
Highly crystalline β-FeOOH(Cl) nanorod catalysts doped with transition metals for efficient water oxidation†
Tomiko M.
Suzuki
 *,
Takamasa
Nonaka
,
Akihiko
Suda
,
Noritomo
Suzuki
,
Yoriko
Matsuoka
,
Takeo
Arai
,
Shunsuke
Sato
and
Takeshi
Morikawa
*
*,
Takamasa
Nonaka
,
Akihiko
Suda
,
Noritomo
Suzuki
,
Yoriko
Matsuoka
,
Takeo
Arai
,
Shunsuke
Sato
and
Takeshi
Morikawa
*
Toyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute, Aichi 480-1192, Japan. E-mail: tomiko@mosk.tytlabs.co.jp; morikawa@mosk.tytlabs.co.jp
First published on 3rd March 2017
Abstract
The application of the water oxidation reaction to extract electrons from water molecules is important for the future sustainable synthesis of useful chemicals such as hydrogen and organic compounds. Therefore, a cost-effective oxygen evolution reaction (OER) in alkaline, neutral or acidic solution is required, based on the use of catalysts incorporating earth abundant elements. This work demonstrates that β-FeOOH(Cl) nanorod catalysts with high crystallinity and small size (an average diameter of 3 nm and a length of 15 nm) provided the best performance in the OER activity among Fe-based oxide and (oxy)hydroxide catalysts. The pristine β-FeOOH(Cl) nanorods with the high crystallinity are realized by a new process enabling one-pot, fast, and room temperature synthesis, which is the key to forming colloidal suspensions of the highly crystallized pure-phase β-FeOOH(Cl) nanorods. The versatility of this process also enabled doping of a wide variety of transition metals. Doping of Ni2+ (at 1.2 at%) improved the OER activity and shifted the threshold potential in the negative direction by 100 mV in an alkaline electrolyte, which was comparable to that of conventional IrOx colloidal nanoparticles.
1. Introduction
The water oxidation process referred to as the oxygen evolution reaction (OER) extracts electrons from water molecules and is a critical step in generating useful chemicals, such as hydrogen or organic compounds, as part of the drive toward a sustainable society. There has long been a need for cost-effective OER catalysts for use in alkaline water electrolysis, electrochemical and photoelectrochemical cells and photocatalysis. Currently, the catalyst cost is considered to be not a major factor in the total cost of the proton exchange membrane (PEM) electrolysis system, while in a large scale, earth-abundant catalysts which are only stable under neutral-to-alkaline conditions will be necessary. Various metal oxides, oxyhydroxides, Co phosphates (Co-Pi) and Co borates (Co-Bi) have been studied extensively as a means of connecting the OER with reductive reactions to synthesize various compounds.1–14 Among these, iridium oxide is recognized as a highly active OER catalyst that functions over a broad pH range,1,2,15–19 and IrOx·nH2O nanocolloids have been shown to be useful because catalysts in colloidal solutions can be easily deposited onto a wide variety of substrates to form electrodes.18,20 However, it is more beneficial to develop low-cost systems based on colloidal solutions containing highly active catalysts comprising abundant elements. For this reason, nickel oxyhydroxide OER catalysts such as NiOOH and Fe-doped NiOOH have recently been reported.5–10 Iron, an element with low toxicity and the fourth highest Clark number (4.70), has been preferred as an alternative. Among Fe compounds, FeOOH is known to exist as a natural component in soil and corrosion products of steels and therefore it is attractive. In fact, amorphous and γ-FeOOH have been found to function as OER catalysts.21–23 Choi et al. demonstrated the photo-oxidation of water using BiVO4 coated with γ-FeOOH,24,25 while Mullins et al. reported the photo-oxidation of water by a SiGe-jn in conjunction with amorphous FeOOH.26 Mullins also determined that the reaction overpotential could be reduced by doping with Ni (5–20%).27 Although these previous studies were limited in scope, the results are extremely significant. β-FeOOH(Cl) (akaganeite, hereafter, β-FeOOH) is another iron oxyhydroxide polymorph28 of ferric oxyhydroxides (FeOOH) with 4 double chains of FeO3(OH)3 octahedra, which form a unique tunnel-type nanoporous structure of 0.5 nm × 0.5 nm in size running along [010] (Fig. 1).29 The tunnel sites are partly occupied by Cl− ions, and the negative charge is balanced by adding H+, which forms hydroxyl (OH−) groups with oxygen. The Cl− ions are believed to play an essential role in stabilizing the structure. β-FeOOH has many applications, such as a precursor for α-Fe2O3 (hematite)30 and an electrode material in lithium batteries.31 Despite this, there have been few reports concerning catalysis of the OER with β-FeOOH, although in one study a hierarchically nanostructured β-FeOOH photoanode showed no particular advantages with regard to onset potential and water oxidation rates.32 Studies dating back half a century have established that the hydrolysis of Fe(III) salts at room temperature gives a precipitate composed of amorphous FeOOH or α-FeOOH, while β-FeOOH particles can be obtained by heat treatment of ferric solutions containing Cl− or F− at 60–90 °C for time spans ranging from 5 h to several days.28 Unfortunately, the crystalline β-FeOOH particles resulting from this process are typically 15 to 30 nm in diameter and 200 to 400 nm in length, and these relatively large dimensions lead to difficulty in forming stable colloidal solutions, which is disadvantageous to practical use due to the high electrical resistivity, etc.28,31,33 Recently, Boettcher and his research group speculated that pure FeOxHy was much more active than previously thought, and that the apparent activity was limited by poor electrical conductivity.34 They showed that FeOOH was electrically insulating and thus OER inactive. Although synthesis of β-FeOOH nanorods with a smaller size (∼5 × ∼20 nm) was conducted recently in the field of biotechnology under low pH conditions (<pH 1.5) over 8 days synthesis, the authors explained that the crystallinity was still poor.35 Addition of Fe is known to activate Ni-based OER catalysts, while OER activities of Fe-based catalysts reported to date were much lower.34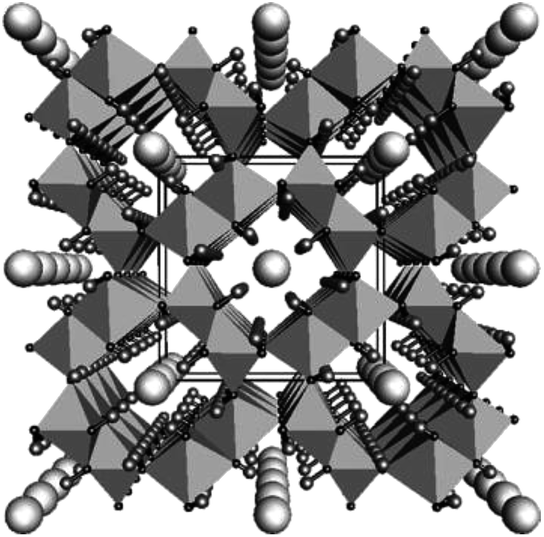 | ||
| Fig. 1 Crystalline structure of β-FeOOH(Cl) (akaganeite) shown viewed down the b-axis. Large spheres in the center of the octahedral tunnels represent chlorides, while small terminal spheres hydrogens. The data are taken from the literature.29 Copyright 2003 Elsevier. | ||
Herein, we present the first ever report of β-FeOOH nanorod catalysts with excellent OER activity. The small size (approximately 3 nm in diameter and 15 nm in length) together with high crystallinity was realized by a new facile and one-pot process at ambient temperature. The new process also enabled doping of transition metals for the enhancement of the electrocatalytic OER. The best-performing Ni (1.2 at%)-doped β-FeOOH nanorods obtained in this work exhibit performance as an Fe-based oxide and (oxy)hydroxide catalyst, which was comparable to that of conventional IrOx nanoparticle catalysts. The present results could support the recent argument that pure FeOxHy might be much more active than previously thought and that the transition metal-doped Fe-based system could be an important candidate for earth-abundant OER catalysts.
2. Experimental
2.1 Materials
All starting materials were used as received. FeCl3·6H2O (99%), CrCl3·6H2O (99.5%), Mn(NO3)2·6H2O (99.9%), Co(NO3)2·6H2O (99.5%), Ni(NO3)2·6H2O (99.9%), Zn(NO3)2·6H2O (99.9%), ethylenediamine, 1 M NaOH, monoethanolamine and 1 M KOH solution were purchased from Wako Pure Chemical Industries (Japan).2.2 Preparation of the metal-doped β-FeOOH colloidal solution
To prepare a metal-doped β-FeOOH colloidal solution, 500 mL of an aqueous solution containing FeCl3·6H2O (0.1 M Fe3+), the dopant and the basic reagent (ethylenediamine, NaOH, or monoethanolamine), adjusted to the appropriate pH, was stirred with a magnetic stirrer at room temperature. After 30 min of continuous stirring, the solution was aged overnight at room temperature.2.3 Preparation of the metal-doped β-FeOOH nanorods on carbon paper (CP)
Metal-doped β-FeOOH nanorods on carbon paper (CP) were prepared by depositing 1000 μL of a given colloidal solution together with a small amount of ethanol on the CP (TORAY, TGP-H-060, 1.8 × 2.2 cm), followed by drying at room temperature for 6 h and further drying under vacuum at 40 °C. The samples were subsequently washed with water and 0.1 M KOH solution and then dried. In this manner, a 1.0 mg quantity of the metal-doped β-FeOOH nanorods was loaded on each 1 cm2 of the CP.2.4 Preparation of IrOx nanocolloids
For comparison purposes, an IrOx nanocolloid solution was also deposited on CP. The IrOx nanocolloid solution was prepared according to a method previously reported by Zhao et al.18 in which 50 mL of a 2 mM aqueous K2IrCl6 solution was adjusted to pH 13 using a 10 wt% NaOH solution and then heated at 110 °C for 20 min. The solution was subsequently placed in an ice bath and adjusted to pH 1 by the addition of 3 M nitric acid, after which the pH was again adjusted, to 12, by the addition of a 1.5 wt% NaOH solution, producing a dark blue color. A 1000 μL quantity of the colloidal solution, together with a small amount of ethanol, was deposited on CP (1.0 × 1.0 cm) followed by drying at 60 °C for 10 min in an oven. In this manner, 7.1 mg of the IrOx nanocolloid was loaded per 1 cm2 of the CP.2.5 Characterization
Measurements of the particle size distributions of the colloidal solutions were conducted using a dynamic light scattering particle size analyzer (Nikkiso, Nanotrac UPA250EX, effective range: 0.8–6000 nm). Dark field STEM (DF-STEM) and TEM images were obtained using a JEM-2100F TEM (Joel) at an acceleration voltage of 200 kV. The average particle sizes in test samples were calculated from the sizes of 60 particles in a STEM image. Scanning electron microscopy (SEM) images were obtained using a S-4300 (Hitachi) microscope. The dopant concentration in the metal-doped β-FeOOH was determined by ICP analysis (Rigaku, CIROS 120 EOP). The crystal structures of the samples were assessed by XRD (Rigaku, Ultima IV) using Cu Kα radiation at 40 kV and 40 mA, while the chemical states and concentrations of Ni and Fe were determined by XPS (ULVAC-PHI, PHI-500 Versa Probe) using monochromated Mg Kα radiation. The chemical state of Ni was evaluated by X-ray absorption fine structure (XAFS) spectroscopy. Fe and Ni K-edge X-ray absorption near edge structure (XANES) spectra were acquired in the transmission mode using the BL33XU beamline at SPring-8 (Hyogo, Japan).2.6 Electrochemical measurements for oxygen evolution by water oxidation
The electrochemical characteristics of a metal-doped β-FeOOH nanorod/CP (1 × 1 cm) electrode were investigated in an aqueous 0.1 M KOH solution (pH 12.8) with a three-electrode configuration using an Ag/AgCl reference electrode and a Pt wire counter electrode. Electrochemical O2 and H2 evolution during water splitting by a metal doped β-FeOOH nanorod/CP (1 × 1 cm) electrode was examined using a Pyrex sealed glass reactor (total volume: 115.5 mL) in an aqueous 0.1 M KOH solution (60 mL). A three-electrode configuration using an Ag/AgCl reference electrode and a Pt wire counter electrode was employed and the applied potential was set at +1.56 V (vs. RHE). The amounts of O2 and H2 generated by water splitting over the metal doped β-FeOOH nanorod anode and Pt cathode were determined using a gas chromatograph equipped with a thermal conductivity detector (Shimadzu, GC-2014).3. Results and discussion
3.1 Synthesis of highly crystalline, transition metal-doped β-FeOOH colloidal nanorods
In order to obtain pure-phase β-FeOOH nanoparticles with high crystallinity, we employed a combination of a spontaneous peptization and Ostwald ripening36 to form colloidal solutions of nanocrystals at room temperature, and the use of coprecipitation for metal doping during the peptization process by pH control. This study demonstrated that colloidal solutions of β-FeOOH nanorods doped with metallic ions can be synthesized by simply stirring a mixture of Fe3+ and the dopant metal ions in an acidic aqueous solution (pH of approximately 2.2) containing Cl− and an amine for 30 min at ambient temperature, followed by aging at ambient temperature under acidic conditions. Fig. 2 shows the characteristic features of Ni-doped β-FeOOH (β-FeOOH:Ni) colloidal nanorods containing 1.2 at% Ni. This material exhibited the highest catalytic activity for the OER by water oxidation. In Fig. 2a, the particle size distribution as measured by dynamic light scattering (DLS) revealed that the mean volume diameter (MVD) in the clear solution was estimated to be 13 nm and that particles larger than 30 nm were not observed. The profile was nearly identical to that of non-doped β-FeOOH (Fig. S1, ESI†). These clear colloidal solutions were found to be very stable and did not generate any visible precipitate even after standing for months at room temperature. Fig. 2b shows scanning transmission electron microscopy (STEM) images of β-FeOOH:Ni. The colloidal solution contained nanorods with an average diameter of 3 nm and a length of 15 nm. These values represent a nanorod volume two orders of magnitude smaller than the volumes previously reported for highly crystalline β-FeOOH nanoparticles.28,31,33 As shown in Fig. 2c, the TEM diffraction pattern of these nanorods revealed crystalline β-FeOOH. Non-doped nanorods were also composed of crystalline β-FeOOH (Fig. S1, ESI†), suggesting that Ni doping did not affect the formation of the highly crystalline nanorods. The β-FeOOH phase in β-FeOOH:Ni colloidal solution was also confirmed by XRD (Fig. S2, ESI†). These results indicate that β-FeOOH colloidal solutions can be obtained at ambient temperature by a process consisting of the formation (spontaneous peptization) and crystal growth (Ostwald ripening36) to form the β-FeOOH nanorods. As was described, iron(III) hydrolysis in the presence of chloride ions yields akaganeite (β-FeOOH) with a tunnel structure stabilized by the inclusion of chloride as described (Fig. 1). The presence of chloride in the tunnel-structure of β-FeOOH is due to a relatively strong binding to the earliest iron oxyhydroxide precursors.37 Here, it was found that ethylenediamine (EDA) acts both as a base promoting the formation of highly crystalline β-FeOOH and to maintain a clear colloidal dispersion at ambient temperature. EDA plays an important role in facilitating the formation of nanocrystalline β-FeOOH and the incorporation of Ni at the early stage of β-FeOOH formation. STEM images indicate that nanorods were similarly synthesized with additives of EDA, monoethanolamine (MEA) and NaOH (generally utilized for the synthesis of β-FeOOH) (Fig. S3 and Table S1, ESI†). The detailed difference in crystallinity between these samples was unclear in XRD, because the major peak of β-FeOOH overlapped with that of the CP support (Fig. S4, ESI†). However, judging from the peak intensity of neighboring Fe–Fe interaction (peaking at an interatomic distance of 2.6–3.2 angstrom) measured by a quick scanning X-ray absorption spectroscopy (Q-XAFS),38 it was clarified that crystallinity is highly dependent on additives and the order of crystallinity was EDA > monoethanolamine (MEA) = NaOH (conventional method) (Fig. S5, ESI†). Thus, though it is difficult clarify the mechanism, it is speculated that amines interact with the chloride ions during the formation of β-FeOOH, which strongly depends on the chemical nature of the additives. As will be described later, the effect of crystallinity of β-FeOOH on the OER activity will be evaluated. | ||
| Fig. 2 (a) Size distribution and photographic image of the β-FeOOH:Ni colloidal solution (1.2 at% Ni, pH 2.2), and (b) STEM image and (c) TEM diffraction image of the same particles. | ||
The MVD was highly dependent on the pH of the solution during the synthesis. When the colloidal solution was synthesized at a pH higher than the upper limit (the precipitation pH of Fe3+, which is 2.1–2.5),39 the MVD was one order of magnitude larger than that of the material synthesized at a lower pH, leading to phase separation due to the aggregation of particles (Fig. S6, ESI†). The extremely different pH values for the precipitation of Ni2+ (6.2–16.2)39 and Fe3+ (less than 2.5) meant that approximately half the Ni2+ ions were incorporated into the β-FeOOH nanorods under acidic conditions, as confirmed by inductively coupled plasma (ICP) analyses (Fig. S6a, ESI†). Similar results were obtained for different transition metal ions (Cr3+, Mn2+, Co2+ and Zn2+) having precipitation pH values higher than 4.5 (Table S2, ESI†).
3.2 Properties of doped β-FeOOH nanorods
Electrodes were synthesized by depositing the colloidal solutions on carbon paper (CP). The X-ray diffraction (XRD) patterns for all samples exhibit broad peaks assignable to β-FeOOH, while no peaks originating from the dopant metals or their composites are observed (Fig. 3a and S7, ESI†). The average crystallite sizes of both the β-FeOOH and β-FeOOH:Ni (1.2 at%) as estimated by Scherrer's equation were 5 nm, a value that is consistent with the results of the STEM observations described above. Mössbauer spectra acquired from β-FeOOH:Ni (1.2 at%) suggest that the pure β-FeOOH hyperfine structure (containing almost no amorphous, α, γ-phases) remained unchanged upon doping (Fig. S8, ESI†). In addition, the Fe 2p3/2 X-ray photoelectron spectroscopy (XPS) data for all Ni-doped samples contain a peak at approximately 711 eV which indicates the formation of Fe3+ compounds, in conjunction with a very small negative shift induced by Ni doping (Fig. S9a, ESI†). In the XPS Ni 2p1/2 spectra, the presence of a small peak at approximately 873 eV suggests that the Ni concentration in β-FeOOH:Ni was 1.4 at%, in good agreement with the results of ICP analysis, although the Ni chemical state could not be classified as Ni2+ or Ni3+ (Fig. S9 and Table S3, ESI†). X-ray absorption fine structure (XAFS) analysis confirmed that the Ni was not trivalent but divalent, suggesting that NiOOH and Ni2O3 were not formed (Fig. 3b). From these structural analyses, it is most likely that the colloidal dispersions contained almost pure β-FeOOH:Ni nanorods (3 × 15 nm in average diameter and length, respectively) in which Ni2+ was doped into the β-FeOOH crystalline phase. SEM images of β-FeOOH:Ni (1.2 at%)/CP shown in Fig. 4 revealed that the β-FeOOH:Ni nanorods were densely packed on the carbon fiber of CP, and the thickness was evaluated to be ca. 200–300 nm (Fig. 4d). The nanorods covered not only the top of the CP but also deep in the CP, the surface of individual carbon fiber. Scanning electron microscopy – energy dispersive X-ray spectroscopy (SEM-EDX) analysis showed that the β-FeOOH:Ni colloidal nanorods contained Fe, O, Cl and Ni (Fig. S10, ESI†). The Cl/Fe atomic ratio of β-FeOOH:Ni was calculated to be 0.18 (an average value of 10 points), which was in good agreement with the results of XPS (0.17, Table S3, ESI†). These values were very close to those reported previously.40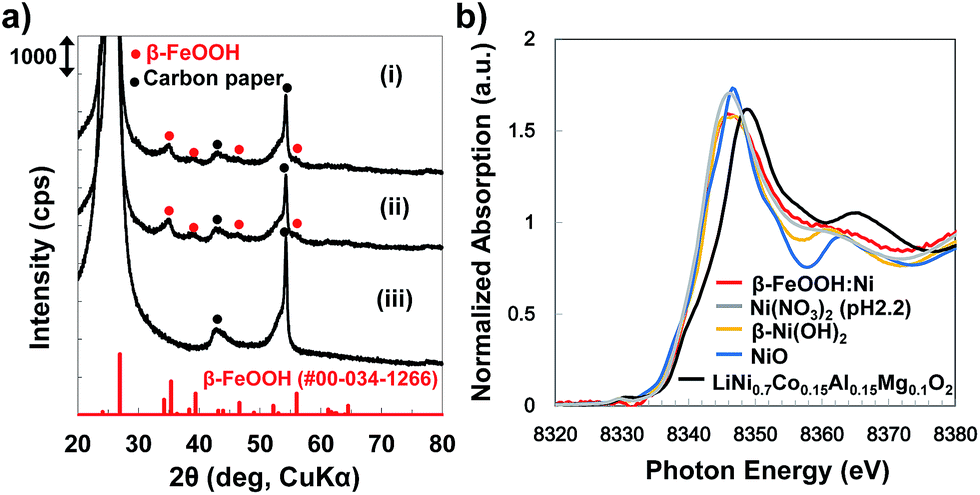 | ||
| Fig. 3 (a) XRD patterns of (i) β-FeOOH nanorods (on carbon paper (CP)), (ii) β-FeOOH:Ni (1.2 at%) nanorod/CP and (iii) the CP support, and (b) Ni-K edge XANES spectra of β-FeOOH:Ni (1.2 at%, on CP, red), Ni(NO3)2 solution/CP (pH adjusted to 2.2, without FeCl3, grey), β-Ni(OH)2 (orange), NiO (blue), and LiNi0.7Co0.15Al0.15Mg0.1O2 (black, Ni valence: 2.97). | ||
 | ||
| Fig. 4 (a) and (b) Top view and (c) and (d) cross-sectional view of SEM images of β-FeOOH:Ni (1.2 at%)/CP. | ||
3.3 Electrocatalytic OER activities with doped β-FeOOH
Electrochemical measurements examining the OER with the metal-doped β-FeOOH nanorod/CP electrodes were performed in a 0.1 M aqueous KOH solution, using a three electrode configuration composed of a platinum wire counter electrode and an Ag/AgCl reference electrode (Fig. 5a). Fig. 5b presents cyclic voltammograms (CVs) for non-doped and Ni-doped (1.2 at%) β-FeOOH/CP samples. An oxidative sweep of the non-doped β-FeOOH/CP specimens generated a small rise in current at approximately +1.5 V vs. reversible hydrogen electrode (RHE) and a sharp increase in the vicinity of +1.58 V, which corresponds to the OER current during water oxidation, as discussed further on. Surprisingly, a small amount (1.2 at%) of Ni doping reduced the threshold potential by 100 mV compared with that of non-doped β-FeOOH. It was also striking that the OER properties of β-FeOOH:Ni/CP were comparable to those of IrOx nanoparticle18,20/CP specimens loaded with an equimolar amount of colloidal nanoparticles. The CV patterns were significantly affected by the dopant species; doping with Co or Zn led to slight enhancements in the current density slope (Fig. S11a, ESI†), while Cr and Mn had no positive effect (data not shown). The time courses of the reaction current measured at +1.56 V (vs. RHE) show a reaction current density of 1.2 mA cm−2 under these alkaline electrolyte conditions, and β-FeOOH:Ni/CP exhibits higher stability than IrOx (Fig. 5c). XRD also confirmed that the crystal structure of the β-FeOOH:Ni did not deteriorate after the current–time measurements (Fig. S12, ESI†).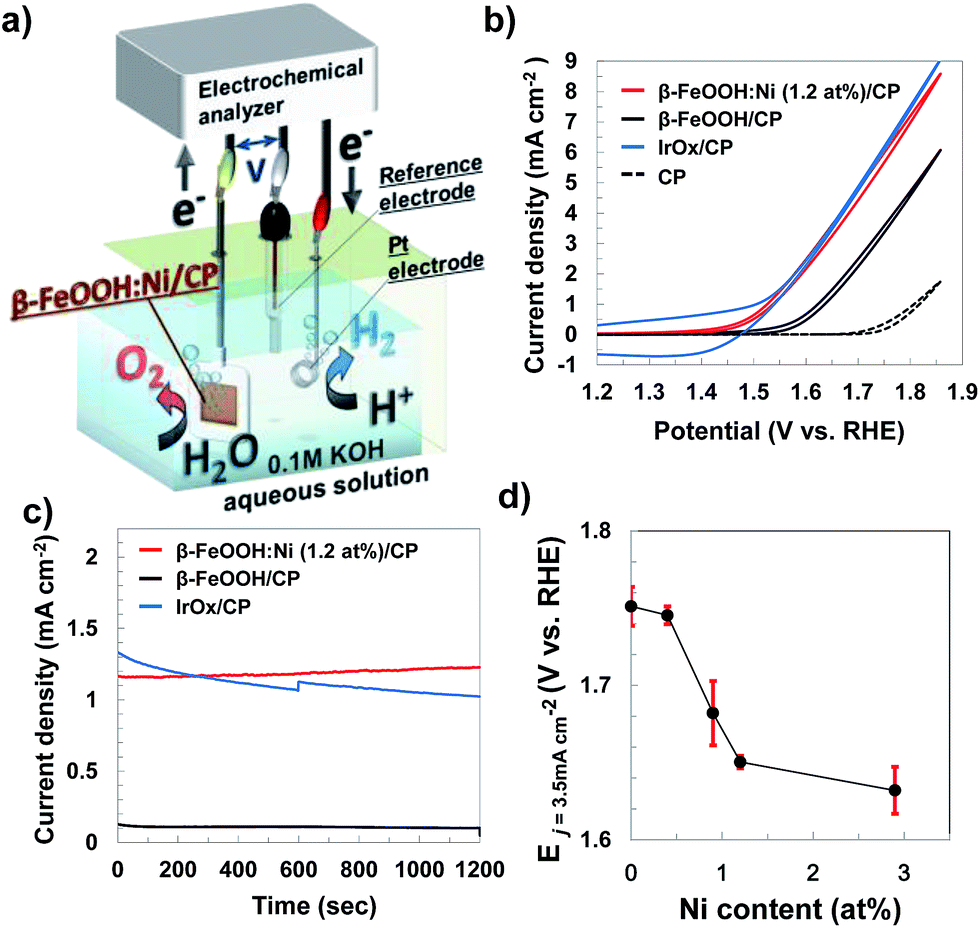 | ||
| Fig. 5 (a) Schematic illustration of electrochemical water oxidation by a β-FeOOH:Ni nanorod/CP specimen with a three electrode system, measured in a 0.1 M aqueous KOH solution (pH 12.8), (b) current–potential characteristics and (c) time courses for the currents (at +1.56 V vs. RHE) of non-doped and Ni-doped β-FeOOH nanorod/CP electrodes and an IrOx nanocolloid/CP electrode, and (d) potentials required to reach a current density (j) of 3.5 mA cm−2 for β-FeOOH:Ni nanorods with different amounts of Ni doping (stored for one month), in which error bars indicate the standard deviations obtained from multiple (3–6) electrodes. | ||
Lowering the overpotential of the reaction system is another important factor to consider. Fig. 5d shows the dependence of the potential required to generate a current density (j) of 3.5 mA cm−2 on the Ni concentration in β-FeOOH:Ni. Here, a j value of E3.5 was selected as sufficient for CO2 photoreduction to organic substances with a solar-to-chemical conversion efficiency of 5% as a device form.20 The threshold potential for water oxidation was also found to be correlated with the amount of Ni doping (Fig. S13, ESI†), and E3.5 improved with increasing Ni concentration, eventually plateauing around 1 to 3% Ni. The potential required to reach 3.5 mA cm−2 was determined to be +1.65 V (vs. RHE) for β-FeOOH:Ni (1.2 at%).
Based on the overpotential–current density plot, the present highly crystalline β-FeOOH enhanced the current density. It is evident that the OER current generated by the β-FeOOH:Ni was very high compared with other Fe-based oxides and various polymorphic oxyhydroxides prepared at higher temperatures or electrodeposition (Fig. 6). We also investigated the conventional synthesis of β-FeOOH nanorods using FeCl3 and NaOH as reported previously (Fig. S14, ESI†),35 which resulted in very slow β-FeOOH formation resulting in low crystallinity (Fig. S15, ESI†) even at 70 °C after 7 days synthesis and the β-FeOOH exhibited poor OER activity (Fig. 6 and S16, ESI,† shown as the “previous method”). It is strongly suggested that the crystallinity of β-FeOOH largely affects OER activity.
 | ||
| Fig. 6 Tafel plots comparing the activity of β-FeOOH:Ni (1.2 at%) to various OER Fe-catalysts in alkaline electrolytes. Solid lines with red and deep blue closed circles show data for highly crystalline β-FeOOH:Ni (1.2 at%) and β-FeOOH, respectively (this work). For comparison, the dashed line with black open circles and blue open triangles show data for poorly crystalline β-FeOOH synthesized according to the report35 and commercial α-FeOOH (Alfa Aesar). Orange (amorphous Fe2O3)4 and gray lines (from the data of current–potential curve 〈CV〉)41 and blue10 and black triangle symbols26 indicate data from the literature. | ||
There are few studies on the electronic structure and transport property of β-FeOOH. Alexandrov and Rosso performed density functional theory based (DFT+U) calculations to examine the electron mobility in goethite (α-FeOOH), akaganéite (β-FeOOH), and lepidocrocite (γ-FeOOH) polymorphs.42 They suggested that their room temperature charge transport should be dominated by the small-polaron hopping type, and clarified that short-range structural topology, not long-range order, dominates the electron-hopping rate.43 Although a polaron has lower mobility and larger effective mass than the original electron, the authors showed that the attendant mobility should be highest for pure α-FeOOH and β-FeOOH among the polymorphs. Experimental estimates of the electrical conductivity in these iron oxyhydroxides are also few but relevant values are 10−8 to 10−9 Ω s−1. Only β-FeOOH contains chloride ions. The Cl atoms partially fill the tunnel sites, suggesting that certain amount of the tunnel site is vacant. Each chloride ion is hydrogen bonded with eight neighboring hydroxyls in the β-FeOOH structure and is charge-balanced by incorporating an extra proton. This proton was suggested to bind to a corner-sharing bare O atom of the Fe–O octahedra,40,44 and molecular dynamic calculations by Song and Boily explained that Cl/Fe molar ratios induce significant changes in the hydrogen bonding environment of bulk hydroxyls.40 β-FeOOH is known to occur naturally in Cl-rich environments and the presence of chloride ions and the associated hydrogen bonds are believed to be very important for the thermal stability and reactivity of β-FeOOH.45 Molecular dynamic calculations revealed that the addition of chloride decreased lattice parameter values within 2% of the crystallographic values, and ATR-FTIR spectra showed change in bulk –OH stretching and bending frequencies depending on the content of chloride.44 Therefore, the electronic structure of β-FeOOH can also be influenced by the Cl ions, and accordingly the OER activity of β-FeOOH could be different from those of α-FeOOH, γ-FeOOH, and amorphous FeOOH.
Coating of OER catalysts on semiconductor photoanodes has been investigated intensively.46–49 Choi et al. has reported that the photodeposition of amorphous NiOOH and subsequent amorphous FeOOH layers onto BiVO4 to make a Bi VO4/FeOOH/NiOOH film enhances photocatalytic water oxidation. The same authors speculated that this improved performance resulted from reduced interface recombination at the BiVO4/OER catalyst junction and the creation of a more favorable Helmholtz layer potential drop at the OER catalyst/electrolyte junction.50 In the present study, we verified that our β-FeOOH:Ni was not identical to this previous FeOOH/NiOOH layered structure. In addition, we measured the activity of β-FeOOH/CP that was post-coated with amorphous NiOxHy, in which approximately 1.6 times larger amount of Ni was loaded onto β-FeOOH/CP compared with β-FeOOH:Ni (1.2 at%)/CP. Although the surface modification of β-FeOOH/CP by amorphous NiOxHy led to improvement of the reaction current, it was ca. 40% of that of β-FeOOH:Ni/CP (Fig. S17, ESI†).
A mechanism by which the Ni doping functions can be proposed based on previous reports concerning Ni-based oxyhydroxides containing Fe and hematite. In the case of Fe-doped γ-NiOOH, Friebel and Bell et al. reported that Fe3+ ions in γ-(FexNi1−xOOH) occupy sites with unusually short Fe–O bond distances, enabling reductions in the adsorption energies associated with OER intermediates such as –OH, –O and –OOH at these Fe sites.10 Swierk and Tilley et al. demonstrated that the faradaic resistances and activation energies of Fe-doped nickel (oxy) hydroxides (FexNi1−xOOH) are lower than those of pure NiOOH or FeOOH based on electrochemical impedance and activation energy measurements.51 Carter et al. predicted that Co- or Ni-doped α-Fe2O3 (hematite) would show effective OER activity and provide a favorable thermodynamic reaction pathway because Co and Ni are less positively charged than Fe, which results in improved binding to OER intermediates.52 In the first-row transition metal (oxy)hydroxides, it is believed that electronic M–O interaction is intrinsic and that surface metal cations (M) are the active sites for the OER. The reaction then proceeds through a series of intermediates (e.g., M–OH, M–O, M–OOH, M–OO), all bound by an M–O bond. Koper, Calle-Vallejo and their research groups demonstrated a systematic study by DFT calculation on Ni-based hydroxides doped with other first-row metals M, the M–O distances were modified by M. They observed that the ligand effect is small on Ni sites, while it is significant on M sites. This is intuitive, as M is embedded in a lattice where the M–O distances are different from those of its pure oxide.53
Based on these reports, we can also speculate that Ni doping of β-FeOOH leads to more favorable intermediate activation and adsorption energies compared to bare β-FeOOH during the OER, despite the differences in lattice parameters and surface acidity. However, further investigations to better elucidate these effects are underway.
Gaseous oxygen evolution over β-FeOOH/CP and β-FeOOH:Ni/CP was also confirmed at +1.56 V vs. RHE using a sealed glass reactor filled with a 0.1 M aqueous KOH solution, as shown in Fig. 7. In both cases, H2 and O2 were generated stoichiometrically (at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio), and the O2 evolution rates based on consuming four electrons (e−/4) remained constant during a period of 4 h together with almost 100% faradaic efficiency (dotted line). The O2 evolution rate was increased by a factor of 8.2 upon doping with 1.2 at% Ni. It was confirmed that the crystal phase, morphology, chemical states, and atomic ratios (Ni/Fe and Cl/Fe) of β-FeOOH:Ni/CP were almost unchanged after the electrochemical water oxidation reaction for 4 h as clarified by XRD, SEM, SEM-EDX and XPS (Fig. S18–21 and Table S3, ESI†). Although Ni XPS peaks were very weak because of its low concentration, the existence of Ni was also confirmed by X-ray fluorescence spectrometry (XRF) (Fig. S22†). Optimization of the material compositions and coatings used in these electrodes could further improve the activity, meaning that the facile synthesis of Fe-based crystalline oxyhydroxide OER catalysts at room temperature is highly feasible. Stoichiometric H2 and O2 formation with reaction current exceeding 10 mA cm−2 was also confirmed in a gas closed-circulation system in a 0.1 M K2B4O7–0.2 M K2SO4 aqueous solution (pH 6.9) at +1.95 V vs. RHE (Fig. S23, ESI†) and 0.1 M KOH at +1.91 V vs. RHE (Fig. S24, ESI†), which suggested that β-FeOOH:Ni/CP can be operated at neutral-to-alkaline pH. These overpotentials at this great current region at neutral-to-alkaline pH can be reduced by further optimization of loading of β-FeOOH:Ni on CP.
:1 molar ratio), and the O2 evolution rates based on consuming four electrons (e−/4) remained constant during a period of 4 h together with almost 100% faradaic efficiency (dotted line). The O2 evolution rate was increased by a factor of 8.2 upon doping with 1.2 at% Ni. It was confirmed that the crystal phase, morphology, chemical states, and atomic ratios (Ni/Fe and Cl/Fe) of β-FeOOH:Ni/CP were almost unchanged after the electrochemical water oxidation reaction for 4 h as clarified by XRD, SEM, SEM-EDX and XPS (Fig. S18–21 and Table S3, ESI†). Although Ni XPS peaks were very weak because of its low concentration, the existence of Ni was also confirmed by X-ray fluorescence spectrometry (XRF) (Fig. S22†). Optimization of the material compositions and coatings used in these electrodes could further improve the activity, meaning that the facile synthesis of Fe-based crystalline oxyhydroxide OER catalysts at room temperature is highly feasible. Stoichiometric H2 and O2 formation with reaction current exceeding 10 mA cm−2 was also confirmed in a gas closed-circulation system in a 0.1 M K2B4O7–0.2 M K2SO4 aqueous solution (pH 6.9) at +1.95 V vs. RHE (Fig. S23, ESI†) and 0.1 M KOH at +1.91 V vs. RHE (Fig. S24, ESI†), which suggested that β-FeOOH:Ni/CP can be operated at neutral-to-alkaline pH. These overpotentials at this great current region at neutral-to-alkaline pH can be reduced by further optimization of loading of β-FeOOH:Ni on CP.
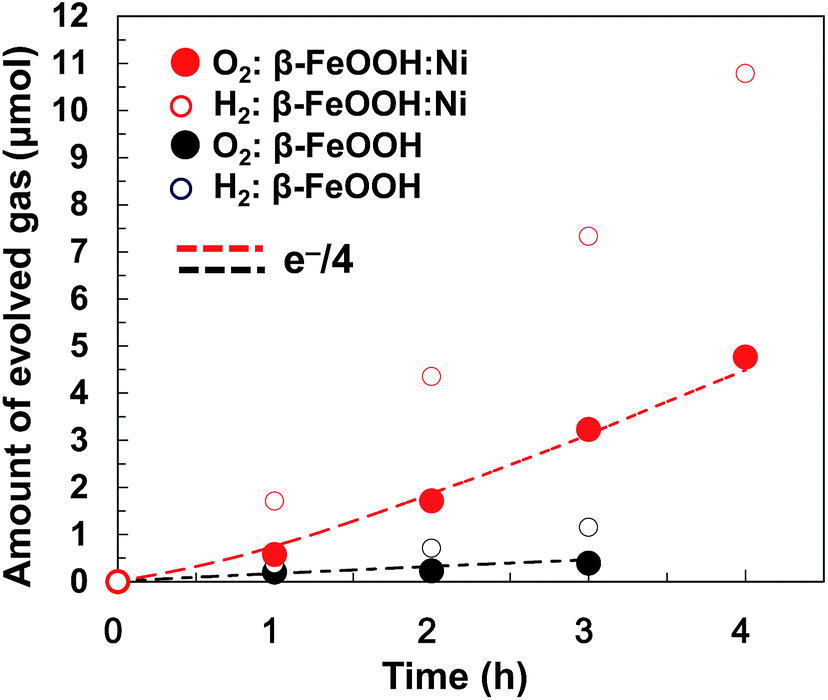 | ||
| Fig. 7 Stoichiometric electrochemical water splitting by β-FeOOH:Ni (1.2 at%) nanorods (stored for one month)/CP (red circles) and β-FeOOH nanorods/CP (blue circles) anodes at +1.56 V (vs. RHE) in a 0.1 M aqueous KOH solution with a Pt cathode. The red dashed lines indicate the theoretical amount of oxygen calculated from the observed current with 100% current efficiency. | ||
4. Conclusions
In summary, highly effective pristine- and metal-doped β-FeOOH nanorod catalysts for water oxidation were synthesized by a simple, scalable and versatile method at ambient temperature. In particular, Ni-doped β-FeOOH catalysts exhibited the highest OER activity among the Fe-based (oxy)hydroxide catalysts, which was probably due to the high crystallinity together with the nanoscale dimensions, and the effect of Ni doping on the catalysis. Additionally, the present new synthesis method in aqueous solution could be extended to other metal hydroxide (or oxide) nanoparticles for providing efficient catalysts.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas Artificial Photosynthesis (AnApple) from the Japan Society for the Promotion of Science (JSPS) for material synthesis (24107005), and by the Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C) from Japan Science and Technology Agency (JST). The XAFS data were acquired at the BL33XU beamline at the SPring-8 facility with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2013B7022 and 2016B7032). The authors wish to thank Mr Ikoma Narita, Mr Makoto Kondo, Ms Kae Yamamura and Ms Madoka Hasegawa for experimental assistance, Dr Yusaku Nishimura and Dr Hideki Takagi for XAFS measurement, Ms Naoko Takashi and Mr Kosuke Kitazumi for XPS measurements, Mr Satoru Kosaka for ICP and XRF measurements and Dr Takeshi Uyama for solution XRD measurement. The authors are also grateful to Dr Keita Sekizawa for fruitful discussions.Notes and references
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 136, 16977–16987 CrossRef PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- D. A. Corrigan and R. M. Bendert, J. Electrochem. Soc., 1989, 136, 723–728 CrossRef CAS.
- E. L. Miller and R. E. Rocheleau, J. Electrochem. Soc., 1997, 144, 3072–3077 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- B. M. Hunter, J. D. Blakemore, M. Deimund, H. B. Gray, J. R. Winkler and A. M. Müller, J. Am. Chem. Soc., 2014, 136, 13118–13121 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- Y. Surendranath, M. Dincă and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS PubMed.
- A. J. Esswein, Y. Surendranath, S. Y. Reece and D. G. Nocera, Energy Environ. Sci., 2011, 4, 499–504 CAS.
- J. W. D. Ng, M. Garcia-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 16053 CrossRef CAS.
- G. S. Nahor, P. Hapiot, P. Neta and A. Harriman, J. Phys. Chem., 1991, 95, 616–621 CrossRef CAS.
- T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958–12961 CAS.
- T. Nakagawa, N. S. Bjorge and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 15578–15579 CrossRef CAS PubMed.
- Y. Zhao, E. A. Heinandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402–406 CrossRef CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 CAS.
- G. L. Elizarova, L. G. Matvienko, V. L. Kuznetsov, D. I. Kochubey and V. N. Parmon, J. Mol. Catal. A: Chem., 1995, 104, 43–50 CrossRef.
- J. W. Schultze, S. Mohr and M. M. Lohrengel, J. Electroanal. Chem., 1983, 154, 57–68 CrossRef CAS.
- M. E. G. Lyons and L. D. Burke, J. Electroanal. Chem., 1984, 170, 377–381 CrossRef CAS.
- J. A. Seabold and K.-S. Choi, J. Am. Chem. Soc., 2012, 134, 2186–2192 CrossRef CAS PubMed.
- K. J. McDonald and K.-S. Choi, Energy Environ. Sci., 2012, 5, 8553–8557 CAS.
- W. D. Chemelewski, H.-C. Lee, J.-F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843–2850 CrossRef CAS PubMed.
- W. D. Chemelewski, J. R. Rosenstock and C. B. Mullins, J. Mater. Chem. A., 2014, 2, 14957–14962 CAS.
- T. Misawa, K. Hashimoto and S. Shimodaira, Corros. Sci., 1974, 14, 131–149 CrossRef CAS.
- K. Ståhl, K. Nielsen, J. Jiang, B. Lebech, J. C. Hanson, P. Norby and J. Lanschot, Corros. Sci., 2003, 45, 2563–2575 CrossRef.
- Y. Xiong, Z. Li, X. Li, B. Hu and Y. Xie, Inorg. Chem., 2004, 43, 6540–6542 CrossRef CAS PubMed.
- X. Wang, X. Chen, L. Gao, H. Zheng, M. Ji, C. Tang, T. Shen and Z. Zhang, J. Mater. Chem., 2004, 14, 905–907 RSC.
- T.-Y. Yang, H.-Y. Kang, K. Jin, S. Park, J.-H. Lee, U. Sim, H.-Y. Jeng, Y.-C. Joo and K. T. Nam, J. Mater. Chem. A., 2014, 2, 2297–2305 CAS.
- A. G. Joly, G. Xiong, C. Wang, D. E. McCready, K. M. Beck and W. P. Hess, Appl. Phys. Lett., 2007, 90, 103504 CrossRef.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. H. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- S. H. Jiang, S. Park, Y. Yoon, J. H. Lee, W. M. Wu, N. P. Dan, M. J. Sadowsky and H. G. Hur, Environ. Sci. Technol., 2013, 47, 10078–10084 CrossRef CAS PubMed.
- M. Kahlweit, Adv. Colloid Interface Sci., 1975, 5, 1–35 CrossRef CAS.
- J. Scheck, T. Lemke and D. Gebauer, Minerals, 2015, 5, 778–787 CrossRef.
- T. Nonaka, K. Domae, T. Araki, Y. Hayashi, Y. Hirose, T. Uruga, H. Yamazaki, T. Mochizuki, H. Tanida and S. Goto, Rev. Sci. Instrum., 2012, 83(8), 083112 CrossRef CAS PubMed.
- K. Sone and M. Tanaka, Teisei Bunseki Kagaku I, Kyoritsu-Shuppan, Tokyo, Japan, 1958, p. 116 Search PubMed.
- X. Song and J. F. Boily, J. Phys. Chem. C, 2012, 116, 2303–2312 CAS.
- W. Luo, C. Jiang, Y. Li, S. A. Shevlin, X. Han, K. Qiu, Y. Cheng, Z. Guo, W. Huang and J. Tang, J. Mater. Chem. A., 2017, 5, 2021–2028 CAS.
- V. Alexandrov and K. M. Rosso, J. Chem. Phys., 2014, 140, 234701 CrossRef PubMed.
- J. E. Katz, X. Zhang, K. Attenkofer, K. W. Chapman, C. Frandsen, P. Zarzycki, K. M. Rosso, R. W. Falcone, G. A. Waychunas and B. Gilbert, Science, 2012, 337, 1200–1203 CrossRef CAS PubMed.
- J. Kim and C. P. Grey, Chem. Mater., 2010, 22, 5453–5462 CrossRef CAS.
- U. Schwertmann and R. M. Cornell, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, Wiley-VCH, Weinheim, Germany, 2003 Search PubMed.
- U. Kang, S. K. Choi, D. J. Ham, S. H. Ji, W. Choi., D. S. Han, A. Abdel-Wahabe and H. Park, Energy Environ. Sci., 2015, 8, 2638–2643 CAS.
- S. K. Pilli, T. E. Furtak, L. D. Brown, T. G. Deutsch, J. A. Turner and A. M. Herring, Energy Environ. Sci., 2011, 4, 5028–5034 CAS.
- M. Zhong, T. Hisatomi, Y. B. Kuang, J. Zhao, M. Liu, A. Iwase, Q. X. Jia, H. Nishiyama, T. Minegeshi, M. Nakabayashi, N. Shibata, R. Niishiro, C. Katayama, H. Shibano, M. Katayama, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 5053–5060 CrossRef CAS PubMed.
- M. G. Ahmed, I. E. Kretschmer, T. A. Kandiel, A. Y. Ahmed, F. A. Rashwan and D. W. Bahnemann, ACS Appl. Mater. Interfaces, 2015, 7, 24053–24062 CAS.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- J. R. Swierk, S. Klaus, L. Trotochaud, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2015, 119, 19022–19029 CAS.
- P. Liao, J. A. Keith and E. A. Carter, J. Am. Chem. Soc., 2012, 134, 13296–13309 CrossRef CAS PubMed.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00043j |
| This journal is © The Royal Society of Chemistry 2017 |