
Ca2+ and Ga3+ doped LaMnO3 perovskite as a highly efficient and stable catalyst for two-step thermochemical water splitting†
Lulu
Wang
a,
Mohammad
Al-Mamun
 a,
Yu Lin
Zhong
a,
Lixue
Jiang
a,
Porun
Liu
a,
Yun
Wang
a,
Hua Gui
Yang
b and
Huijun
Zhao
*ac
a,
Yu Lin
Zhong
a,
Lixue
Jiang
a,
Porun
Liu
a,
Yun
Wang
a,
Hua Gui
Yang
b and
Huijun
Zhao
*ac
aCentre for Clean Environment and Energy, Griffith University, Gold Coast Campus, QLD 4222, Australia. E-mail: h.zhao@griffith.edu.au; Fax: +61 7 5552 8067; Tel: +61 7 5552 8261
bKey Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
cCentre for Environmental and Energy Nanomaterials, Institute of Solid State Physics, Chinese Academy of Sciences, Hefei 230031, P. R. China
First published on 27th March 2017
Abstract
High performance and stable catalysts for two-step thermochemical water splitting are key to synthesising direct fuels in the form of H2 or liquid hydrocarbon fuels by the Fischer–Tropsch process. Herein, we designed and synthesised LaMnO3 perovskite structured oxides doped on both the A and B sites for two-step thermochemical water splitting. First, Ca2+, Sr2+ and Ba2+ divalent cations were successfully doped on the A site of LaMnO3 and the thermochemical water splitting performances were analysed. After that, Al3+ and Ga3+ ions were doped on the B site of the perovskites produced in the first step. Through this strategy, a novel perovskite composition (La0.6Ca0.4Mn0.8Ga0.2O3) was found with remarkable water splitting performance, producing 401 μmol g−1 of H2 at low thermochemical cycle temperatures between 1300 and 900 °C. The as-prepared perovskite exhibits twelve times higher H2 production than the benchmark CeO2 catalyst under the same experimental conditions. This novel perovskite is also capable of maintaining steady-state redox activity during the water splitting cycles.
The thermochemical water splitting process represents an efficient way to convert solar energy into chemically storable fuels in the form of H2, which is an important precursor for synthesising liquid hydrocarbon-based fuels.1–3 Since the direct thermolysis of water takes place at extremely high temperatures4–6 (>3000 °C), efficient catalysts are required to lower this processing temperature for practical applications. Typically, the catalysed thermochemical process involves two steps: a higher temperature endothermic reduction step and a subsequent lower temperature exothermic gas splitting step.7–9 The processing temperature and solar fuel production performance of the two-step thermochemical method greatly depend on the redox properties of catalysts, typically metal oxides. Over the past several decades, hundreds of metal oxides have been tested but only a few (e.g. Fe3O4,10–12 ZnO,13–15 and CeO2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16–18) were reasonably active for thermochemical solar fuel production. Currently, non-stoichiometric CeO2 is regarded as the benchmark catalyst due to its rapid reaction kinetics and high stability.19 However, the high operation temperature (≥1500 °C) and relatively small oxygen exchange capacity of CeO2 limit its large-scale utilisation. Being a versatile material, perovskite type oxides, ABO3, have received widespread attention, particularly as a promising alternative catalyst for thermochemical water splitting. In principle, the perovskite-structured ABO3 can release its lattice oxygen through a high-temperature thermal reduction process resulting in the highly reduced ABO3−δ (eqn (1)). Subsequently, the ABO3−δ can effectively reduce introduced H2O to molecular H2 (eqn (2)), while recovering back to its original ABO3 state.
16–18) were reasonably active for thermochemical solar fuel production. Currently, non-stoichiometric CeO2 is regarded as the benchmark catalyst due to its rapid reaction kinetics and high stability.19 However, the high operation temperature (≥1500 °C) and relatively small oxygen exchange capacity of CeO2 limit its large-scale utilisation. Being a versatile material, perovskite type oxides, ABO3, have received widespread attention, particularly as a promising alternative catalyst for thermochemical water splitting. In principle, the perovskite-structured ABO3 can release its lattice oxygen through a high-temperature thermal reduction process resulting in the highly reduced ABO3−δ (eqn (1)). Subsequently, the ABO3−δ can effectively reduce introduced H2O to molecular H2 (eqn (2)), while recovering back to its original ABO3 state.| ABO3 + thermal energy → ABO3−δ + δ/2O2 | (1) |
| ABO3−δ + δH2O → ABO3 + δH2 | (2) |
To date, a number of perovskite oxides have been studied for two-step thermochemical gas splitting.20–27 These include reports demonstrating the effectiveness of LaMnO3 based perovskite catalysts, e.g. LaxSr1−xMnO3,28–30 LaxCa1−xMnO3,31 and LaxSr1−xMnyAl1−yO3,32 in thermochemical processes. Based on the available scientific evidence, the catalytic performance of perovskite oxides is closely related to their inherent catalytic properties and corresponding chemical compositions.33,34 The thermochemical gas splitting behaviour of LaMnO3 perovskite oxide can be purposely tuned by doping different metal cations onto its A and B sites. Very recently, Demont et al. reported improved thermochemical CO2 splitting performances via the successful incorporation of different chemical elements into the LaMnO3 perovskite oxide.35 To date, however, no work has investigated the thermochemical H2O splitting performance of LaMnO3 based perovskites with different elemental doping, especially the main group metal ions on the A and B sites.
Herein, for the first time, we have investigated the two-step thermochemical H2O splitting performances of LaMnO3 based perovskites with Ca2+, Sr2+ and Ba2+ doped on the A site and Al3+ and Ga3+ doped on the B site. The doped LaMnO3 exhibits significantly enhanced O2 and H2 production as well as re-oxidation yields compared to the parent LaMnO3. More importantly, through this process, a novel La0.6Ca0.4Mn0.8Ga0.2O3 perovskite oxide is developed which shows excellent H2O splitting performance at a low thermal reduction temperature (1300 °C) with superb stability.
La1−xAxMn1−yByO3 (A = Ca2+, Sr2+, and Ba2+; x = 0 and 0.4; B = Al3+ and Ga3+; y = 0 and 0.2) perovskite oxides were prepared via a modified Pechini method36 (refer to the ESI† for details). To facilitate effective testing and increase H2O splitting activity, the freshly prepared perovskite powders (∼0.5 g) were transformed into a solid porous monolith structure by mixing with isopropanol (1.5 ml) and then heating at 1300 °C for 6 h in air.29,37Fig. 1a shows the X-ray diffraction (XRD) pattern of La0.6Ca0.4Mn0.8Ga0.2O3 (LCMGO) as a representative of all the samples and the main diffraction peaks are characteristic of the typical orthorhombic perovskite structure. However, when the Ga3+ doping content was further increased to 0.3, the impurity of Ga2O3 was detected in the XRD pattern (Fig. S1, ESI†). A similar observation was reported by Dey et al.38 Compared to those of the parent structure of LaMnO3 (LMO), the Bragg peaks of LMO perovskites doped on both the A and B sites were noticeably shifted due to the distinct ionic radii of the introduced metal ions (Fig. S2, ESI†). The partial substitution of La3+ (1.36 Å) with a smaller Ca2+ (1.34 Å) causes the decrease of lattice parameters, which is reflected by the shifted main Bragg peak (110) towards a higher diffraction angle. Similar upshift trends in the XRD results of LCMAO and LCMGO were also detected because of the introduced Al3+ (0.535 Å) and Ga3+ (0.62 Å) ions, which also have smaller ionic radii in comparison with Mn3+ (0.645 Å). However, the doping of Sr2+ and Ba2+ on the A site of LMO shows a downshift of XRD patterns due to the fact that the ionic radii of Sr2+ (1.44 Å) and Ba2+ (1.61 Å) are larger than that of La3+ (1.36 Å). Fig. 1b shows the scanning electron microscopy (SEM) image of LCMGO, revealing a porous structure with interconnected nano-sized particles (<100 nm, see the inset of Fig. 1b). Although the introduction of metal ions into the A and B sites of LMO leads to varied crystal lattice parameters, the surface morphologies of the perovskites remain similar, reflecting the retention of the parent structural morphology (Fig. S3, ESI†). The transmission electron microscopy (TEM) image (Fig. 1c) further confirms that the particle size of LCMGO is smaller than 100 nm. The high-resolution transmission electron microscopy (HRTEM) image (bottom inset of Fig. 1c) shows a lattice spacing of 0.27 nm corresponding to the (110) reflection plane of the crystal structure of LCMGO, which is consistent with the XRD result (Fig. 1a). Furthermore, the elemental distributions in the perovskites were mapped by the energy dispersive X-ray (EDX) spectroscopic technique. As shown in Fig. 1d, a homogeneous distribution of the corresponding Ca, Mn and Ga elements in the LCMGO sample was observed. Furthermore, the BET surface areas of LaMnO3 based perovskites before and after thermochemical measurements were also investigated. As summarised in Table S1,† LCMGO showed the largest surface area among the tested perovskite samples, which could promote reactant transportation during the thermochemical process and lead to higher solar fuel production yield. Based on the XRD, SEM, HRTEM, EDX and BET analyses, it is evident that LCMGO perovskite was successfully synthesised through the manipulation of metal ion doping on both A and B sites of the parent LMO perovskite.
 | ||
| Fig. 1 (a) XRD pattern, (b) SEM images, (c) TEM, and (d) EDX elemental mapping of Ca, Mn and Ga distribution of LCMGO perovskite. The insets of (b) and (c) show the high magnification SEM and local HRTEM images. | ||
The thermochemical H2O splitting performances were evaluated in a laboratory-scale fixed-bed reactor (Fig. S4, ESI†) and the O2 and H2 produced during the thermochemical process were monitored using a mass spectrometer (MS). First, we compared the thermochemical performances of LMO with the A site doped La0.6A0.4MnO3 (A = Ca2+, Sr2+, and Ba2+, labelled LCMO, LSMO and LBMO, respectively) perovskites. The extents of oxygen vacancies generated by the prepared samples in high-temperature thermal reduction processes were investigated by measuring O2 production, which in turn will determine the maximum solar fuel production. As shown in Fig. 2a, LMO and the A site doped La0.6A0.4MnO3 perovskites started to release O2 at around 950 °C and the O2 evolution process was completed after holding at 1300 °C for 1 h. The figure demonstrates that compared to the parent LMO, the A site doped La0.6A0.4MnO3 perovskites exhibit significantly improved O2 evolution profiles. In particular, LCMO required the shortest time to reach the peak rate due to its unique orthorhombic perovskite structure, which can significantly promote oxygen transportation during the thermal reduction process.31 As a result, LCMO (167 μmol g−1) has the highest O2 evolution performance among all the tested A site doped La0.6A0.4MnO3 perovskites, which is 2.19 times higher than that produced by using the parent LMO (76 μmol g−1). Thus we can conclude that partial substitution of La3+ with alkali earth metal ions (Ca2+, Sr2+ and Ba2+) on the A site of LMO can greatly promote O2 evolution during the thermal reduction process and thereby achieve a larger extent of O2 non-stoichiometry.
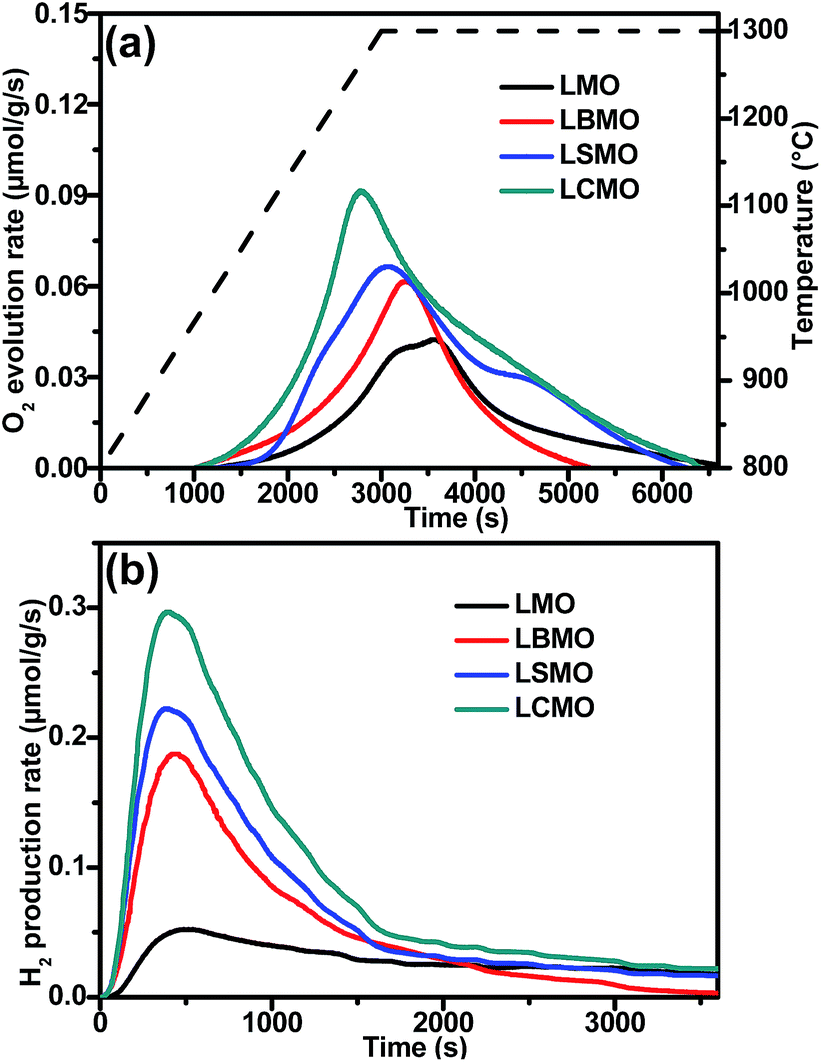 | ||
| Fig. 2 (a) O2 and (b) H2 production curves of LaMnO3 and A site doped La0.6A0.4MnO3 (A = Ba2+, Sr2+ and Ca2+) perovskites. | ||
In the second step, H2O splitting tests were conducted at 900 °C and H2 production was immediately detected after the aspiration of H2O vapour into the system. Fig. 2b shows the H2 production curves of the LMO and the A site doped La0.6A0.4MnO3 perovskites. The H2 production had not completely ceased after 60 min of reaction, leaving a long tail after 30 min, which is consistent with the results reported elsewhere.29,38,39 The sluggish H2O splitting reaction kinetics of perovskites may arise from several factors, such as limited chemical diffusion and decreased surface reaction constants.29 Nevertheless, over 80% of the total H2 production was achieved within 30 min. The H2 production trend of the A site doped perovskite concurs with the trend obtained for O2 production (Fig. 2a). As expected, LCMO shows significantly improved O2 production (312 μmol g−1), which is almost 6 times higher than that of LMO (48 μmol g−1). From the final results summarised in Table 1, it is clear that both O2 and H2 production follow the same trend, i.e. LMO < LBMO < LSMO < LCMO. In addition, their re-oxidation yields also follow the same trend. The better thermochemical performance of LCMO may be attributed to the lower reduction enthalpy and lighter atomic weight of Ca2+ when compared to those of Sr2+ and Ba2+ at the same doping level.35 To confirm the effectiveness of transforming perovskite powder into a porous monolith structure, we compared their thermochemical performances under the same conditions. The O2 evolution results (Fig. S5, ESI†) obtained for the porous monolith and perovskite powder are very close. This confirms the identical material composition of both powder and porous monolith structured perovskites. However, a significant difference in the H2 production performance was observed which is mainly due to the increased surface area of the monolith structure compared to that of the powder sample.
| Redox materials | O2 (μmol g−1) | H2 (μmol g−1) | Re-oxidation yield (%) |
|---|---|---|---|
| LMO | 76 | 48 | 31 |
| LBMO | 113 | 179 | 79 |
| LSMO | 126 | 215 | 85 |
| LCMO | 167 | 312 | 93 |
| LCMAO | 184 | 339 | 92 |
| LCMGO | 212 | 401 | 95 |
The B site doping of perovskites can also significantly affect their thermochemical performances. Trivalent metal ion doping on the B site of LMO has enhanced the extent of oxygen vacancies and redox behaviour for some catalytic reactions.38,40,41 Therefore, to further improve the thermochemical performance of LCMO, two different trivalent metal ions, Al3+ and Ga3+, were partially doped on the B site (La0.6Ca0.4Mn0.8Al0.2O3 and La0.6Ca0.4Mn0.8Ga0.2O3, labelled LCMAO and LCMGO, respectively). As shown in Fig. 3, both LCMAO and LCMGO exhibited higher thermochemical performances than LCMO, confirming the positive effect of the B site doping strategy. More importantly, the doping of Ga3+ on the B site of LCMO showed better promotion of H2 production performance than Al3+ doping. As summarised in Table 1, LCMGO exhibits a H2 production of 401 μmol g−1, which is 30% higher than that achieved by LCMO (312 μmol g−1), 20% higher than that of LCMAO (339 μmol g−1) and twelve times higher than that of the benchmark CeO2 under the same experimental conditions (Fig. S6, ESI†).
 | ||
| Fig. 3 (a) O2 and (b) H2 production curves of LCMAO and LCMGO perovskites. The thermochemical process was carried out under the same conditions as those used for LCMO, operated between 1300 °C and 900 °C. | ||
The noteworthy thermochemical performance of LCMGO may arise from its outstanding BET surface area, which greatly promoted the solid–gas reaction during the thermochemical catalytic process with remarkable H2 production. Furthermore, as reported by Patrakeev et al., the doping of Ga3+ in similar La1−xSrxFeO3 perovskite systems was proved to decrease the reduction enthalpy and resulted in enhanced oxygen vacancy formation.41 To the best of our knowledge, the H2 production of LCMGO is at the top level of all the reported perovskite type catalysts (Table S2†). It is worth mentioning that the thermal reduction temperature (1300 °C) we applied in this study is lower than those used for most of the reported perovskites (≥1400 °C, refer to Table S2†). The lowered thermal reduction temperature is beneficial for the ease of solar reactor design and may result in higher energy conversion efficiency. The H2 yield of LCMGO was further improved to 473 μmol g−1 (Fig. S7, ESI†), which is the highest H2 production amount reported to date when the thermal reduction temperature was increased to 1400 °C during the thermochemical H2O splitting process. However, serious sintering and aggregation phenomena were detected in LCMGO when thermally reduced at 1400 °C (Fig. S8, ESI†) and the re-oxidation yield was found to drop from 95% (1300 °C) to 74% (1400 °C), which will significantly decrease the catalytic durability for long-term thermochemical application.
Durability is another critical parameter for assessing the viability of redox materials for thermochemical solar fuel production. Thus, multiple cycles of thermochemical H2O splitting tests were conducted on LCMGO between 1300 °C and 900 °C. As shown in Fig. 4a, stable O2 and H2 evolution profiles were observed during the cycling measurements. This trend can also be seen in the summarised O2 and H2 production amount per cycle (Fig. 4b). Moreover, the molar ratio of H2 to O2 is quite close to the theoretical value of 2, which means that almost all the O2 released during the thermal reduction process is recovered during the re-oxidation process. Furthermore, the sample exhibited reliable stability and no crystal structure change (Fig. S9, ESI†) or severe sintering phenomena were observed after the cycling test (Fig. S10, ESI†).
 | ||
| Fig. 4 (a) Six consecutive O2 and H2 production profiles and (b) O2 and H2 yields and their corresponding ratio as a function of cycle number. | ||
Conclusions
A systematic investigation of the effects of doping LaMnO3 based perovskites on both the A and B sites on their thermochemical H2O splitting performances was carried out at a low operating temperature of 1300 °C. Our results demonstrate that doping on either A (Ca2+, Sr2+, and Ba2+) or B (Al3+ and Ga3+) sites of LaMnO3 perovskites has a positive impact on the two-step thermochemical H2O splitting performances. More importantly, the partial doping of Ca2+ on the A site and Ga3+ on the B site produces a novel perovskite composition (La0.6Ca0.4Mn0.8Ga0.2O3) which shows a much higher H2 production (401 μmol g−1) than the benchmark CeO2. Furthermore, La0.6Ca0.4Mn0.8Ga0.2O3 perovskite exhibited superb cycling stability during the thermochemical H2O splitting process. We, therefore, believe that this doping strategy can help develop highly efficient and stable perovskites for other energy conversion and storage applications.Acknowledgements
This work was financially supported by the Australian Research Council (ARC) and the National Natural Science Foundation of China (Grant no. 51372248 and 51432009).Notes and references
- S. Z. Baykara, Int. J. Hydrogen Energy, 2004, 29, 1451 CrossRef CAS.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797 CrossRef CAS PubMed.
- T. Kodama and N. Gokon, Chem. Rev., 2007, 107, 4048 CrossRef CAS PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29 CrossRef CAS.
- M. Romero and A. Steinfeld, Energy Environ. Sci., 2012, 5, 9234 CAS.
- J. E. Miller, A. H. McDaniel and M. D. Allendorf, Adv. Energy Mater., 2014, 4, 1300469 CrossRef.
- S. Abanades and G. Flamant, Sol. Energy, 2006, 80, 1611 CrossRef CAS.
- S. Abanades, P. Charvin, G. Flamant and P. Neveu, Energy, 2006, 31, 2805 CrossRef CAS.
- P. Furler, J. R. Scheffe and A. Steinfeld, Energy Environ. Sci., 2012, 5, 6098 CAS.
- T. Kodama, Y. Nakamuro and T. Mizuno, J. Sol. Energy Eng., 2004, 128, 3 CrossRef.
- P. Charvin, S. Abanades, G. Flamant and F. Lemort, Energy, 2007, 32, 1124 CrossRef CAS.
- M. E. Gálvez, P. G. Loutzenhiser, I. Hischier and A. Steinfeld, Energy Fuels, 2008, 22, 3544 CrossRef.
- K. Wegner, H. C. Ly, R. J. Weiss, S. E. Pratsinis and A. Steinfeld, Int. J. Hydrogen Energy, 2006, 31, 55 CrossRef CAS.
- P. G. Loutzenhiser, A. Meier and A. Steinfeld, Materials, 2010, 3, 4922 CrossRef.
- A. Stamatiou, P. G. Loutzenhiser and A. Steinfeld, Chem. Mater., 2010, 22, 851 CrossRef CAS.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797 CrossRef CAS PubMed.
- P. Furler, J. Scheffe, M. Gorbar, L. Moes, U. Vogt and A. Steinfeld, Energy Fuels, 2012, 26, 7051 CrossRef CAS.
- W. C. Chueh and S. M. Haile, ChemSusChem, 2009, 2, 735 CrossRef CAS PubMed.
- W. C. Chueh and S. M. Haile, Philos. Trans. R. Soc., A, 2010, 368, 3269 CrossRef CAS PubMed.
- Q. Q. Jiang, J. H. Tong, G. L. Zhou, Z. X. Jiang, Z. Li and C. Li, Sol. Energy, 2014, 103, 425 CrossRef CAS.
- A. Demont, S. Abanades and E. Beche, J. Phys. Chem. C, 2014, 118, 12682 CAS.
- Z. Zhang, L. Andre and S. Abanades, Sol. Energy, 2016, 134, 494 CrossRef CAS.
- C. L. Muhich, B. D. Ehrhart, I. Al-Shankiti, B. J. Ward, C. B. Musgrave and A. W. Weimer, Wiley Interdiscip. Rev.: Energy Environ., 2016, 5, 261 CrossRef CAS.
- M. M. Nair and S. Abanades, ChemistrySelect, 2016, 1, 4449 CrossRef CAS.
- M. E. Galvez, R. Jacot, J. Scheffe, T. Cooper, G. Patzke and A. Steinfeld, Phys. Chem. Chem. Phys., 2015, 17, 6629 RSC.
- T. Cooper, J. R. Scheffe, M. E. Galvez, R. Jacot, G. Patzke and A. Steinfeld, Energy Technol., 2015, 3, 1130 CrossRef CAS.
- A. M. Deml, V. Stevanović, A. M. Holder, M. Sanders, R. O'Hayre and C. B. Musgrave, Chem. Mater., 2014, 26, 6595 CrossRef CAS.
- J. R. Scheffe, D. Weibel and A. Steinfed, Energy Fuels, 2013, 27, 4250 CrossRef CAS.
- C. K. Yang, Y. Yamazaki, A. Aydin and S. M. Haile, J. Mater. Chem. A, 2014, 2, 13612 CAS.
- A. Demont and S. Abanades, RSC Adv., 2014, 4, 54885 RSC.
- S. Dey, B. S. Naidu, A. Govindaraj and C. N. R. Rao, Phys. Chem. Chem. Phys., 2015, 17, 122 RSC.
- A. H. McDaniel, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O'Hayre, W. C. Chueh and J. H. Tong, Energy Environ. Sci., 2013, 6, 2424 CAS.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981 CrossRef.
- L. G. Tejuca, J. L. G. Fierro and J. M. D. Tascon, Adv. Catal., 1989, 36, 237 CAS.
- A. Demont and S. Abanades, J. Mater. Chem. A, 2015, 3, 3536 CAS.
- Y.-S. Han and H.-G. Kim, J. Power Sources, 2000, 88, 161 CrossRef CAS.
- Y. Hao, C. K. Yang and S. M. Haile, Chem. Mater., 2014, 26, 6073 CrossRef CAS.
- S. Dey, B. S. Naidu and C. N. R. Rao, Dalton Trans., 2016, 45, 2430 RSC.
- S. Dey, B. S. Naidu and C. N. R. Rao, Chem.–Eur. J., 2015, 21, 7077 CrossRef CAS PubMed.
- S. Cimino, L. Lisi, S. De Rossi, M. Faticanti and P. Porta, Appl. Catal., B, 2003, 43, 397 CrossRef CAS.
- M. V. Patrakeev, E. B. Mitberg, A. A. Lakhtin, I. A. Leonidov, V. L. Kozhevnikov, V. V. Kharton, M. Avdeev and F. M. B. Marques, J. Solid State Chem., 2002, 167, 203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6se00097e |
| This journal is © The Royal Society of Chemistry 2017 |