
Colloidal synthesis of iridium-iron nanoparticles for electrocatalytic oxygen evolution†
Luhong
Fu
a,
Ping
Cai
*a,
Gongzhen
Cheng
a and
Wei
Luo
 *ab
*ab
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, China. E-mail: applecaiping@163.com; wluo@whu.edu.cn; Tel: +86-27-68752366
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China
First published on 17th May 2017
Abstract
The design of high-performance electrocatalysts for oxygen evolution reactions (OER) in acidic condition is highly desirable for the development of the proton exchange membrane (PEM)-based water electrolyzer. Herein, we report the colloidal synthesis of iridium-iron alloy nanoparticles with an average diameter of 2.4 nm. The as-synthesized IrFe alloy produces a current density of 10 mA cm−2 in acidic and alkaline conditions at overpotentials of 278 and 286 mV, respectively.
Introduction
Hydrogen has been regarded as one of the most promising energy carriers for practical alternative energy sources.1,2 Most hydrogen is obtained from the reforming of natural gas, which releases a large amount of CO2, thus restricting its extensive application.3,4 In recent years, electrochemical water splitting into hydrogen and oxygen has been well known to be an alternative carbon-free strategy.5–8 However, efficient electrochemical overall water splitting is severely hindered by the sluggish kinetics of oxygen evolution reaction (OER) at the anode.9–12 Currently, the expensive and scarce IrO2 and RuO2 are still considered as the state-of-the-art OER catalysts.13–16 Therefore, a great deal of effort has been made to design non-precious-metal-based OER catalysts that are efficient and stable.17 However, the catalytic performance of these catalysts is still below the benchmarks of RuO2 and IrO2.18–20 Adding insult to injury, most studies on these non-precious-metal-based OER catalysts are carried out in alkaline electrolyzers, which hinders their possible use in proton exchange membrane (PEM)-based water electrolyzers.21–24 Therefore, in view of practical application, the development of highly efficient and long-term stable OER catalyst in strongly acidic condition is highly desirable, but still a great challenge.It is well known that bimetallic nanoparticles (NPs) can promote the catalytic activity and stability of the corresponding monometallic NPs.25–27 It is reasonable to understand that alloying may lead to modification of the catalyst surface, probably due to the synergistic effect derived from two metals.28 Moreover, the Sabatier principle claims that catalytic activity could be controlled by the surface of the catalysts.29 Thus, it is expected that alloying Ir with 3d transition metals might boost their activity and stability, as well as reduce the cost. However, to the best of our knowledge, there are only few reports about the controlled synthesis of IrNi and IrCu nanostructures.30,31 Despite significant progress having been achieved, the acidic OER performance of these catalysts is still restricted by their low activity and stability.32–34 Herein, for the first time, we report the colloidal synthesis of Ir–Fe alloy NPs with diameters of approximately 2.4 nm. Compared to single Ir NPs, the IrFe alloy nanocatalysts exhibit much higher OER activity with exceptional overpotentials of 278 and 286 mV at a current density of 10 mA cm−2 in acidic and alkaline conditions, respectively. To the best of our knowledge, the catalytic performance of the as-synthesized IrFe nanocatalysts is better than that of most reported OER catalysts in both acidic and alkaline conditions.
Experimental
Chemicals and materials
Iridium chloride [IrCl3, 99%], iron pentacarbonyl [Fe(CO)5, >99.9%], sodium oleate, oleylamine (OAm, >70%), 1-octadecene (ODE, 90%), hexane (98.5%), isopropanol (99.5%), ethanol, acetic acid (99%), and Nafion solution (5%) were purchased from Sigma-Aldrich. Carbon black (Vulcan, XC-72R) was used as support.Physical characterizations
The morphologies and sizes of the samples were observed by using a Tecnai G20 U-Twin transmission electron microscope (TEM) equipped with an energy-dispersive X-ray detector (EDX) at an acceleration voltage of 200 kV. Powder X-ray diffraction (XRD) patterns were measured by a Bruker D8-Advance X-ray diffractometer using Cu Kα radiation source (λ = 0.154178 nm) with a velocity of 10° min−1. X-ray photoelectron spectroscopy (XPS) measurement was performed with a Thermo Fischer ESCALAB 250Xi spectrophotometer. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was performed on IRIS Intrepid II XSP.Synthesis of Ir–Fe alloy NPs
Under gentle nitrogen (N2) flow and magnetic stirring, 0.3 g of sodium oleate and 4 mL of ODE were mixed and heated to 160 °C to make a clear solution. The solution was cooled down to room temperature, and 0.1 mmol of IrCl3 dissolved in 4 mL of OAm was added. Under a blanket of N2, the solution was re-heated to 110 °C. Then, 0.1 mmol of Fe(CO)5 was injected into the solution. The solution was heated to 280 °C at a heating rate of 4–5 °C min−1 and kept at this temperature for 2 h. The reaction mixture was cooled to room temperature, and 20 mL of hexane and 20 mL of ethanol were added to precipitate the product. The product was separated by centrifugation at 9500 rpm for 8 min. The as-prepared NPs were dispersed in hexane for further use.Catalyst preparation
5 mg of NPs and 20 mg of XC-72 carbon support were mixed in 30 mL of hexane and sonicated for 1 h. The catalyst was separated by centrifugation, suspended in 30 mL of acetic acid, and heated at 70 °C for 10 h to remove the surfactant.35 Then, the product was separated by centrifugation, washed with water three times, and dried under vacuum for 2 h.Preparation of working electrodes
The NP/C catalyst was suspended in a mixture solvent containing isopropanol and 5% Nafion (v/v = 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to form a 5 mg mL−1 suspension. 18 μL catalyst ink was deposited on the surface of a glassy carbon (GC) electrode that was first polished with 1.0, 0.5 and 0.05 μm gamma alumina powders and rinsed with ultrapure water and ethanol. Then, the catalyst was dried at ambient condition.
:1) to form a 5 mg mL−1 suspension. 18 μL catalyst ink was deposited on the surface of a glassy carbon (GC) electrode that was first polished with 1.0, 0.5 and 0.05 μm gamma alumina powders and rinsed with ultrapure water and ethanol. Then, the catalyst was dried at ambient condition.
Electrochemical measurements
To test the OER activity of the as-synthesized catalysts, a conventional three-electrode system was used with a CHI 760E electrochemical analyzer (CH Instruments, Chenhua Co., Shanghai, China). Glassy carbon (GC, ϕ = 5 mm) was used as the working electrode, platinum sheet (dimension: 1 cm × 1 cm × 0.02 cm) was selected as the counter electrode, and the reference electrode of this work was the Hg/HgO electrode (MOE) in an alkaline system or the Hg/Hg2Cl2 electrode (SCE) in an acidic system. The temperature was kept at 30 °C using a water bath. All the potentials were translated versus the reversible hydrogen electrode (RHE) by the reference electrode (MOE or SCE) to perform the electrochemical measurements on the glassy carbon rotating disk electrode (RDE).Before the electrochemical measurement, the catalysts were first activated at a scan rate of 500 mV s−1 in argon – saturated alkaline solution or acidic solution. The oxygen evolution reaction (OER) polarization curves were obtained by scanning the potentials from 0.3 to 0.9 V in O2-saturated 1.0 M KOH alkaline solution at 1600 rpm with a scan rate of 5 mV s−1, or by scanning the potentials from 0.4 to 1.5 V in O2-saturated 0.5 M HClO4 solution.
Results and discussion
The Ir–Fe alloy NPs were prepared through thermal decomposition of iron pentacarbonyl [Fe(CO)5] and the reduction of iridium chloride (IrCl3). In this reaction, [Fe(CO)5] was used as a reducer and a Fe precursor for alloying with Ir. The composition of IrFe NPs was controlled by varying the initial molar ratio of IrCl3 and [Fe(CO)5] at 1:1, 3:1, and 1:3. Furthermore, Ir NPs were also synthesized for comparison. The microstructures of the IrFe NPs and Ir NPs were characterized by transmission electron microscopy (TEM). As shown in Fig. 1 and S1,† the IrFe NPs have a narrow size distribution with a mean particle size of ∼2.4 nm, which is smaller than that of Ir NPs (∼2.8 nm) (Fig. S1†). Fig. 1f shows the powder X-ray diffraction (XRD) patterns of the as-synthesized catalysts. Ir NPs show two prominent diffraction peaks at 40.7° and 69.1°, corresponding to the (111) and (220) reflections of Ir (JCPDS no. 46-1044), suggesting the successful reduction of the Ir ions. After the introduction of [Fe(CO)5], the primary diffraction peak at 40.7° shifts positively, and the second diffraction peak disappears, indicating the formation of Ir–Fe alloy. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) combined with the corresponding EDS mapping (Fig. S2†) further indicated the uniform distribution of Ir and Fe.
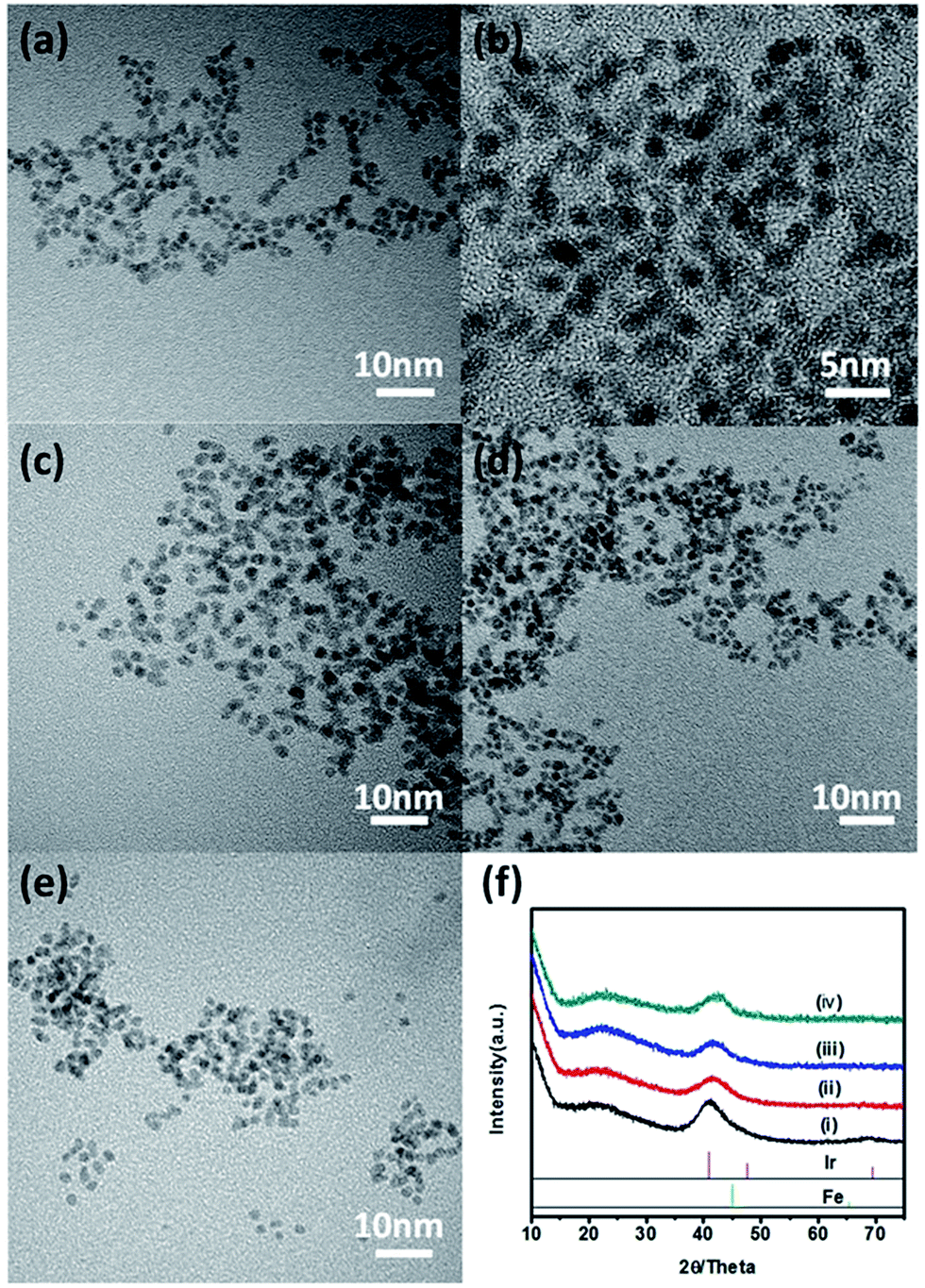 | ||
| Fig. 1 TEM image of as-prepared Ir–Fe alloy nanoparticles with initial molar ratios of the Ir/Fe precursors at (a and b) 1:1, (c) 1:0, (d) 1:3, and (e) 3:1, respectively; (f) XRD patterns for IrFe NPs with initial molar ratios of Ir/Fe precursors at 1:0 (i), 3:1 (ii), 1:1 (iii) and 1:3 (iv), respectively. | ||
The as-synthesized IrFe NPs were studied as catalysts toward OER. During the preparation of the catalyst, we first mixed the hexane dispersion of IrFe NPs and activated carbon (Vulcan XC72R carbon) under sonication to assemble IrFe NPs on carbon, and then the IrFe/C were washed with acetic acid (AA) at 70 °C to remove the surfactant and unstable species on the surface.35 The final compositions of the catalysts were changed from Ir1Fe1, Ir3Fe1, and Ir1Fe3 to Ir1Fe0.41, Ir1Fe0.11, and Ir1Fe1.47, respectively, as confirmed by inductively coupled plasma-atomic emission spectroscopy (ICP-AES, Table S1†). TEM image in Fig. S3† indicates no visible change for the Ir1Fe0.41 NPs, and the energy-dispersive X-ray spectroscopy (EDX) spectrum in Fig. S4† further proves that the atomic ratios of Ir and Fe in the catalysts are in accordance with ICP-AES results.
X-ray photoelectron spectroscopy (XPS) measurement was carried out to further analyze the surface of Ir1Fe0.41. The high-resolution spectra in Fig. 2a show four types of Ir 4f peaks, located at 60.34 eV, 61.26 eV, 63.41 eV and 64.59 eV, which are respectively assigned to Ir(0) 4f7/2, Ir4+ 4f7/2, Ir(0) 4f5/2 and Ir4+ 4f5/2. There are four principal types of Fe 2p peaks in Fig. 2b, among which the binding energies of the mainly double peaks of Fe 2p3/2 and Fe 2p1/2 emerge at 706.56, 709.61 eV, and 720.01, 723.41 eV, respectively. The peak at 706.56 eV represents Fe(0) in Ir–Fe, whereas the peaks at 709.61, 720.01 and 723.41 eV are associated with iron oxide as a result of the surface oxidation of Fe due to exposure to air. In addition to the four main peaks, two different Fe satellite peaks could be found as well. Compared to the standard binding energy, the peaks of Ir(0) 4f in Ir–Fe alloy at 60.34 eV and 63.41 eV both shift negatively about 0.4 eV, indicating that the electronic structure of Ir could be modified by the introduction of iron, resulting in the enhancement of electrochemical activity (vide infra). Furthermore, iron oxides are also conducive to improving the catalytic activity, which have been confirmed in several relevant reports.36,37
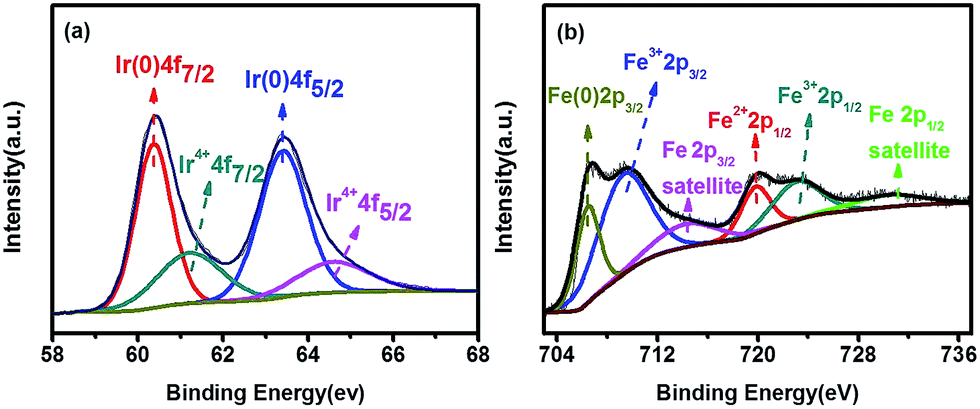 | ||
| Fig. 2 The XPS spectra of (a) Ir 4f and (b) Fe 2p for Ir1Fe0.41. | ||
To evaluate the OER activity of as-synthesized IrFe catalysts, we redispersed them into a mixed solution with isopropanol and 5% Nafion (v/v = 49/1) to reach a concentration of 5 mg mL−1 ink. 18 μL of these dispersions were deposited on the surface of a glassy carbon electrode (loading of 0.092 mg cm−2) and dried under room temperature. First of all, cyclic voltammetry (CV) was used to activate the as-prepared catalysts by repeating 50 cycles with a scan rate of 500 mV s−1 at the range of 1.0–1.5 V (vs. RHE) in Ar-saturated 0.5 M HClO4 solution. Fig. 3a shows the OER polarization curves performed at a scan rate of 5 mV s−1 in O2-saturated 0.5 M HClO4 acidic solution (pH ≈ 0.28). The IrFe alloys exhibit almost the same onset potential of about 1.46 V vs. RHE, which is smaller than that of Ir/C catalyst (1.48 V vs. RHE). Among all the catalysts tested, Ir1Fe0.41/C exhibits the best activity with a low overpotential of 278 mV to reach a current density of 10 mA cm−2, followed by Ir1Fe0.11/C (η10 = 286 mV), Ir1Fe1.47/C (η10 = 293 mV), and Ir/C (η10 = 313 mV). This result highlights the synergistic effect between Ir and Fe at the molecular level. It is worth noting that the OER electrocatalytic performance of Ir1Fe0.41/C is better than most of the other reported catalysts in acidic condition, as shown in Table S2.† Furthermore, as shown in Fig. 3b, the Tafel slope of the Ir1Fe0.41/C is calculated to be 56 mV dec−1, which is lower than that of Ir1Fe0.11/C (62 mV dec−1), Ir1Fe1.47/C (66 mV dec−1), and Ir/C (70 mV dec−1). We further test the stability of the as-synthesized Ir1Fe0.41/C catalysts by a chronopotentiometric method. As shown in Fig. 3c, there is almost no difference in the current density after 500 cycles of CV for Ir1Fe0.41/C. However, Ir/C exhibits obvious overpotential increase during the measurement. Moreover, after 14000 s of continuous operation at a static current density of 10 mA cm−2, the overpotential of Ir/C catalyst increased by 50 mV, while the Ir1Fe0.41/C shows a negligible change (Fig. 3d), suggesting the superior stability of the Ir1Fe0.41/C catalyst in strongly acidic condition.
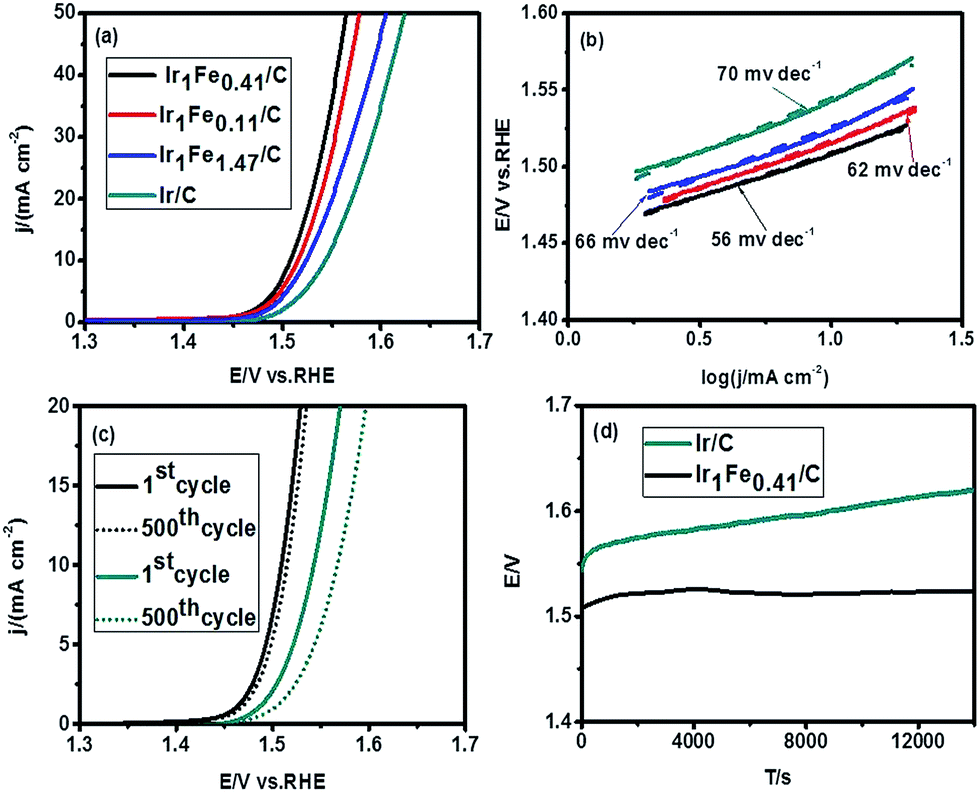 | ||
| Fig. 3 (a) The OER polarization curves performed at a scan rate of 5 mV s−1 for Ir1Fe0.41/C, Ir1Fe0.11/C, Ir1Fe1.47/C and Ir/C in O2-saturated 0.5 M HClO4 acidic solution (pH ≈ 0.28) with a rotating rate of 1600 rpm; (b) the corresponding Tafel slopes of Ir1Fe0.41/C, Ir1Fe0.11/C, Ir1Fe1.47/C, and Ir/C; (c) LSV curves for Ir1Fe0.41/C (black) and Ir/C (dark cyan) with a sweep rate of 100 mV s−1 in 0.5 M HClO4 solution, the solid line is the first cycle and the dotted line is the 500th cycle; (d) the chronopotentiometry test of Ir1Fe0.41/C in contrast to Ir/C in acidic solution at a current density of 10 mA cm−2. | ||
We also examined the OER activities of the as-synthesized IrFe catalysts in 1.0 M KOH solution (pH ≈ 13.9). As shown in Fig. 4a, both Ir1Fe0.41/C and Ir1Fe0.11/C catalysts show the same sharp onset potential of about 1.45 V versus RHE, which is 40 mV smaller than that of Ir/C catalyst. At the current density of 10 mA cm−2, Ir1Fe0.41/C exhibits the highest activity with a low overpotential of 286 mV, which is smaller than that of Ir1Fe0.11/C (η10 = 296 mV), Ir1Fe1.47/C (η10 = 328 mV), Ir/C (η10 = 351 mV), and most of the reported catalysts, as shown in Table S3.†Fig. 4b shows the corresponding Tafel slopes. Among all the catalysts tested, Ir1Fe0.41/C exhibits the smallest Tafel slope of 65 mV dec−1. Fig. 4c shows the corresponding chronopotentiometry test of the as-synthesized Ir1Fe0.41/C catalyst. As shown in Fig. 4c, compared with Ir/C catalyst with poor stability, Ir1Fe0.41/C shows a smaller overpotential change after 500 cycles. The chronopotentiometry test shown in Fig. 4d further confirms the long-term stability of the as-synthesized Ir1Fe0.41/C. The measurement of products is vital to the catalyst, so we measured the faradaic efficiency of O2 production. In order to maximize O2 production, we conducted the experiment on nickel foam, and the results displayed in Fig. S5† indicate that the IrFe alloy catalysts yield nearly 100% faradaic efficiency over the course of 30 min at the current of 100 mA.
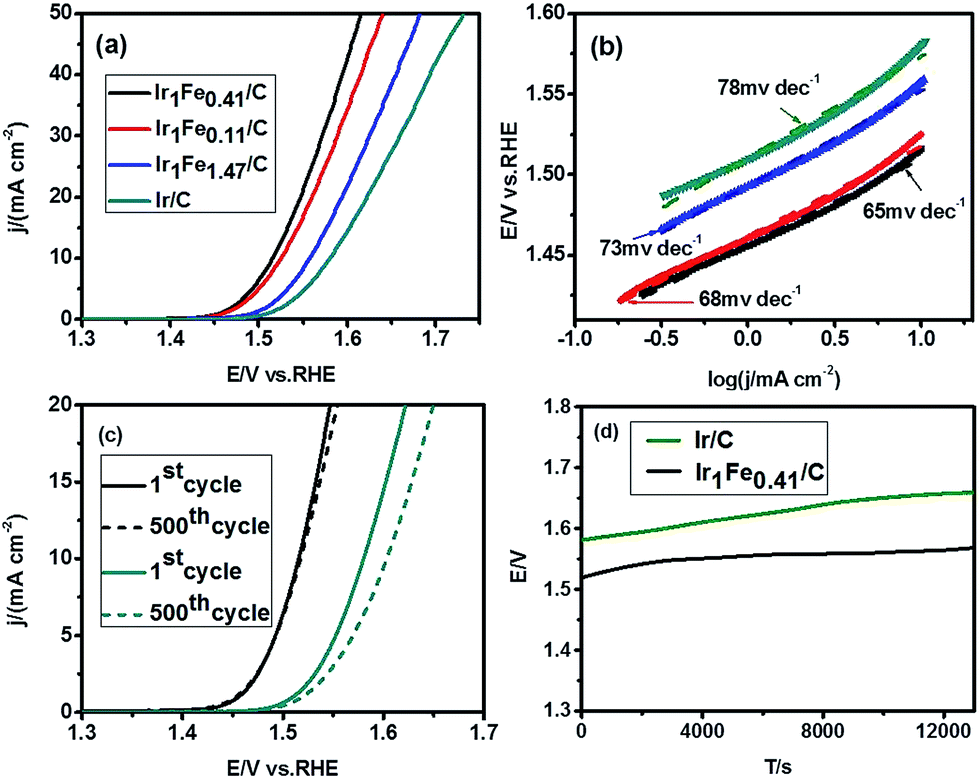 | ||
| Fig. 4 (a) The OER polarization curves performed at a scan rate of 5 mV s−1 for Ir1Fe0.41/C, Ir1Fe0.11/C, Ir1Fe1.47/C and Ir/C in O2-saturated 1.0 M KOH alkaline solution (pH ≈ 13.9) at a rotating rate of 1600 rpm; (b) the corresponding Tafel slopes of Ir1Fe0.41/C, Ir1Fe0.11/C, Ir1Fe1.47/C, and Ir/C; (c) LSV curves for Ir1Fe0.41/C (black) and Ir/C (dark cyan) with a sweep rate of 100 mV s−1 in 1.0 M KOH alkaline solution, the solid line is the first cycle and the dotted line is the 500th cycle; (d) the chronopotentiometry test of Ir1Fe0.41/C in contrast to Ir/C in basic solution at a current density of 10 mA cm−2. | ||
To figure out the real catalyst state, we analyzed the XPS spectra of the Ir–Fe alloy nanoparticles after anodic scan treatments in alkaline solution and acid solution, respectively. It is clear that the nanoparticles were almost oxidized (Fig. S6 and S7†). Compared with the XPS spectra of Ir 4f (a) and Fe 2p (b) for Ir1Fe0.41 in Fig. 2, the metal peaks almost disappeared, while the oxidation peaks became evident, which might be regarded as IrOx and FeO(OH), respectively. Through consulting relevant literature, we found that the peak located at 61.26 eV mainly belongs to Ir4+, while the higher binding energy such as 62.5 eV manifested an oxidation state higher than Ir4+.38,39 In Fig. S6(a) and S7(a),† the oxidation peaks of Ir 4f could be separated into two types through peak-fit processing and were assigned to Ir4+ and Ir>4+, respectively. Several studies have suggested that both Ir4+ and Ir>4+ play an important role in enhancing the OER electrocatalytic process.39–41 In 1.0 M KOH, the IrOx mainly provided the OER catalytic site, and FeO(OH) might also be the OER active site, consistent with previous reports.42–49 In 0.5 M HClO4 condition, only the Ir 4f peaks could be observed, indicating that the catalytic capability of IrFe for OER in acidic solution originates from the IrOx.
Conclusions
In summary, IrFe alloy NPs with diameters of approximately 2.4 nm have been synthesized through a colloidal method. When used as electrochemical catalyst for OER, the as-synthesized Ir1Fe0.41/C exhibits much higher OER activity and stability, with exceptional overpotentials of 278 and 286 mV at a current density of 10 mA cm−2 in acidic and alkaline conditions, respectively. The excellent electrocatalytic activity might be attributed to the excellent IrOx and FeO(OH) active sites and the synergistic effect between Ir and Fe at the molecular level. The extremely high catalytic activity and stability of IrFe alloy NPs toward OER, especially in the acidic condition, is believed to strongly promote the practical application of proton exchange membrane (PEM)-based water electrolyzers.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21571145, 21633008), the Creative Research Groups of Hubei Province (2014CFA007) and Large-Scale Instrument and Equipment Sharing Foundation of Wuhan University.Notes and references
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909 CrossRef CAS PubMed.
- S. Y. Lin, M. Harada, Y. Suzuki and H. Hatano, Fuel, 2002, 81, 2079 CrossRef CAS.
- M. Oertel, J. Schmitz, W. Weirich, D. Jendryssek-Neumann and R. Schulten, Chem. Eng. Technol., 1987, 10, 248 CrossRef.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC.
- M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942 CAS.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- D. H. Yoon, S. G. Bang, J. S. Park, J. C. Kim, H. S. Baik, H. S. Yang and K. Y. Lee, CrystEngComm, 2015, 17, 6843 RSC.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. J. Mayrhofer, ChemCatChem, 2014, 6, 2219 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC.
- H. N. Nong, H. S. Oh, T. Reier, E. Willinger, M. G. Willinger, V. Petkov, D. Teschner and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 2975 CrossRef CAS PubMed.
- A. Abdelhafiz, A. Vitale, C. Joiner, E. Vogel and F. M. Alamgir, ACS Appl. Mater. Interfaces, 2015, 7, 6180 CAS.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102 CrossRef CAS PubMed.
- S. Liu, L. Hu, X. Xu, A. A. Al-Ghamdi and X. Fang, Small, 2015, 11, 4267 CrossRef CAS PubMed.
- D. Jeong, K. Jin, S. E. Jerng, H. Seo, D. Kim, S. H. Nahm, S. H. Kim and K. T. Nam, ACS Catal., 2015, 5, 4624 CrossRef CAS.
- M. T. Zhang, Z. F. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048 CrossRef CAS PubMed.
- H. Wang and H. Dai, Chem. Soc. Rev., 2013, 42, 3088 RSC.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81 CrossRef CAS PubMed.
- D. Yoon, S. Bang, J. Park, J. Kim, H. Baik, H. Yang and K. Lee, CrystEngComm, 2015, 17, 6843 RSC.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlogl, K. J. J. Mayrhofer and P. Strasser, J. Am. Chem. Soc., 2015, 137, 13031 CrossRef CAS PubMed.
- C. Chen, Y. J. Kang, Z. Y. Huo, Z. W. Zhu, W. Y. Huang, P. D. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, J. Catal., 2002, 209, 275 CrossRef.
- J. Lim, S. Yang, C. Kim, C.-W. Roh, Y. Kwon, Y.-T. Kimb and H. Lee, Chem. Commun., 2016, 52, 5641 RSC.
- J. Pei, J. Mao, X. Liang, C. Chen, Q. Peng, D. Wang and Y. Li, Chem. Commun., 2016, 52, 3793 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955 RSC.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765 CrossRef CAS.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588 CrossRef CAS PubMed.
- C. Tang, H.-F. Wang, X.-L. Zhu, B.-Q. Li and Q. Zhang, Part. Part. Syst. Charact., 2016, 33, 473 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744 CrossRef CAS PubMed.
- T. Kwon, H. Hwang, Y. J. Sa, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2017, 27, 1604688 CrossRef.
- H. G. S. Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169 CrossRef PubMed.
- P. Lettenmeier, L. Wang, U. Golla-Schindler, P. Gazdzicki, N. A. CaÇas, M. Handl, R. Hiesgen, S. S. Hosseiny, A. S. Gago and K. A. Friedrich, Angew. Chem., 2016, 128, 752 CrossRef.
- W. Sun, Y. Song, X.-Q. Gong, L. Cao and J. Yang, Chem. Sci., 2015, 6, 4993 RSC.
- M. K. Bates, Q. Jia, H. Doan, W. Liang and S. Mukerjee, ACS Catal., 2016, 6, 155 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305 CrossRef CAS PubMed.
- M. E. G. Lyons and M. P. Brandon, Part III-Iron, Int. J. Electrochem. Sci., 2008, 3, 1463 CAS.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253 CrossRef CAS PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452 CrossRef CAS PubMed.
- T. Grewe, X. Deng and H. Tüysüz, Chem. Mater., 2014, 26, 3162 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00113d |
| This journal is © The Royal Society of Chemistry 2017 |