enzyme is also able to dephosphorylate DOPE.
MS analysis of the excess buffer of the bulk cubic phase samples did not detect the PEA moiety removed from the DOPE lipids during the enzymatic reaction and we propose that the PEA moiety (mass ∼123 g mol−1) is attached to an OH group of MO. We have termed this lipid MOPE, and it has a mass of ∼480 g mol−1 (Table 1). The RP-HPLC data contains a peak at a retention time of ∼18–19 minutes (∼78% methanol), that increases in intensity with reaction time (Fig. 3). The peak corresponds to m/z = 480 and the appearance of this peak can also be seen in the ESI-TOF MS data of the samples after 1, 12 and 24 hours (Fig. 2). Additional ESI-TOF MS data for the relevant fractions of the RP-HPLC result from the sample with MO, DOPE, and NmEptA after 24 hours is shown in ESI,† Fig. S1.2. In the absence of lipid A, we propose that the PEA moiety was attached to MO, confirming the lack of specificity of accepting lipids for this reaction and illustrating that the acceptor is not restricted to lipids that are present in the cell membrane of the Gram-negative bacteria.
For NmEptA samples incubated with DMPE, a small amount of MOPE was formed after 1 hour, as shown by the ESI-TOF MS data in Fig. 4(B–E) indicating that the production of MOPE reaction product was independent of the alkyl chain identity of the substrate, consistent with our suggestion that the PEA moiety has been attached to MO. In this case, the relative intensity of MOPE increased at 12 hours and 24 hours, while the relative intensity of DMPE decreased. The dephosphorylated DMG was observed after 12 hours but not after 24 hours, possibly due to the low sensitivity of the ESI-TOF MS analysis with uncharged compounds.
 | ||
| Fig. 4 Masses detected in the NmEptA assay with DMPE by ESI-TOF: (A) MO, DMPE, DMG, (B) MO, DMPE, buffer/DDM after 24 hours, and MO, DMPE and NmEptA samples after (C) 1 hour, (D) 12 hours, (E) 24 hours. | ||
In summary, the NmEptA assay showed that the enzyme was active in the bulk cubic phase of MO, even though the enzyme is expected to be confined due to the geometrical mismatch of the hydrophilic domain of NmEptA (∼45 × 68 Å) in the water channels of the cubic phase (Dw = 51 Å), Fig. 1. After 24 hours the PEA moiety was predominantly attached to MO rather than DOPE or DMPE. The reverse dephosphorylation reaction of MOPE by NmEptA might also occur but this cannot be concluded from this experiment. The rate of transfer of the PEA group by NmEptA is of the same order of magnitude as enzymatic reaction rates of other membrane bound enzymes,25,46 with full transformation occurring over a 24 hour time period: the overall conversion reaction rate is ∼1 μM s−1. This is surprising considering the suggested limited flexibility of the NmEptA enzyme to form different conformations within the nanoscale cubic phase water channels. Within the cubic phase disordered nanopockets to accommodate the NmEptA enzyme might have formed.47,48 The diffusion rate of lipids within the lipid bilayer (the substrate) is typically an order of magnitude slower than soluble components in the water channels,49 which is also expected to affect the reaction rate. It was found that the enzyme was not specific to native saturated phospholipids, as the PEA moiety of the singly unsaturated lipid DOPE could be transferred to MO with an efficiency similar to the saturated DMPE lipid. Additionally, in the absence of lipid A the PEA moiety was transferred onto the cubic phase forming lipid MO to form MOPE.
High-throughput NmEptA assay: effect of enzyme and substrate concentration
To investigate the effect of NmEptA and substrate (PEA) concentration, a high-throughput assay was performed with ∼100 times smaller samples (∼0.3 mg) than for the MS and RP-HPLC analysis. High-throughput analysis of enzyme activity is also of prospective use as a screening tool for inhibitors, which could lead to novel treatments of diseases such as gonorrhea and could be applied more readily as an experimental setup for enzyme screening in the cubic phase. Samples were prepared using robotics and the analysis was performed by investigating changes in the nanostructure of the cubic phase using SAXS at room temperature and in excess (∼70% w/w) hydration. Samples were prepared in duplicate in the top and bottom subwells of 96-well plates. Reproducibility between the subwells was confirmed, as shown in Supplementary Information 2 (ESI†), with data found to be reproducible (lattice parameters less than 5 Å different) in ∼87% of wells. Irreproducible wells, which were more likely for samples with high NmEptA concentration presumably due to the significant impact of the enzyme on the cubic phase nanostructure, were not considered further.Under these conditions, the lattice parameter of the MO diamond cubic QDII phase with water was found to be 105.7 Å, in agreement with the literature.32,33NmEptA was purified in 50 mM Hepes (pH 7.0) and 100 mM NaCl with 0.023% w/v DDM to solubilize the hydrophobic transmembrane domain. For this reason, the lattice parameter of the diamond cubic QDII phase of MO was also investigated with excess buffer, and buffer with DDM as a background for comparison. The lattice parameter with 50 mM Hepes (pH 7.0) and 100 mM NaCl was 104.0 Å, and with buffer containing 0.023% w/v DDM at 104.4 Å. This reduction in lattice parameter compared to the value with water can be explained by the presence of 100 mM of NaCl, since NaCl is a kosmotropic salt. Kosmotropic salts can cause a reduction of the water activity,43,50 resulting in a slightly less hydrated cubic phase. High concentrations of the detergent DDM are known to cause an increase in lattice parameters of the MO cubic phase,51 but this was not observed here. The Hepes and NaCl components of the buffer were found to dominate the effect of the buffered detergent solution, probably due to the low concentration of DDM in the sample (0.023% w/v). Together these data show the changes in the lipid system due to the buffer components are small and enable subsequent application of the high-throughput system.
 | ||
| Fig. 5 Lattice parameter in angstrom of the MO diamond cubic QDII phase (obtained from SAXS analysis) as a function of (A) NmEptA concentration in buffer/DDM as determined after 12 hours using the high-throughput setup. (B) DOPE and DOG concentration in water, buffer and buffer/DDM. In some wells the inverse hexagonal HII phase was observed, as indicated in the graph (lattice parameters ∼60 Å). (C) DMPE and DMG concentration in water, buffer and buffer/DDM. (B and C) Were analyzed after 24 hours using the same high-throughput setup. | ||
SAXS results for MO with 0–10% w/w DOPE and 0–18 mg mL−1NmEptA show an increase in lattice parameter with increasing NmEptA concentration for all DOPE concentrations (Fig. 6), providing an indication of a change in the mass fraction of substrate (DOPE) and product (DOG) and progress of the NmEptA enzymatic reaction. The magnitude of this increase was reduced, however, in the presence of DOPE as shown in Fig. 6(A). i.e. the presence of a higher concentration of substrate (DOPE) leads to higher NmEptA enzyme activity, leading to a higher concentration of product (DOG) and a decrease in the lattice parameter, as DOG reduces MO lattice parameters more significantly than DOPE. This reduction in lattice parameter at higher DOPE concentrations is consistent with NmEptA being active and forming DOG by 12 hours.
 | ||
| Fig. 6 SAXS results for high-throughput NmEptA assay with DOPE. In (A and B) the lattice parameters in angstrom for the MO diamond cubic QDII phase (obtained from SAXS analysis) are shown as a function of NmEptA concentration with different concentrations of DOPE (% w/w). Data in (A) are after 12 hours and data in (B) are after 20 hours. In (C) the slope of linear fit results of the data in (A) are shown as a function of DOPE and DOG concentration: fits of the data in (A) all had R2 > 0.92. A linear fit through the data is shown in (C) and this linear decrease is statistically significant: P-value = 0.007 (<0.05) as determined using regression analysis in Excel. | ||
A linear fit to the slope of all reproducible data as shown in Fig. 6(A) shows the substrate dependence of the change in lattice parameters and reaction progress and strongly suggests dephosphorylation of DOPE occurred with formation of DOG after 12 hours in the high-throughput assay with samples of ∼0.5 μL (Fig. 6(C), all fits of the data in (A) with coefficient of determination or R2 > 0.92), as shown in Fig. 6(C). The effect of forming MOPE on the cubic phase nanostructure is unknown but is expected to be minor. The effect of an additive lipid on phase behavior depends on the concentration of the lipid and its average “shape” as determined by the CPP. The addition of the PEA moiety to the MO lipid should not change the CPP for this lipid significantly. In addition, as the concentration of MOPE is low (<10% w/w), the effect of this lipid on MO phase behavior is expected to be minor.
Results after 20 hours are shown in Fig. 6(B); the QDII lattice parameters at high NmEptA concentrations were significantly reduced compared to the results after 12 hours. Small pockets of disorder to accommodate the NmEptA enzyme may have formed within the cubic phase,47,48 allowing the bulk cubic phase to adapt lattice parameters closer to values without the NmEptA protein. It is also possible that the protein conformation changed within these small samples and after storage at room temperature for one day.
Results obtained under identical conditions after 2 hours were found to be irreproducible, consistent with a previous observation that a time period of several hours is required for full reconstitution of the protein into the cubic phase.28
SAXS results for MO with 0–10% w/w DMPE and 0–18 mg mL−1NmEptA show an increase in lattice parameter with increasing NmEptA concentration for all DMPE concentrations (Fig. 7), again indicating a change in the mass fraction of substrate (this time DMPE) and product (DOG) and progress of the NmEptA enzymatic reaction. The magnitude of this increase scales with increasing concentration of DMPE, consistent with the formation of the enzymatic product DMG, which reduces the MO lattice parameters (note that DMPE causes an increase). A linear fit through all reproducible data points as shown for each of the DMPE concentrations in Fig. 7(A), with R2 > 0.94, shows a substrate dependence for reaction progress and the lattice parameters and again strongly suggests that DMPE was dephosphorylated forming DMG by 12 hours (Fig. 7(C)). The linear decrease from 0 to 10% w/w DMPE/DMG may not be statistically significant in this case and the reduction in slope was much greater for DOPE, as could be expected. The limited solubility of DMPE and DMG in the cubic phase of MO is consistent with the observation that trends with concentrations of 6% w/w DMPE and higher are less significant (data from 0 to 4% w/w DMPE/DMG do show a statistically significant linear decrease) than with DOPE. This is expected to be due to the limited solubility of the saturated C14 alkyl chains of DMPE and DMG with the singly unsaturated C18 alkyl chains of MO. In contrast, DOPE and DOG have the same alkyl chains as MO, and are therefore expected to be more soluble within the MO bilayer.
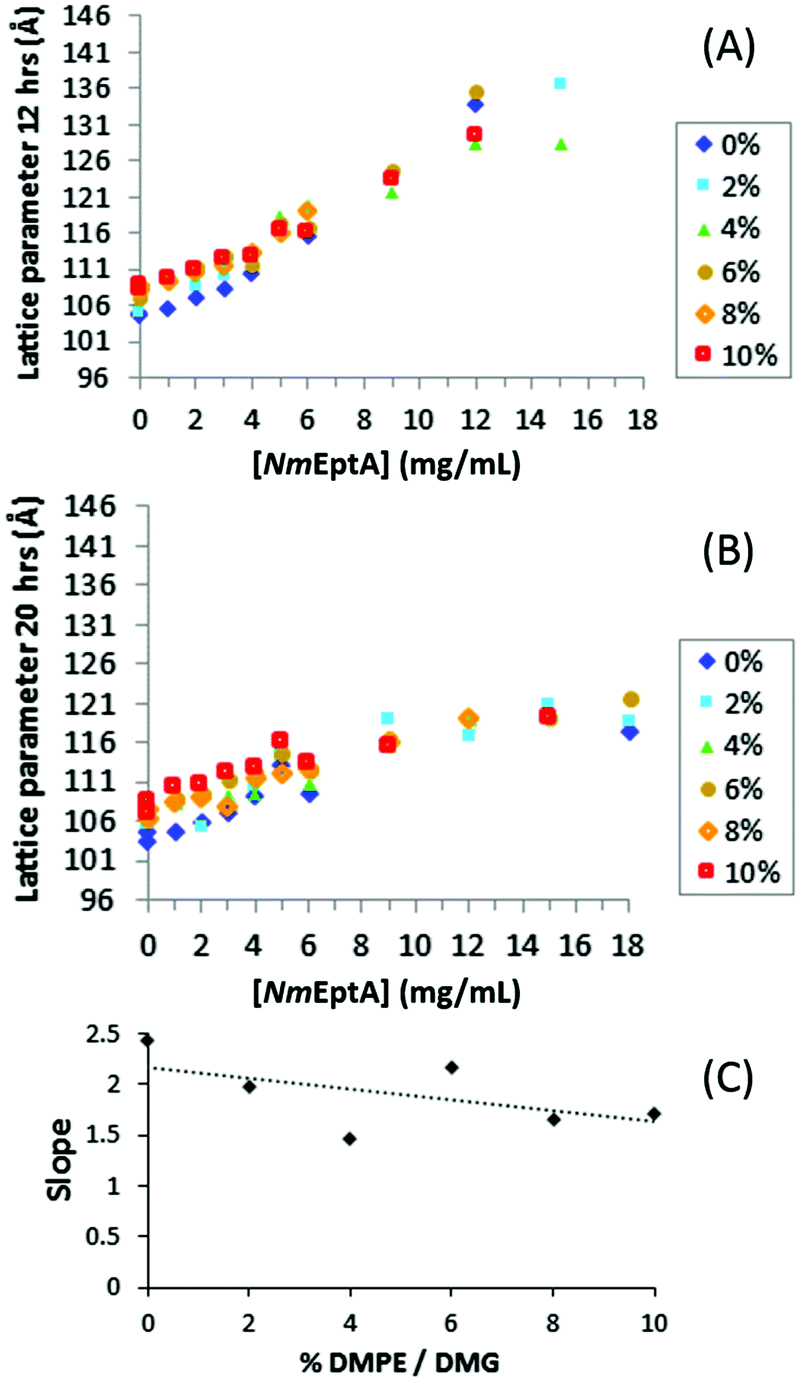 | ||
| Fig. 7 SAXS results for the high-throughput NmEptA assay with DMPE. In (A and B) the lattice parameters in angstrom for the MO diamond cubic QDII phase (obtained from SAXS analysis) are shown as a function of NmEptA concentration with different concentrations of DMPE (% w/w). Data in (A) are after 12 hours and data in (B) are after 20 hours. In (C) the slope of linear fit results of data in (A) are shown as a function of DMPE and DMG concentration: fits of the data in (A) all had R2 > 0.94. A linear fit through the data is shown in (C) as a guide for the eye, but the linear decrease is not statistically significant: P-value = 0.25 (>0.05) as determined using regression analysis in Excel. This is explained by the limited solubility of DMPE and DMG in the cubic phase of MO at more than 6% w/w; the linear decrease from 0 to 4% w/w DMPE/DMG is statistically significant (P-value = 0.02). | ||
Results with DMPE after 20 hours are shown in Fig. 7(B), and the QDII lattice parameters at high NmEptA concentrations were again significantly reduced compared to the results after 12 hours, consistent with the results with DOPE. The results after 2 hours were again irreproducible confirming a time frame of more than 2 hours was needed for the samples to equilibrate.
The high-throughput NmEptA assay results confirmed conversion of a significant amount of the DOPE and DMPE to DOG and DMG respectively after 12 hours in samples that were ∼100 times smaller than in the initial bulk phase NmEptA assay. Enzyme conversion rates might differ depending on the sample size. At the lowest NmEptA concentrations studied enzyme activity could already be detected. At the highest NmEptA concentration used in this study, 18 mg mL−1, no saturation of enzyme activity was observed. This shows that high-throughput methods can be used to screen a wide range of NmEptA enzyme concentrations. For DOPE and DMPE we can observe the dephosphorylation at all PEA concentrations, even though at low concentrations the diffusion of the protein and the substrate within the lipid bilayer is slow. Detection of enzyme activity in 96-well plates using SAXS reinforces the prospective nature of high-throughput sample preparation and analysis as a screening tool for drug discovery and in biosensors.
Conclusions
In this study, the enzyme activity of the large amphiphilic NmEptA enzyme upon encapsulation in the nanostructured bicontinuous cubic phase was studied using ESI-TOF MS and HPLC analysis, as well as high-throughput techniques including SAXS analysis. We showed that the NmEptA enzyme was active when encapsulated in the cubic phase. Transfer of the PEA group from DOPE or DMPE to MO, the cubic phase forming lipid, was shown. These data demonstrate that the reaction of NmEptA is not specific to the alkyl chain identity of the donor or receiver substrate. In vivo the donor substrate is typically a phospholipid, such as DMPE, and the receiver substrate the lipid A containing lipooligosaccharide. SAXS analysis additionally showed that NmEptA caused a swelling of the diamond cubic QDII phase, shown by a ∼25% increase in lattice parameter, due to the presence of the large hydrophilic domain in the nanoscale water channels. At the lowest NmEptA concentrations used, enzyme activity could be detected. At the highest NmEptA concentrations used in this study enzyme activity was still observed, potentially due to the relatively large surface area to volume ratio of the nanostructured bicontinuous cubic phase. This shows that high-throughput methods can be used for both high and low NmEptA concentrations, reinforcing the prospect of using high-throughput sample preparation and analysis as a drug screening tool and in biosensors. The observed rate of enzyme activity was comparable to other membrane bound enzymes, with complete transfer of the PEA group occurring in vitro, under the conditions studied, over a 24 hour timescale. This is surprising considering the large hydrophilic domain must be confined within the nanoscale water channels of the cubic phase, and disordered nanopockets to accommodate the NmEptA enzyme might have formed.Acknowledgements
We thank Drs Adrian M. Hawley, Stephen T. Mudie, Nigel M. Kirby and Timothy M. Ryan for their assistance with SAXS experiments, and we acknowledge use of the SAXS/WAXS beamline at the Australian Synchrotron. We thank Dr Janet Newman for help with the robotic setup of plates at the C3 Collaborative Crystallization Centre, CSIRO, Parkville, Australia. We acknowledge Dr Ching-Seng Ang for help with RP-HPLC and ESI-TOF MS measurements. Ms Leonie van ‘t Hag is supported by a Melbourne International Research Scholarship and a CSIRO PhD scholarship. Ms Anandhi Anandan acknowledges support through a Fay Gale Fellowship from the University of Western Australia. A/Prof. Sally L. Gras is supported by the ARC Dairy Innovation Hub IH120100005. Funding support to Prof. Alice Vrielink from the National Health and Medical Research Council of Australia is acknowledged (APP1003697 and APP1078642). Dr Charlotte Conn is the recipient of an ARC DECRA Fellowship DE160101281.References
- C. E. Conn and C. J. Drummond, Soft Matter, 2013, 9, 3449–3464 RSC.
- V. Luzzati, A. Tardieu, T. Gulikkrz, E. Rivas and F. Reisshus, Nature, 1968, 220, 485–488 CrossRef CAS PubMed.
- C. Fong, T. Le and C. J. Drummond, Chem. Soc. Rev., 2012, 41, 1297–1322 RSC.
- E. M. Landau and J. P. Rosenbusch, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14532–14535 CrossRef CAS.
- X. Mulet, B. J. Boyd and C. J. Drummond, J. Colloid Interface Sci., 2013, 393, 1–20 CrossRef CAS PubMed.
- A. Zabara and R. Mezzenga, J. Controlled Release, 2014, 188, 31–43 CrossRef CAS PubMed.
- A. Angelova, B. Angelov, V. M. Garamus, P. Couvreur and S. Lesieur, J. Phys. Chem. Lett., 2012, 3, 445–457 CrossRef CAS PubMed.
- S. M. Sagnella, X. Gong, M. J. Moghaddam, C. E. Conn, K. Kimpton, L. J. Waddington, I. Krodkiewska and C. J. Drummond, Nanoscale, 2011, 3, 919–924 RSC.
- J. Zhai, J. A. Scoble, N. Li, G. Lovrecz, L. J. Waddington, N. Tran, B. W. Muir, G. Coia, N. Kirby, C. J. Drummond and X. Mulet, Nanoscale, 2015, 7, 2905–2913 RSC.
- J. J. Vallooran, S. Handschin, S. M. Pillai, B. N. Vetter, S. Rusch, H.-P. Beck and R. Mezzenga, Adv. Funct. Mater., 2016, 26, 181–190 CrossRef CAS.
- E. Nazaruk, S. Smolinski, M. Swatko-Ossor, G. Ginalska, J. Fiedurek, J. Rogalski and R. Bilewicz, J. Power Sources, 2008, 183, 533–538 CrossRef CAS.
- Y. L. Liang, C. E. Conn, C. J. Drummond and C. Darmanin, J. Colloid Interface Sci., 2015, 441, 78–84 CrossRef CAS PubMed.
- L. van't Hag, X. Li, T. G. Meikle, S. V. Hoffmann, N. C. Jones, J. S. Pedersen, A. M. Hawley, S. L. Gras, C. E. Conn and C. J. Drummond, Langmuir, 2016, 32, 6882–6894 CrossRef PubMed.
- L. van't Hag, H.-H. Shen, T.-W. Lin, S. L. Gras, C. J. Drummond and C. E. Conn, Langmuir, 2016, 32, 12442–12452 CrossRef PubMed.
- L. van't Hag, H. H. Shen, J. X. Lu, A. M. Hawley, S. L. Gras, C. J. Drummond and C. E. Conn, Langmuir, 2015, 31, 12025–12034 CrossRef PubMed.
- A. Zabara, R. Negrini, O. Onaca-Fischer and R. Mezzenga, Small, 2013, 9, 3602–3609 CrossRef CAS PubMed.
- B. Angelov, A. Angelova, S. K. Filippov, M. Drechsler, P. Stepanek and S. Lesieur, ACS Nano, 2014, 8, 5216–5226 CrossRef CAS PubMed.
- M. Caffrey and V. Cherezov, Nat. Protoc., 2009, 4, 706–731 CrossRef CAS PubMed.
- A. Misiunas, Z. Talaikyte, G. Niaura, V. Razumas and T. Nylander, Biophys. Chem., 2008, 134, 144–156 CrossRef CAS PubMed.
- T. Nylander, C. Mattisson, V. Razumas, Y. Miezis and B. Hakansson, Colloids Surf., A, 1996, 114, 311–320 CrossRef CAS.
- V. Razumas, J. Kanapieniene, T. Nylander, S. Engstrom and K. Larsson, Anal. Chim. Acta, 1994, 289, 155–162 CrossRef CAS.
- W. Sun, J. J. Vallooran, A. Zabara and R. Mezzenga, Nanoscale, 2014, 6, 6853–6859 RSC.
- W. J. Sun, J. J. Vallooran and R. Mezzenga, Langmuir, 2015, 31, 4558–4565 CrossRef CAS PubMed.
- E. Nazaruk, R. Bilewicz, G. Lindblom and B. Lindholm-Sethson, Anal. Bioanal. Chem., 2008, 391, 1569–1578 CrossRef CAS PubMed.
- W. J. Sun, J. J. Vallooran, W. K. Fong and R. Mezzenga, J. Phys. Chem. Lett., 2016, 7, 1507–1512 CrossRef CAS PubMed.
- C. Speziale, L. Salvati Manni, C. Manatschal, E. M. Landau and R. Mezzenga, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7491–7496 CrossRef CAS PubMed.
- N. H. Joh, T. Wang, M. P. Bhate, R. Acharya, Y. B. Wu, M. Grabe, M. Hong, G. Grigoryan and W. F. DeGrado, Science, 2014, 346, 1520–1524 CrossRef CAS PubMed.
- C. Darmanin, S. Sarkar, L. Castelli and C. E. Conn, Cryst. Growth Des., 2016, 16, 5014–5022 CAS.
- C. G. Tate and G. F. X. Schertler, Curr. Opin. Struct. Biol., 2009, 19, 386–395 CrossRef CAS PubMed.
- A. Wise, K. Gearing and S. Rees, Drug Discovery Today, 2002, 7, 235–246 CrossRef CAS PubMed.
- C. V. Kulkarni, W. Wachter, G. Iglesias-Salto, S. Engelskirchen and S. Ahualli, Phys. Chem. Chem. Phys., 2011, 13, 3004–3021 RSC.
- J. Briggs, H. Chung and M. Caffrey, J. Phys. II, 1996, 6, 723–751 CrossRef CAS.
- H. Qiu and M. Caffrey, Biomaterials, 2000, 21, 223–234 CrossRef CAS PubMed.
- T. G. Meikle, C. E. Conn, F. Separovic and C. J. Drummond, RSC Adv., 2016, 6, 68685–68694 RSC.
- L. van‘t Hag, L. de Campo, C. J. Garvey, G. C. Feast, A. E. Leung, N. R. Yepuri, R. Knott, T. L. Greaves, N. Tran, S. L. Gras, C. J. Drummond and C. E. Conn, J. Phys. Chem. Lett., 2016, 7, 2862–2866 CrossRef PubMed.
- C. Wanty, A. Anandan, S. Piek, J. Walshe, J. Ganguly, R. W. Carlson, K. A. Stubbs, C. M. Kahler and A. Vrielink, J. Mol. Biol., 2013, 425, 3389–3402 CrossRef CAS PubMed.
- P. Sperandeo, G. Deho and A. Polissi, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2009, 1791, 594–602 CrossRef CAS PubMed.
- C. M. John, M. F. Liu, N. J. Phillips, Z. J. Yang, C. R. Funk, L. I. Zimmerman, J. M. Griffiss, D. C. Stein and G. A. Jarvis, Infect. Immun., 2012, 80, 4014–4026 CrossRef CAS PubMed.
- V. Cherezov, J. Clogston, Y. Misquitta, W. Abdel-Gawad and M. Caffrey, Biophys. J., 2002, 83, 3393–3407 CrossRef CAS PubMed.
- C. Darmanin, C. E. Conn, J. Newman, X. Mulet, S. A. Seabrook, Y. L. Liang, A. Hawley, N. Kirby, J. N. Varghese and C. J. Drummond, ACS Comb. Sci., 2012, 14, 247–252 CrossRef CAS PubMed.
- N. M. Kirby, S. T. Mudie, A. M. Hawley, D. J. Cookson, H. D. T. Mertens, N. Cowieson and V. Samardzic-Boban, J. Appl. Crystallogr., 2013, 46, 1670–1680 CrossRef CAS.
- J. M. Seddon, A. M. Squires, C. E. Conn, O. Ces, A. J. Heron, X. Mulet, G. C. Shearman and R. H. Templer, Philos. Trans. R. Soc., A, 2006, 364, 2635–2655 CrossRef CAS PubMed.
- L. van't Hag, C. Darmanin, T. C. Le, S. Mudie, C. E. Conn and C. J. Drummond, Cryst. Growth Des., 2014, 14, 1771–1781 Search PubMed.
- N. B. Cech and C. G. Enke, Mass Spectrom. Rev., 2001, 20, 362–387 CrossRef CAS PubMed.
- P. Juaneda and G. Rocquelin, Lipids, 1986, 21, 239–240 CrossRef CAS PubMed.
- S. J. Ullrich, U. A. Hellmich, S. Ullrich and C. Glaubitz, Nat. Chem. Biol., 2011, 7, 263–270 CrossRef CAS PubMed.
- A. Angelova, B. Angelov, R. Mutafchieva, S. Lesieur and P. Couvreur, Acc. Chem. Res., 2011, 44, 147–156 CrossRef CAS PubMed.
- A. Angelova, M. Ollivon, A. Campitelli and C. Bourgaux, Langmuir, 2003, 19, 6928–6935 CrossRef CAS.
- P. O. Eriksson and G. Lindblom, Biophys. J., 1993, 64, 129–136 CrossRef CAS PubMed.
- D. F. Parsons, M. Bostrom, P. Lo Nostro and B. W. Ninham, Phys. Chem. Chem. Phys., 2011, 13, 12352–12367 RSC.
- X. Ai and M. Caffrey, Biophys. J., 2000, 79, 394–405 CrossRef CAS PubMed.
- C. E. Conn, C. Darmanin, S. M. Sagnella, X. Mulet, T. L. Greaves, J. N. Varghese and C. J. Drummond, Soft Matter, 2010, 6, 4828–4837 RSC.
- R. H. Templer, K. H. Madan, N. A. Warrender and J. M. Seddon, in The Structure and Conformation of Amphiphilic Membranes: Proceedings of the International Workshop on Amphiphilic Membranes, Jülich, Germany, September 16–18, 1991, ed. R. Lipowsky, D. Richter and K. Kremer, Springer Berlin Heidelberg, Berlin, Heidelberg, 1992, pp. 262–265, DOI:10.1007/978-3-642-84763-9_51.
Footnote |
| † Electronic supplementary information (ESI) available: (1) ESI-TOF MS data of RP-HPLC fractions as shown in Fig. 3(A and D). (2) Figures showing reproducibility of the SAXS results as shown in Fig. 5–7. (3) High-throughput data with DMPE, presented in individual graphs for every % w/w DMPE. See DOI: 10.1039/c6sm02487d |
| This journal is © The Royal Society of Chemistry 2017 |