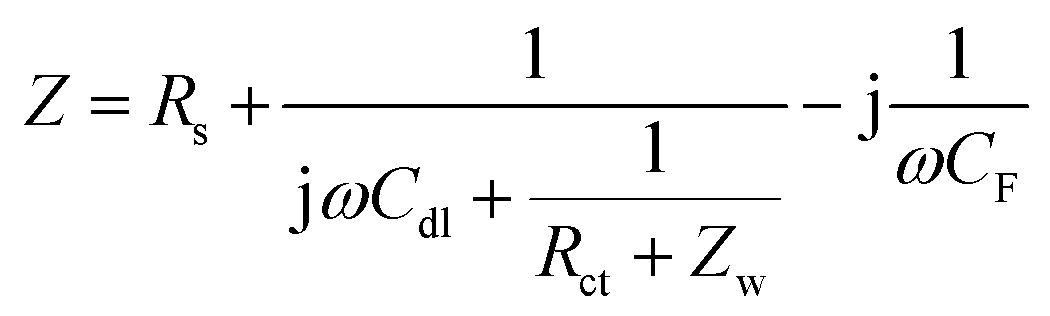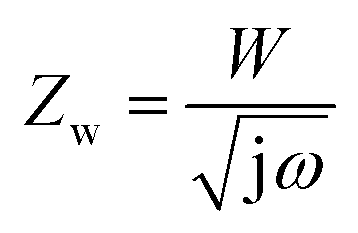Enhanced electrochemical activity of perforated graphene in nickel-oxide-based supercapacitors and fabrication of potential asymmetric supercapacitors†
Received
2nd December 2016
, Accepted 26th January 2017
First published on 3rd February 2017
Abstract
We synthesize a hierarchically porous electrode material with a high specific capacity composed of nickel oxide (NiO) nanosheets and perforated graphene (PG) sheets grown on a three-dimensional (3D) macroporous nickel foam via a facile hydrothermal method. Employing perforated graphene instead of non-perforated graphene greatly improves the electrochemical performance of the composite by increasing the specific surface area of the NiO/PG composite owing to the small perforations in the graphene and by improving the mechanical strength and electrical conductivity of NiO due to the graphene covering layer. The PG also provides (a) the accessibility and diffusion of ions onto NiO and PG surfaces through the perforations; (b) the electrolyte cages in the spaces below the perforations to allow ion-reversible adsorption to the inner surface of the graphene; (c) the inevitable edge defects around the holes causing reactivity with ions. After assembling an asymmetric supercapacitor coin cell composed of NiO/PG as the positive electrode, a separator, PG as the negative electrode, and a 1 M KOH electrolyte, the coin cell exhibits a high energy density of 57.8 W h kg−1 at a power density of 1030.9 W kg−1 and excellent cycling stability (82.1%) after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
1. Introduction
Because of the sharp increase in demand for portable clean energy devices over the past few decades and the capabilities of supercapacitors with high power density, fast charge and discharge rates, and high reliability, supercapacitors (SCs) have been attracting attention as alternatives to lithium-ion batteries (which in fact deliver a higher energy density (150–250 W h kg−1)1 than SCs). Enhancing the energy density of SCs without sacrificing their high power density would make these devices competitive with lithium batteries. Generally, SCs can be divided into two types according to their electron-storage mechanisms: electrical double-layer capacitors, which rely on ion adsorption/desorption, and supercapacitors,2–6 wherein rapid surface redox reactions are dominant. Electrical double-layer capacitors, which normally utilize carbonaceous materials as the double-layer electrode, exhibit high cyclability, but their specific capacitances are not very high. Supercapacitors generally use transition metal oxides/hydroxides/layered double hydroxides, etc., as the electrode materials, and they exhibit better specific capacitances but are less durable than devices with double-layer carbonaceous materials.7 Among the transition metal oxide materials used thus far in SC devices, nickel oxide (NiO) has been widely utilized as a faradaic capacitive electrode because of its high theoretical capacity, low production cost, excellent chemical/thermal stability, low environmental impact, etc.,8–10 and it is currently one of the promising electrode materials used in SCs.10–13 However, it is still the critical problem of its low electrical conductivity and poor rate capability caused by the large volume change during the charge–discharge processes.14 In addition, its weak mechanical strength and poor cycling stability impede its direct use as an advanced electrode material in high-performance SC devices.15 To capitalize on its strengths and compensate for its weaknesses, many active materials, such as polymers, graphene, etc., have been combined with NiO materials as supercapacitor electrodes.16,17
Because graphene has many extraordinary properties such as a high specific surface area, high electrical conductivity, mechanically robust strength, and high chemical stability,18–20 various NiO/graphene composite systems have been suggested in the literature. Cao et al. reported the use of a 3D NiO-deposited CVD-growth graphene composite electrode with a specific capacitances of 816 F g−1 (at a scan rate of 5 mV s−1),21 and Wu et al. prepared a novel 3D NiO/ultrathin derived graphene/Ni foam hybrid material with a specific capacitance of 425 F g−1 (at a current density of 2 A g−1).17 Jiang et al. synthesized a porous thermally carbonized graphene/NiO composite which was capable of delivering a capacitance of 342.9 F g−1 at a current density of 1 A g−1 and retained 86.1% of its capacitance after 2000 cycles.22 Notably, the specific capacitance values of NiO/graphene composite electrodes in SCs are generally low, especially compared with that of pristine NiO electrodes (the theoretical specific capacitance value within the potential window of 0–0.5 V is 2584 F g−1).23 This low capacitance might be because planar or less perforated graphene sheets block the nanopores created on the NiO nanostructure, minimizing the electrolyte percolation pathways and thereby decreasing the ion diffusion rate/amount.24 To develop an SC with a high specific capacitance, good rate capability, and a long cycle life, we design and synthesize a high-surface-area advanced composite electrode system with vertically grown NiO nanosheets covered with a perforated graphene sheet (PG). In the composite system, vertically grown NiO nanosheets provide a large surface area and porous architecture, which lead to the system's high capacity. In addition to improving the weak mechanical strength of the NiO material, the PG covering the NiO nanosheets can compensate for NiO's poor electrical conductivity by providing new paths for electron flow. Remarkably, the perforations in the graphene sheet can play several roles. They provide electrolyte ions easy access to the NiO surface while also providing electrolyte cages in the voids, which act as ion-reversible physisorption sites on the inner graphene surfaces as well as redox sites for the electrolyte ions due to the reactivity of the inevitable edge defects around the graphene holes.
In this work, a chemical etching method25 is used to form the PG. The vertically grown NiO nanosheets and the NiO/PG composite are characterized for their structures, morphologies, chemical components, specific surface areas for both their physical and chemical properties, and their electrochemical performance. Moreover, an asymmetric supercapacitor (ASC) composed of NiO/PG composite as the positive electrode and PG as the negative electrode (NiO/PG//PG) is fabricated and demonstrated for effectively reversible cycling at a high potential of 1.5 V, an energy density of 57.8 W h kg−1 at a power density of 1030.9 W kg−1 (an energy density of 15.9 W h kg−1 with a high power density of 6.4 kW kg−1), and excellent cycling stability at a high current density of 4 A g−1 (i.e., 81.2% retention over 10000 cycles). Lastly, a red light-emitting diode (LED) is driven by using two as-fabricated ASCs connected in series to demonstrate the performance. The LED light is sustained for 600 s until the light evanesces due to the complete discharge of the ASCs under the operating voltage of the LED.
2. Experimental section
2.1 Materials
Concentrated hydrochloric acid (HCl, 37%), and concentrated nitric acid (HNO3, 70%) were purchased from Daejung Chem (Republic of Korea). Acetone and anhydrous ethanol were obtained from SK Chem. Hydrogen peroxide (H2O2, 30.0–35.5%) was received from Samchun chemical (Republic of Korea). All other chemicals, including nickel nitrate hexahydrate (>97%, Ni(NO3)2·6H2O), potassium hydroxide (KOH), hexamethylenetetramine (HMT), sodium nitrate (NaNO3), potassium permanganate (KMnO4), hydrazine hydrate, and graphite, were analytical grade and used as received from Sigma Aldrich.
2.2 Preparation of perforated graphene oxide
Graphene oxide was prepared from graphite powder according to the modified Hummer's method.17,26,27 Generally, 8 g of graphite powder was gradually added into a concentrated H2SO4 (180 mL) solution containing NaNO3 (4 g) and KMnO4 (23 g) at 0 °C (i.e., in an ice bath). After increasing the temperature to 35 °C, the mixture was stirred vigorously for 180 min. Then, 375 mL distilled water was added gradually at 0 °C, and then, the solution was stirred for 5 h until the temperature reached 90 °C. Finally, the solution was diluted with 1000 mL DI water and treated with hydrogen peroxide (H2O2, 30 wt%) until no additional gas was produced. During this process, the colour of solution changed from dark brown to yellow. At this stage, the solution was filtered through a glass filter, washed with a 5% HCl solution in DI water, and dried under vacuum at 60 °C overnight to obtain GO powder. Then, the as-synthesized GO powder was again dispersed in DI water at a concentration of 1 mg mL−1 and 70% concentrated HNO3 (at a mass ratio of GO:HNO3 = 1:13) in a sealed glass bottle under sonication for 1 h to obtain perforated graphene oxide (PGO). Subsequently, the solution was poured into a centrifuge tube, centrifuged 5 times, and dialyzed in a dialysis bag (with a molecular weight cut-off of 2000 Da) for 1 week to remove the acid. Then, the PGO powder was dried overnight under vacuum at 60 °C.
2.3 Preparation of PG negative electrode
In order to measure the electrochemical performance of PG as a negative electrode, PG was synthesized from PGO powder via a one-pot solvothermal process. First, 60 mg of PGO powder was added into 30 mL of DI water and then sonicated for 1 h. The solution was poured into a 50 mL Teflon-lined autoclave and heated at 180 °C for 12 h. The synthesized PG was collected by centrifugation and desiccated at 50 °C overnight in a vacuum oven. The working electrode was constructed by mixing as-synthesized PG, acetylene black, and a poly(tetrafluoroethylene) (PTFE) suspension (60 wt%) binder in a weight ratio of 8:1:1 and then smearing the mixture onto Ni foam to serve as a current collector. The electrode was dried at 50 °C overnight in a vacuum oven.
2.4 Preparation of NiO/PG composite electrode
Before use, the Ni foam (1 cm × 1 cm, with a pore density of 110 PPI and a mass density of 320 g m−2, Artenano Company Limited, Hong Kong) was pre-treated via the following steps: (a) degreasing by immersion in acetone for 30 min, (b) etching with dilute HCl (3.0 mol L−1) for 15 min, (c) rinsing with deionized water, and (d) drying. After pre-treating the Ni foam, the precursor for the growth of nickel hydroxide, Ni(OH)2, was prepared using a method as described in our previous report.28 First, 100 mmol of Ni(NO3)2·6H2O, 25 mmol of HMT, and 30 μL of hydrazine hydrate were dissolved in 50 mL of a 0.1 mg mL−1 PG solution and mixed homogeneously by ultrasonication for 30 min at room temperature. The cleaned Ni foam (2 cm × 4 cm) was placed vertically into the above 50 mL solution and maintained at 90 °C for 5 h in a Teflon-lined autoclave. The Ni(OH)2/PG-Ni foam substrate was washed with de-ionized water and ethanol to remove the surface ions and molecules and then dried in a vacuum at 50 °C for 12 h. Subsequently, the as-obtained Ni(OH)2/PG composites were annealed in an Ar atmosphere at 400 °C for 1 h to generate the NiO/PG composite electrode. The mass of the NiO/PG composite electrode on the Ni foam was confirmed by subtracting the mass of the pure Ni foam before the deposition process. The loaded densities of the active materials were approximately 1.0 mg cm−2 for all NiO/PG electrodes. For comparison, two control electrodes were also prepared using the same method except for the graphene conditions, one with NiO but without carbon and another with NiO and un-etched graphene (non-perforated graphene, NG).
2.5 Material characterization
The samples' structures and present phases were characterized by X-ray diffraction (XRD, D8-Discovery Bruker, Cu Kα, 40 kV, 40 mA) in a 2θ scanning range of 5–80° with a 0.02° step size. Field-emission electron microscope (FESEM, Hitachi, S-4800, 15 kV) digital plane-view images were used to examine the surface morphologies. To confirm the presence of PG on the NiO, high-resolution transmission electron microscope (HRTEM) digital images and selected area electron diffraction (SAED) patterns were recorded using an HRTEM Tecnai F20 unit operated at 200 kV. The Brunauer–Emmett–Teller (BET) surface area was obtained using a Micromeritics ASAP 2010 analyser. The N2 adsorption–desorption isotherm and pore size distribution of the samples were measured on scratched-off powder from the Ni foam. After scraping the NiO/PG powder off of the Ni foam, X-ray photoelectron spectroscopy (XPS) was carried out by a surface analysis system (VG Scientifics ESCALAB250) using a monochromatic Al Kα source (1486.6 eV) to analyse the elemental compositions and chemical states of the NiO/PG composite electrode. The XPS spectra were calibrated to the carbon C 1s peak at 284.6 eV.
2.6 Electrochemical measurements
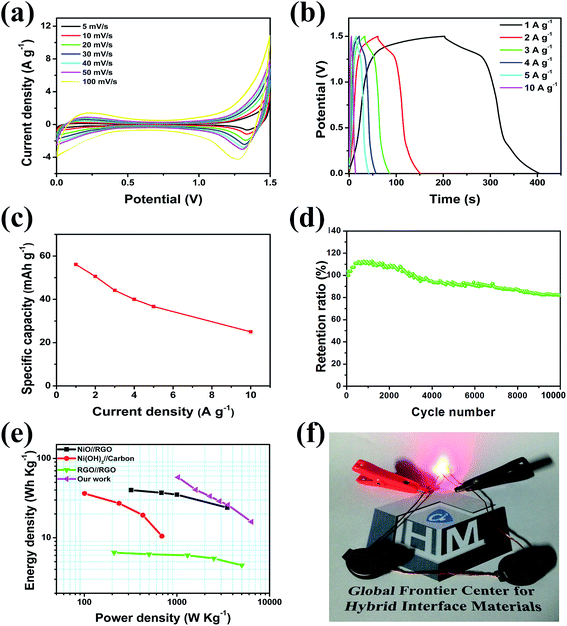
Electrodes composed of PG only, NiO only, NiO/NG, and NiO/PG were electrochemically characterized using a three-electrode electrochemical system in a 1 M KOH electrolyte solution at room temperature (25 °C). A platinum plate and mercury/mercury oxide were used as the counter and reference electrodes, respectively. Cyclic voltammetry (CV) measurements and galvanostatic charging–discharging (GCD) tests were performed on an Ivium-n-Stat electrochemical workstation (Ivium, Netherlands). The specific discharge capacity value of the electrode was calculated using the following eqn (1)2| | 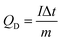 | (1) |
where QD is in mA h g−1, I (A) is the applied current, m (g) is the designated mass (of the active materials), and Δt (s) is the discharge durations.
In order to observe the potential change of each electrode, a NiO/PG material (positive electrode) and a PG material (negative electrode) were placed face-to-face and measured in 1 M KOH. The mass ratio of the active materials (NiO/PG:PG) was estimated to be 1:3.63. The GCD curves of asymmetric cells with the potential change of each individual electrode were monitored in situ by an auxiliary channel and a reference electrode (mercury/mercury oxide).29
2.7 Fabrication of the ASC
The NiO/PG//PG ASC was assembled with NiO/PG and PG as the positive and negative electrodes, respectively, with//representing a polypropylene cellulose paper separator (∼16 μm) used for ionic exchange. According to the charge balance theory, the mass ratio of the active materials (NiO/PG:PG) was estimated to be 1:3.63. A coin cell packaging (CR2032) process was adopted to assemble the ASC device. The electrolyte (1 M KOH) was added into each electrode compartment and sealed in the coin cell using a coin cell hydraulic sealing machine (Shenzhen Pengxiangyunda machinery, PX-HS-20) by pressing at 80 kg cm−2. The specific discharge capacity (mA h g−1) of the ASC device (QD) was calculated from the GCD curves according to eqn (2),| | 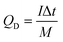 | (2) |
where I (A) is the applied current, Δt (s) is the time required for discharge, and M (g) is the total mass of the active materials i.e., the positive and negative electrodes. The energy density (W h kg−1) and the power density (W kg−1) of the NiO/PG//PG ASC device were derived from the GCD curves according to eqn (3) and (4),2| | 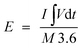 | (3) |
where E (W h kg−1) is the energy density, I (A) is the applied current, M (g) is the total active mass of both positive and negative electrodes, 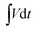 is the galvanostatic discharge current area, P (W kg−1) is the power density, and Δt (s) is the discharge time.
is the galvanostatic discharge current area, P (W kg−1) is the power density, and Δt (s) is the discharge time.
3 Results and discussion
3.1 Surface morphology change
To prepare NiO/PG on 3D Ni foam, 70% concentrated HNO3 was added into the GO suspension in deionized water while stirring. Then, the homogeneous mixture was subjected to bath sonication for 1 h at room temperature. At sufficient acoustic pressures, ultrasound creates high strain rates and frictional forces in cavitation bubbles that attack the carbonaceous surface and break the framework.30,31 Then, the strong acid HNO3 reacted with both the edge and in-plane defects in the graphene sheets, resulting in the PG precursor.25 The suspended PG precursor, hydrazine hydrate as a reduction agent, and the Ni2+ precursor were mixed homogeneously. Then, Ni(OH)2/PG materials were grown on the Ni foam using a one-pot hydrothermal method at an ambient temperature i.e., 90 °C. After sufficiently washing the composite with alcohol and water to remove unbound and unreacted materials, the Ni(OH)2/PG was converted to NiO/PG by annealing at 400 °C for 1 h under an argon gas atmosphere, which is schematically presented in Scheme 1. The NiO/NG composite was used as a reference to confirm the difference between the performance of the perforated and non-perforated graphene sheet (discussed later). The digital images of plane-view FESEM, HRTEM, and SAED patterns (insets) of the NiO and NiO/PG electrodes grown on the 3D macro-porous Ni foam are shown in Fig. 1. After the HNO3 treatment, the GO sheets were etched at random locations to form perforated GO with void sizes ranging from a few nanometres to hundreds of nanometres, as shown in Fig. S1 of the ESI.† The FESEM and HRTEM images of NiO nanosheets are presented in Fig. 1a and b. Fig. 1a verifies the presence of NiO nanosheets vertically grown onto the Ni foam. The average length of the nanosheets is 300 (±100) nm. Further characterization by HRTEM shows the honeycomb structure of NiO (Fig. 1b) with the lattice spacing of around 0.21 nm, corresponding to the (200) plan of NiO. The inset SAED pattern exhibits concentric rings, suggesting the polycrystalline structure of the developed NiO nanosheets. Fig. 1c and d presents the FESEM and HETEM images of NiO/PG electrode grown on the Ni foam. Fig. 1c and its inset show that the surface of the NiO/PG is quite rough, with many more small holes due to the perforated graphene on the NiO composite than on the bare NiO nanosheets (Fig. 1a). The HRTEM image of NiO/PG in Fig. 1d illustrates the lattice fringe with a 0.24 nm spacing, corresponding to the (111) plane of NiO, which is consistent with the XRD data. The yellow solid line in Fig. 1d highlights wrinkles that may be attributed to the elastic deformation of graphene upon the exfoliation and reassembling procedure.32 The d-spacing between the stacked graphene nanosheets is measured to be 0.41 nm, which is quite different from the (002) d-spacing of graphite, i.e., 0.34 nm,33 indicating that the graphene interlayer distances increase after the exfoliation and reassembly process. The SAED pattern of the NiO/PG composite (inset in Fig. 1d) demonstrates the polycrystalline structure of the NiO/PG.
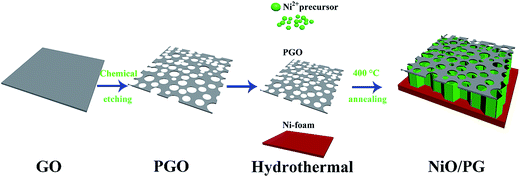 |
| | Scheme 1 Schematic of the preparation of NiO/PG. | |
 |
| | Fig. 1 Low- and high-magnification (insets) FESEM images of (a) NiO and (c) NiO/PG. HRTEM images and corresponding SAED patterns (insets) of (b) NiO and (d) NiO/PG. | |
3.2 Structural elucidation and elemental analysis
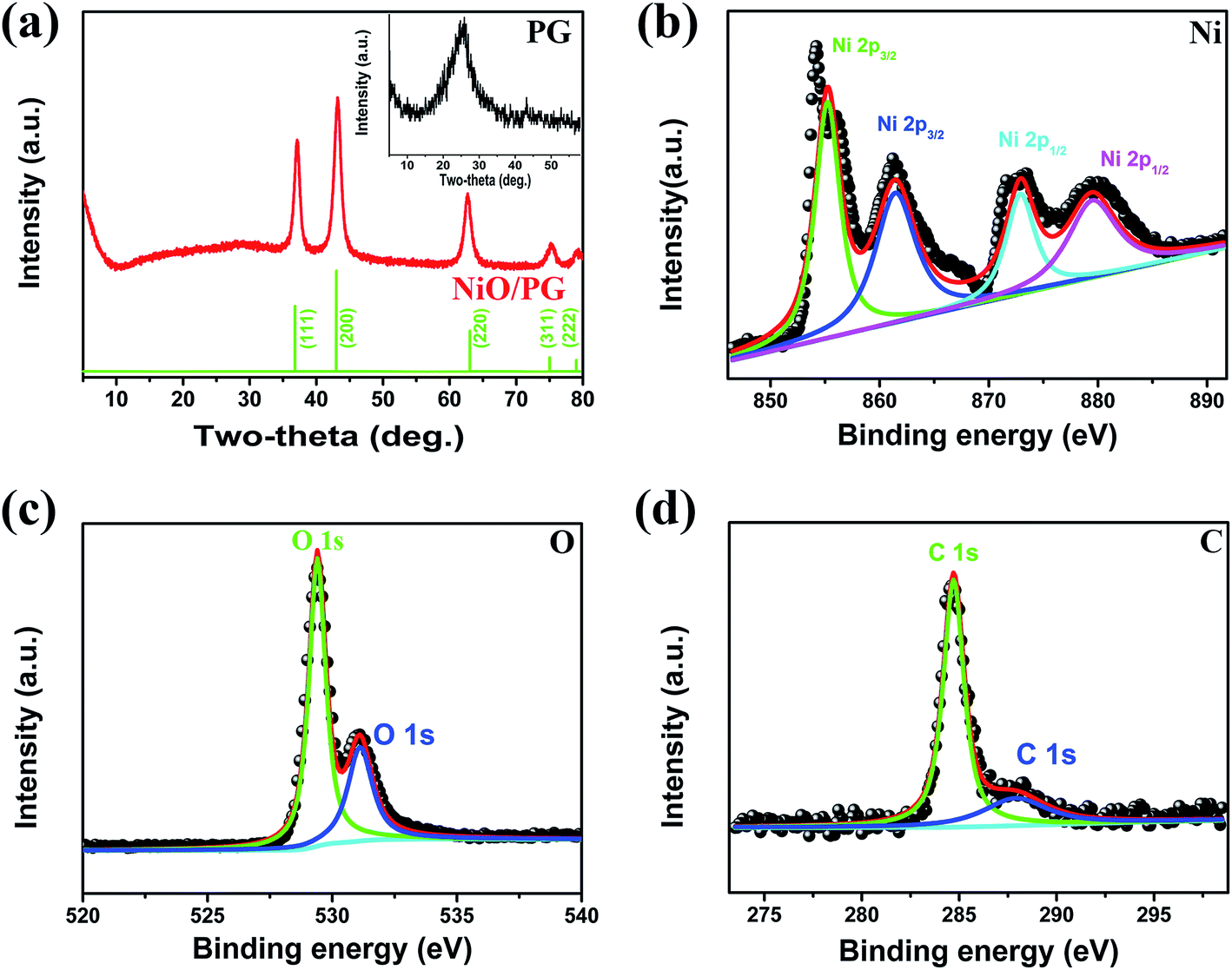
A representative XRD pattern of the as-prepared NiO/PG composite is shown in Fig. 2a. The well-defined diffraction peaks at 2θ = 37.2, 43.2, 62.8, 75.5, and 79.1° can be identified as the (111), (200), (220), (311), and (222) crystal planes of NiO phase, respectively, in agreement with the standard values (JCPDS 01-078-0643). There is a slight peak near 2θ = 20–30° indicative of the presence of graphene, which may be attributed to the hindrance of graphene restacking due to an interaction between the attached graphene sheets and the metal oxide surface34 and the eclipse of the graphene-stacking peak (inset of Fig. 2a) owing to high crystallinity of NiO.35 The presence of the broad (002) hump near 26° signifies the π–π stacking between the PG sheets, which results in an ordered crystalline structure.36 XPS measurements were conducted to examine the composition of the NiO/PG (Fig. 2b–d). In the Ni 2p spectrum (Fig. 2b), two satellite peaks for Ni 2p3/2 (855.3 eV) and Ni 2p1/2 (872.9 eV) are confirmed at 861.4 eV and 879.6 eV, respectively, which agree with previously reported values.37 In Fig. 2c, the O 1s peak at 529.4 eV is attributed to the metal–oxygen bond (Ni–O), whereas the peak located at 531.2 eV is assigned to residual Ni hydroxide.38 In Fig. 2d, the C 1s peak at 284.6 eV is the characteristic peak of graphene, whereas the peak at 288.1 eV is ascribed to residual GO resulting from incomplete reduction.19
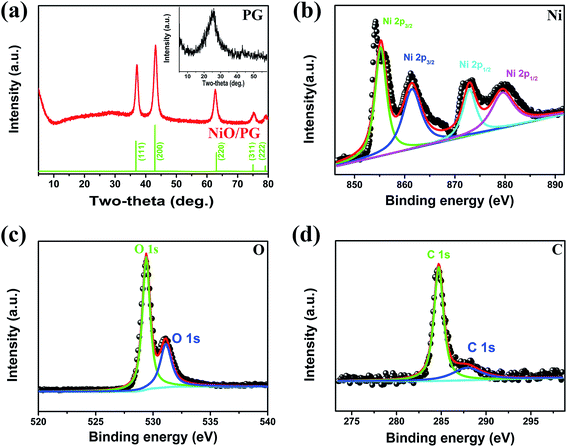 |
| | Fig. 2 (a) XRD patterns of NiO/PG and PG (inset). XPS spectra for (b) Ni 2p, (c) O 1s, and (d) C 1s obtained from NiO/PG. | |
3.3 Surface area and pore-size distribution studies
To confirm that the specific surface area of the PG composite is higher than that of the NG composite, the specific surface area and pore-size distribution of the NiO/PG and NiO/NG powders (scraped from the Ni foam) were investigated by plotting the BET adsorption–desorption isotherms and the Barrett–Joyner–Halenda (BJH) pore-size distributions (Fig. 3). Powders from both electrodes exhibit a type IV isotherm with an H3-type hysteresis according to the Brunauer and IUPAC classifications, with a lot of mesopores in the structure. The estimated specific BET surface area, pore volume, and average pore-size values for NiO/NG and NiO/PG are 34.8 m2 g−1 and 77.0 m2 g−1, 0.17 cm3 g−1 and 0.27 cm3 g−1, and 4.6 nm and 2.4 nm, respectively. The results confirm that the surface area and pore volume of NiO/PG are both higher than those of NiO/NG, which can provide many ion-transport pathways, thus leading to excellent electrochemical performance.
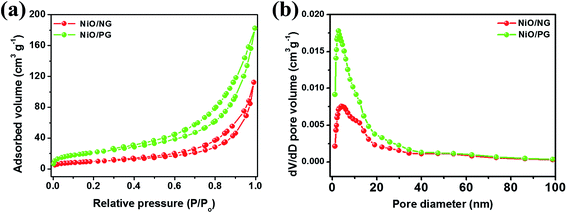 |
| | Fig. 3 (a) N2 adsorption–desorption isotherms of NiO/NG and NiO/PG, and (b) plot of pore volume vs. pore size of NiO/NG and NiO/PG. | |
3.4 Electrochemical properties of NiO/PG and NiO/NG
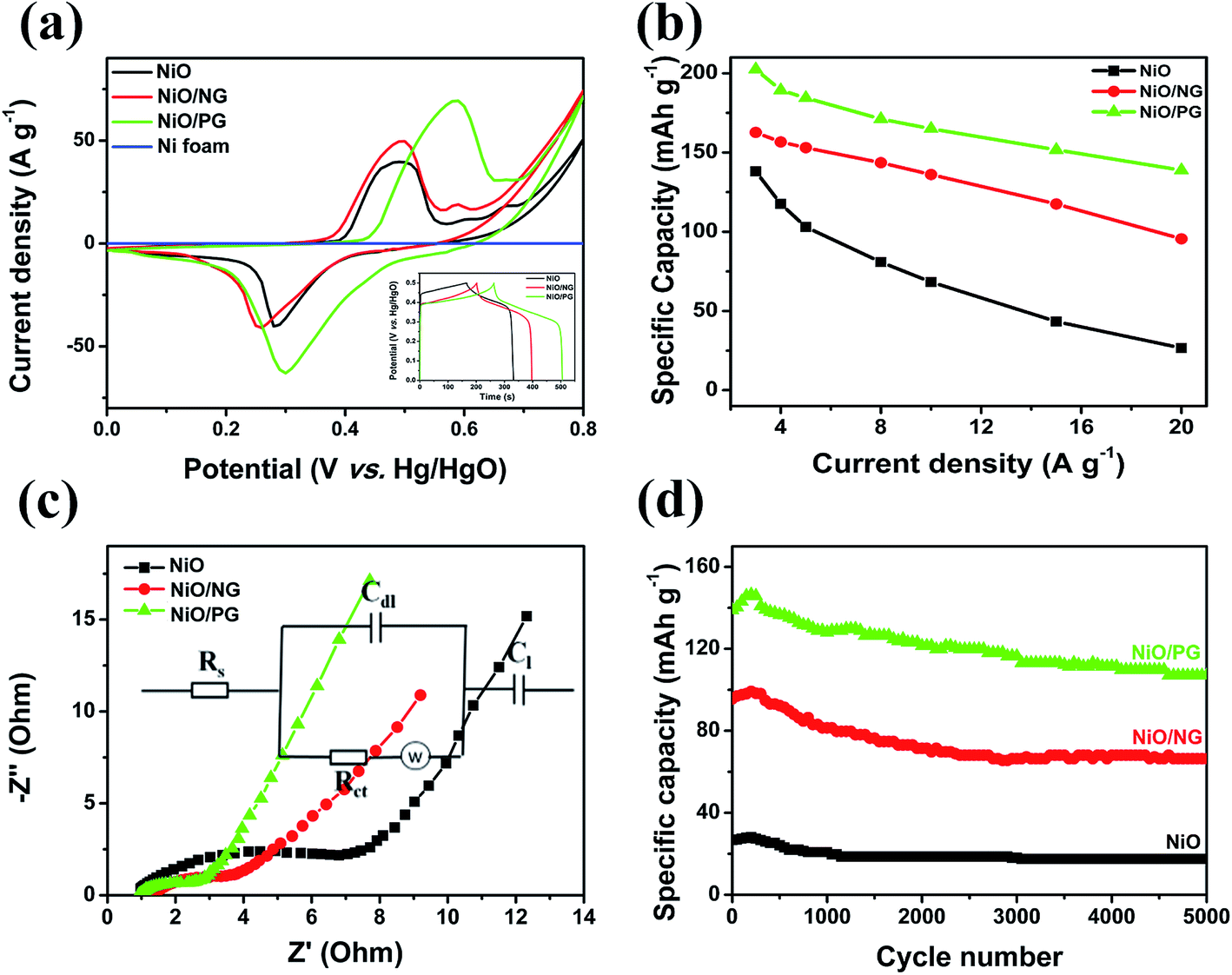
To investigate the effect of PG on the electrochemical performance, the electrochemical properties of the NiO, NiO/PG, and NiO/NG electrode materials were examined using CV, electrochemical impedance spectroscopy (EIS), and GCD and stability measurements in a 1 M KOH aqueous electrolyte and a three-electrode cell with a Hg/HgO reference electrode. We postulated that, compared with the NiO/NG, NiO/PG might serve as a promising electrochemical electrode material (Fig. S2a and b, ESI†) by (a) providing shorter electrolyte ion diffusion paths, (b) ensuring NiO nanosheets in contact with electrolyte, (c) contributing additional redox/storage sites for efficient electrosorption of ions around graphene holes, and (d) improving mechanical and electrical performances with tightly bonded NiO and graphene together. Due to the atomic-scale and two-dimensional structure of NG sheets, NG blocks much of the porous NiO architecture and impedes the access of liquid or metal ions to the NiO,39 thereby decreasing the reversible sorption of ions in an electrolyte, i.e., the ion adsorption and desorption on and in the NiO nanostructure. This indicates that lowering the specific surface area by the surface block is a factor to decrease the capacity capability of a NiO/NG electrode (Fig. S2c, ESI†). On the other hand, the PG of the NiO/PG increases the specific surface area, thus offering many pathways for electrolyte ions to access the metal oxide surface under the PG, resulting in enhanced electrochemical performance (Fig. S2d, ESI†). Fig. 4a shows the CV curves of the NiO, NiO/NG, and NiO/PG electrodes at a scan rate of 10 mV s−1, which exhibit redox anodic and cathodic peaks at ∼0.49 V and ∼0.28 V, respectively, for the NiO electrode. Meanwhile, the NiO/NG electrode exhibits anodic and cathodic peaks at ∼0.50 V and at ∼0.25 V, respectively, and the NiO/PG electrode exhibits a pair of redox peaks with anodic and cathodic peaks at ∼0.59 V and ∼0.30 V, respectively. As the redox peaks shift to a higher potential, indicating lower internal diffusion resistance and more active redox reactions, i.e., an increasing number of ions exchanging electrons on the electrode surface. These results confirm that all of the electrodes are strongly the faradaic capacitive type, and the NiO/PG electrode exhibits the highest specific capacity. The possible faradaic redox reaction is described in the following equation:23| | | NiO + OH− ↔ NiOOH + e− | (5) |
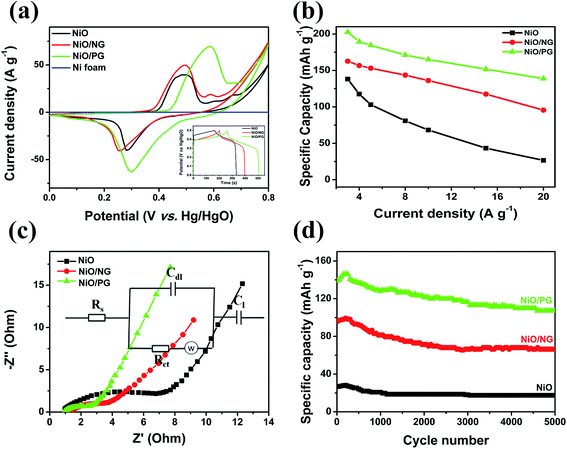 |
| | Fig. 4 Electrochemical characterizations comparing the NiO, NiO/PG and NiO/NG electrodes. (a) CV with galvanostatic charge–discharge (inset) at a scan rate of 10 mV s−1 and at a current density of 3 A g−1, respectively, (b) specific capacity at different current densities, (c) Nyquist plots before cycling tests with the equivalent model circuit (inset), and (d) cycling measurements at a current density of 20 A g−1. | |
In Fig. 4a, the CV curve of Ni foam shows a flat line, indicating that the contribution of Ni foam to the electrochemical performance of the NiO, NiO/NG, and NiO/PG electrodes can be ignored.40,41 When the CV curves of NiO, NiO/NG, and NiO/PG electrodes were measured at different scan rates (Fig. S3a, c, and e†), the current responses of all three electrodes also increase clearly and proportionally with similar curve shapes, indicating its good rate ability.42 The GCD measurements in the potential range of 0–0.5 V at a current density of 3 A g−1 were measured and are given in the inset of Fig. 4a. The charge and discharge curves were almost symmetrical for all electrodes. All three curves showed faradaic capacitive behaviour, whereas the NiO/PG displayed the longest discharge time, thus supporting its highest specific capacity value, which is in good agreement with the CV results. The improved performance could be attributed to (a) accessible electrolyte ions onto NiO through the PG holes, and (b) ion adsorption and desorption to the edge defects around perforated holes in graphene sheets, acting as a charge-storage site, thus contributing to the specific capacity.43 In contrast, the electrochemical performance of the NiO/NG is relatively inferior to that of the NiO/PG because of adversely affecting the percolation of the electrolyte ions by NG. The specific capacity values calculated from the GCD curves (Fig. S3†) within a potential window of 0–0.5 V are given in Fig. 4b. Indeed, the NiO/PG composite electrode shows a maximum capacity of 202.5 mA h g−1 at a current density of 3 A g−1 (the specific capacity values for NiO/NG and NiO are 162.7 mA h g−1 and 138.0 mA h g−1, respectively), which is much higher than those of previously reported NiO/graphene composite electrodes at the same current density.21,44–47 As the current density increased from 3 to 20 A g−1, moreover, the NiO/PG electrode exhibited an excellent high-rate performance in which the specific capacity was reduced by only 31.4%. EIS is commonly used to examine the ion-transport properties of electrode materials for SC devices. To this end, EIS plots for NiO, NiO/NG, and NiO/PG electrodes were measured, and the results are shown in Fig. 4c. The proposed equivalent circuit for the measured EIS data is composed of elements representing the Rs (internal resistance), Rct (faradaic charge transfer resistance), Cdl (double-layer capacitance), Zw (Warburg diffusion element), and CF (faradaic capacitive), as shown in Fig. 4c. Eqn (6) and (7) denote the overall impedance, Z, of the equivalent circuit in Fig. 4c:48
| | 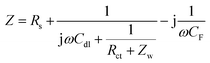 | (6) |
| | 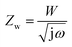 | (7) |
where j is the imaginary unit,
ω is the angular frequency (Hz), and
W is the Warburg parameter in units of Ω s
−1/2. At sufficiently high frequencies, the overall impedance can be reduced to
eqn (8), corresponding to a locus showing a semicircle that intercepts the real axis at
Rs and
Rs +
Rct in the Nyquist plot.
49| | 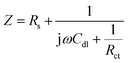 | (8) |
Each curve includes a semicircle intersecting the real axis in the high-frequency region, and a steep line at low frequencies reveals a diffusion-dominated phenomenon. The semicircle is related to the charge-transfer resistance (Rct) caused by the faradaic reactions. The diameters of the semicircles of pristine the NiO, NiO/NG, and NiO/PG electrodes are 7.0, 3.6, and 2.8 Ω, respectively, indicating the superior interfacial contacts between NiO and PG, which contributes to the efficient diffusion of electric charges during the faradaic reaction processes.46 Moreover, in the low-frequency region, the slope of the curve represents the Warburg resistance (Zw), which corresponds to the electrolyte diffusion in the porous electrode and proton diffusion in the active material electrode. The Nyquist plot of the NiO/PG electrode exhibited the steepest slope of the three electrodes, suggesting that ions easily diffuse through this electrode material. Moreover, this electrode exhibits the excellent capacitive behaviour, possibly because PG provides abundant ion-diffusion paths, facilitating the almost effortless movements of ions through the perforated voids. Electrochemical properties of the NiO, NiO/NG, and NiO/PG electrodes were further characterized by long-term cycling tests at a high current density of 20 A g−1 over 5000 cycles (Fig. 4d). The specific capacity value of the NiO/PG electrode rapidly increases from 138.9 mA h g−1 to 145.8 mA h g−1 for the first 250 cycles, then gradually decreases to 107.0 mA h g−1 with a retention rate of 77% up to 5000 cycles. Likewise, for the cycling performances of respective NiO and NiO/NG electrodes, the specific capacity values initially increase slightly and then approach to 18 mA h g−1 and 66 mA h g−1 with retention rates of 65.6% and 69.5%, of which each retention rates are 65.6% and 69.5% at the end of 5000 cycles. These experiments demonstrate that the capacity retention of the NiO/PG electrode is superior to those of the other electrodes.
3.4 Electrochemical performance of PG as the negative electrode
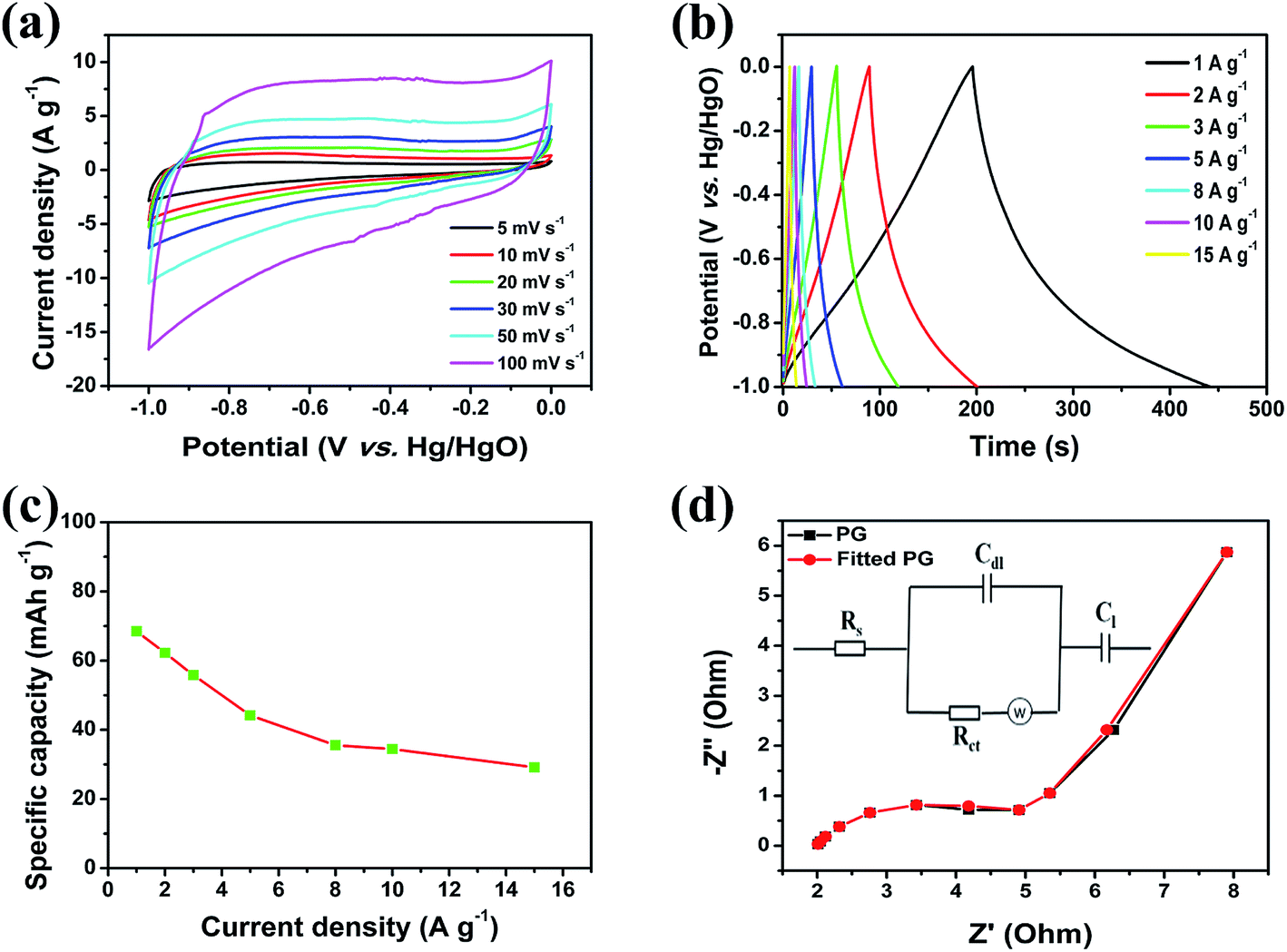
Graphene has shown promise as a material for energy storage and conversion devices because of its high surface area, good chemical stability, and excellent electrical conductivity.50–52 Recently, graphene was employed as an electrode material for SCs, but its capacity was unsatisfactory. However, the low capacity behaviour of graphene as a negative electrode material is incompatible with the high specific capacity of NiO/PG materials as the positive electrode, resulting in poor energy density values for ASCs using these materials. Therefore, to develop negative electrode materials with for high specific capacity values and moderate stability, researchers have attempted to engineer the physical and chemical properties of graphene via chemical functionalization.53 One effective approach has been to inject in-plane carbon vacancy defects into graphene to improve its capacity and rate capability using a chemical etching process. The resulting PG contained high-density ion-diffusion channels, which facilitated rapid electrolyte ion transport through the sheet, thus exposing the other side of the surface and its additional redox sites (due to perforation) and utilizing the inner active electrode sites for more electro-sorption reactions.24,54 Here, the electrochemical properties of the fabricated PG were also investigated for its use as a negative electrode in ASC devices. Fig. 5a presents the electrochemical performance of the PG electrode in a 1 M KOH solution at various scan rates in a three-electrode system. The PG-based negative electrode exhibited no remarkable peaks in any of the curves and showed ideal rectangular CV curves at various scan rates, indicating its excellent double-layer capacitive behaviour with good reversibility. The specific capacity value calculated from GCD curves (Fig. 5b) at various current densities i.e., 1.0, 2.0, 3.0, 5.0, 8.0, 10.0, and 15.0 A g−1 were 68.5, 62.2, 55.8, 44.2, 35.6, 34.4, and 29.2 mA h g−1, respectively, as summarized in Fig. 5c. Notably, the highest specific capacity value recorded (68.5 mA h g−1 at 1.0 A g−1) in the present work is superior to those previously reported on graphene-based or modified graphene-based negative electrodes.55,56 EIS was used to evaluate the electron transport properties of this negative electrode material. The experimental Nyquist plot of a PG electrode fits well the simulated curve of an equivalent circuit model. The simulated parameters obtained from the equivalent circuit model are shown in Table S1.† The impedance results of the PG electrode exhibit low Rs and Rct values of 1.86 Ω and 3.36 Ω, which can be attributed to the excellent conductivity and large porous openings of the PG/Ni foam electrode, respectively. The low charge transfer resistance of the PG electrode indicates its potential as a negative electrode for a full cell ASC device.
 |
| | Fig. 5 Electrochemical characterizations of the PG electrode. (a) CV curves at different scan rates, (b) galvanostatic charge–discharge curves at different current densities, (c) specific capacities at different current densities, and (d) Nyquist plot before cycling tests showing both experimental (black) and simulated (red) results. | |
3.5 Asymmetric supercapacitor: electrochemical properties
In order to improve the capacitive properties of the positive electrode in a hybrid asymmetric cell, it would be highly desirable to boost the performance of the negative electrode. With the purpose of the capacity balance of a positive electrode and a negative electrode, it is necessary to be a suitable mass ratio of the positive electrode:negative electrode. The GCD curves of asymmetric cells with the potential change of each individual electrode in situ were monitored by an auxiliary channel and a reference electrode (mercury/mercury oxide) (Fig. S4†). The applied current for each electrode was kept at 3 mA. The positive electrode (NiO/PG) was swept between 0 to 0.5 V, while the negative electrode (PG) was scanned at 0 to −1 V. When a mass ratio of the positive and negative electrodes was 1:3.6, both electrodes exhibited similar capacity.
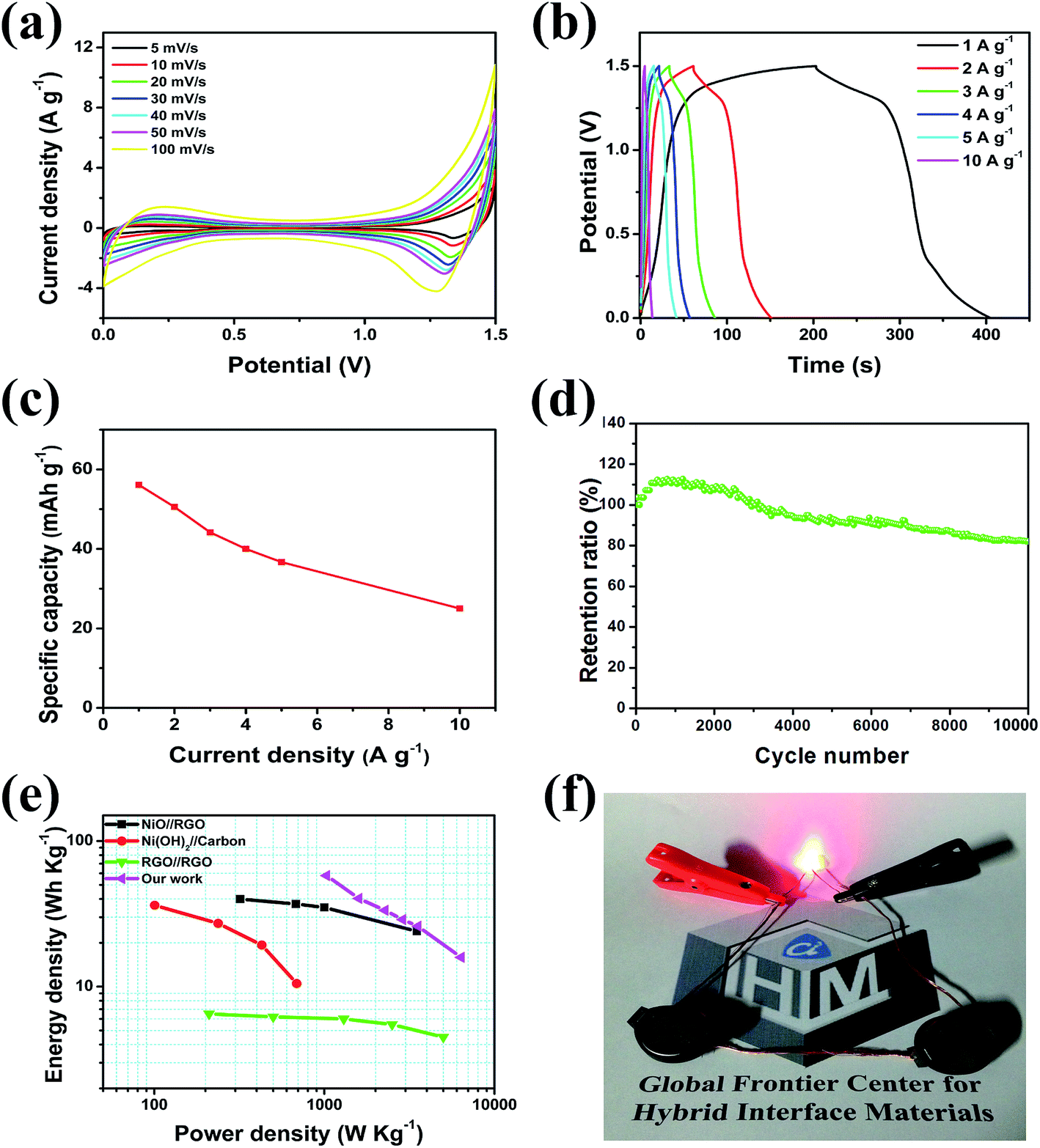
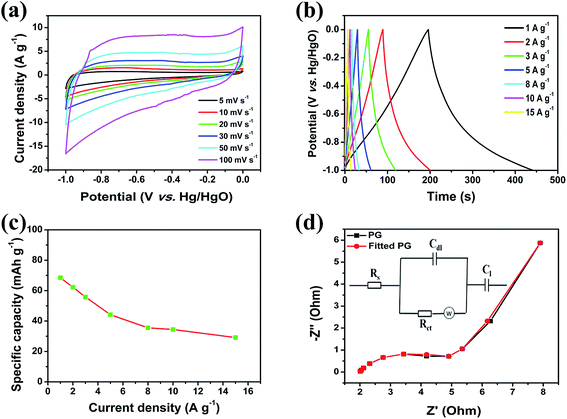
On account of the favourable electrochemical performance of the as-obtained NiO/PG as a positive electrode material and PG as a negative electrode material, an ASC device with a NiO/PG//PG configuration was fabricated using a 1 M KOH electrolyte. The CV curves of the full cell NiO/PG//PG ASC device between 0 and 1.5 V are shown in Fig. 6a. The presence of redox peaks suggests the faradaic capacitive behaviour of a NiO/PG//PG ASC. Fig. 6b shows the GCD behaviour of the NiO/PG//PG ASC device under different current densities. The capacity values of the NiO/PG//PG ASC device at current densities of 1.0, 2.0, 3.0, 4.0, 5.0, and 10.0 A g−1 are respectively 56.1, 50.6, 44.2, 40.0, 36.7, and 25.0 mA h g−1 (Fig. 6c). The GCD was used to determine the cycling performance of a NiO/PG//PG ASC. As shown in Fig. 6d, a NiO/PG//PG ASC possesses remarkable cycling stability, retaining ∼82.1% of its specific capacity after 10000 cycles at a current density of 4 A g−1, which is comparable to some previously reported NiO-based ASCs.57–60 The energy and power densities values for the NiO/PG//PG ASC device calculated from the GCD curves are presented in the Ragone plot in Fig. 6e. In the plot, the device performances were observed with an energy density of 57.8 W h kg−1 at a power density of 1030.9 W kg−1 and an energy density of 15.9 W h kg−1 at the highest power density of 6359.1 W kg−1. These values are higher than the energy densities of most of the previously reported NiO and NiO/carbon or Ni(OH)2/carbon composite-based ASCs.61–63 To further demonstrate the practical applicability of the fabricated laboratory scale NiO/PG//PG ASC, two ASC devices were connected in series, and tested for a red LED light for a minute (Fig. 6f). To observe the discharge level, we kept the LED light on for a few hundred seconds and confirmed that it lasted for up to 600 s (Fig. S5, ESI†). Thus, when the capacitor is turned both on and off, the charge is well sustained, suggesting the future commercialization potential for the device.
 |
| | Fig. 6 Characterization of the NiO/PG//PG ASC (a) CV curves under different scan rates, (b) galvanostatic charge–discharge curves, (c) specific capacity at various current densities, (d) cycling performance at a current density of 4 A g−1, (e) Ragone plot of the NiO/PG//PG ASC were compared with the NiO//RGO,58 Ni(OH)2//carbon,62 and RGO//RGO;63 and (f) a digital photograph of a red LED powered by the double ASC coin cells connected in series. | |
4. Conclusions
NiO/PG was grown uniformly on 3D Ni foam using a facile hydrothermal method. The resulting NiO/PG composed a combination of two morphologies, i.e., upright standing NiO nanosheets covered with a planar perforated graphene sheet. We inferred that this combination of different categories of materials i.e., one that exhibits faradaic capacitive (NiO) and one with electrical double-layer capacity (PG), promotes the overall specific capacity by the synergistic effects of the electron-flow paths in the graphene sheet together with the accessibility and diffusion of ions to the NiO nanosheet facilitated by the graphene perforations. The electrochemical performance and BET measurements demonstrated that the NiO/PG had a relatively higher surface area and electrical conductivity as well as a smaller pore-size distribution than those of NiO/NG. The NiO/PG exhibited a specific capacity as high as 202.5 mA h g−1 at a current density of 3 A g−1 and a good specific capacity retention of 77% after 5000 cycles at a high current density of 20 A g−1. Additively, an ASC composed of PG as the negative electrode and NiO/PG as the positive electrode was investigated for its energy performances at 1.5 V and was confirmed respectively with energy densities of 57.8 and 15.9 W h kg−1 at power densities of 1030.9 and 6359.1 W kg−1, which are certainly superior to the values reported thus far. A red LED powered by two as-fabricated ASCs connected in series was sustained for nearly 600 seconds until the light disappeared. The experimental results of the present work demonstrate the NiO/PG and PG electrodes' potential for practical device applications.
Acknowledgements
This study was supported by Global Frontier Program through the Global Frontier Hybrid Interface Materials (GFHIM) of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013M3A6B1078874).
References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- A. Laheäär, P. Przygocki, Q. Abbas and F. Béguin, Electrochem. Commun., 2015, 60, 21–25 CrossRef.
- S. Zhang and N. Pan, Adv. Energy Mater., 2015, 5, 1401401–1401420 CrossRef.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917–918 CrossRef CAS PubMed.
-
G. Ragoisha and Y. Aniskevich, arXiv preprint arXiv:1604.08154, 2016.
- S. Y. Zhang, L. Ren and S. J. Peng, CrystEngComm, 2014, 16, 6195–6202 RSC.
- C. Wang, J. Xu, M. F. Yuen, J. Zhang, Y. Li, X. Chen and W. Zhang, Adv. Funct. Mater., 2014, 24, 6372–6380 CrossRef CAS.
- Y. Bai, M. Du, J. Chang, J. Sun and L. Gao, J. Mater. Chem. A, 2014, 2, 3834–3840 CAS.
- S. I. Kim, J. S. Lee, H. J. Ahn, H. K. Song and J. H. Jang, ACS Appl. Mater. Interfaces, 2013, 5, 1596–1603 CAS.
- C. Y. Cao, W. Guo, Z. M. Cui, W.-G. Song and W. Cai, J. Mater. Chem., 2011, 21, 3204–3209 RSC.
- S. Vijayakumar, S. Nagamuthu and G. Muralidharan, ACS Appl. Mater. Interfaces, 2013, 5, 2188–2196 CAS.
- D. Han, P. Xu, X. Jing, J. Wang, P. Yang, Q. Shen, J. Liu, D. Song, Z. Gao and M. Zhang, J. Power Sources, 2013, 235, 45–53 CrossRef CAS.
- W. Ji, J. Ji, X. Cui, J. Chen, D. Liu, H. Deng and Q. Fu, Chem. Commun., 2015, 51, 7669–7672 RSC.
- L. Zhang, J. Wang, J. Zhu, X. Zhang, K. San Hui and K. N. Hui, J. Mater. Chem. A, 2013, 1, 9046–9053 CAS.
- S. Hosogai and H. Tsutsumi, J. Power Sources, 2009, 194, 1213–1217 CrossRef CAS.
- C. Wu, S. Deng, H. Wang, Y. Sun, J. Liu and H. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 1106–1112 CAS.
- S. Chen, J. Duan, Y. Tang and S. Zhang Qiao, Chem.–Eur. J., 2013, 19, 7118–7124 CrossRef CAS PubMed.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- Y. Xu, X. Huang, Z. Lin, X. Zhong, Y. Huang and X. Duan, Nano Res., 2013, 6, 65–76 CrossRef CAS.
- X. Cao, Y. Shi, W. Shi, G. Lu, X. Huang, Q. Yan, Q. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- Y. Jiang, D. Chen, J. Song, Z. Jiao, Q. Ma, H. Zhang, L. Cheng, B. Zhao and Y. Chu, Electrochim. Acta, 2013, 91, 173–178 CrossRef CAS.
- K. Liang, X. Tang and W. Hu, J. Mater. Chem., 2012, 22, 11062–11067 RSC.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 5554–5562 CrossRef PubMed.
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, ACS Nano, 2011, 5, 8739–8749 CrossRef CAS PubMed.
- C. H. A. Tsang, K. N. Hui, K. S. Hui and L. Ren, J. Mater. Chem. A, 2014, 2, 17986–17993 CAS.
- M. C. Hu, K. S. Hui and K. N. Hui, Chem. Eng. J., 2014, 254, 237–244 CrossRef CAS.
- Q. Xia, K. S. Hui, K. Hui, D. Hwang, S. Lee, W. Zhou, Y. Cho, S. Kwon, Q. Wang and Y. Son, Mater. Lett., 2012, 69, 69–71 CrossRef CAS.
- Z. Li, Z. Xu, H. Wang, J. Ding, B. Zahiri, C. M. Holt, X. Tan and D. Mitlin, Energy Environ. Sci., 2014, 7, 1708–1718 CAS.
- S. Wang, L. A. L. Tang, Q. Bao, M. Lin, S. Deng, B. M. Goh and K. P. Loh, J. Am. Chem. Soc., 2009, 131, 16832–16837 CrossRef CAS PubMed.
- G. A. Forrest and A. J. Alexander, J. Phys. Chem. C, 2007, 111, 10792–10798 CAS.
- S. M. Paek, E. Yoo and I. Honma, Nano Lett., 2008, 9, 72–75 CrossRef PubMed.
- R. Andrews, D. Jacques, D. Qian and E. Dickey, Carbon, 2001, 39, 1681–1687 CrossRef CAS.
- Z. Ji, G. Zhu, X. Shen, H. Zhou, C. Wu and M. Wang, New J. Chem., 2012, 36, 1774–1780 RSC.
- D. T. Dam, X. Wang and J. M. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 8246–8256 CAS.
- P. Sahoo, R. Aepuru, H. S. Panda and D. Bahadur, Sci. Rep., 2015, 5, 17726–17738 CrossRef CAS PubMed.
- B. Varghese, M. Reddy, Z. Yanwu, C. S. Lit, T. C. Hoong, G. Subba Rao, B. Chowdari, A. T. S. Wee, C. T. Lim and C. H. Sow, Chem. Mater., 2008, 20, 3360–3367 CrossRef CAS.
- X. Ren, C. Guo, L. Xu, T. Li, L. Hou and Y. Wei, ACS Appl. Mater. Interfaces, 2015, 7, 19930–19940 CAS.
- D. E. Jiang, V. R. Cooper and S. Dai, Nano Lett., 2009, 9, 4019–4024 CrossRef CAS PubMed.
- W. Wang, S. Guo, M. Penchev, I. Ruiz, K. N. Bozhilov, D. Yan, M. Ozkan and C. S. Ozkan, Nano Energy, 2013, 2, 294–303 CrossRef CAS.
- Q. X. Xia, K. San Hui, K. N. Hui, S. D. Kim, J. H. Lim, S. Y. Choi, L. J. Zhang, R. S. Mane, J. M. Yun and K. H. Kim, J. Mater. Chem. A, 2015, 3, 22102–22117 CAS.
- Y. G. Wang, H. Q. Li and Y. Y. Xia, Adv. Mater., 2006, 18, 2619–2623 CrossRef CAS.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- M. Huang, C. Gu, X. Ge, X. Wang and J. Tu, J. Power Sources, 2014, 259, 98–105 CrossRef CAS.
- X. Ren, C. Guo, L. Xu, T. Li, L. Hou and Y. Wei, ACS Appl. Mater. Interfaces, 2015, 7, 19930–19940 CAS.
- L. Wang, H. Tian, L. Zhang, F. Gao, J. Gu, L. Hou, Y. Jiang and C. Wu, RSC Adv., 2015, 5, 60141–60147 RSC.
- X. Feng, J. Zhou, L. Wang, Y. Li, Z. Huang, S. Chen, Y. Ma, L. Wang and X. Yan, New J. Chem., 2015, 39, 4026–4034 RSC.
- C. W. Huang and H. Teng, J. Electrochem. Soc., 2008, 155, A739–A744 CrossRef CAS.
- K. P. Wang and H. Teng, J. Electrochem. Soc., 2007, 154, A993–A998 CrossRef CAS.
- Z. J. Fan, J. Yan, L. J. Zhi, Q. Zhang, T. Wei, J. Feng, M. L. Zhang, W. Z. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- Y. B. Tan and J. M. Lee, J. Mater. Chem. A, 2013, 1, 14814 CAS.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 5226 CrossRef CAS.
- Z. H. Wen, X. C. Wang, S. Mao, Z. Bo, H. Kim, S. M. Cui, G. H. Lu, X. L. Feng and J. H. Chen, Adv. Mater., 2012, 24, 5610–5616 CrossRef CAS PubMed.
- X. Han, M. R. Funk, F. Shen, Y.-C. Chen, Y. Li, C. J. Campbell, J. Dai, X. Yang, J. W. Kim and Y. Liao, ACS Nano, 2014, 8, 8255–8265 CrossRef CAS PubMed.
- J. W. Lee, J. M. Ko and J. D. Kim, Electrochim. Acta, 2012, 85, 459–466 CrossRef CAS.
- K. Wang, L. W. Li, T. Z. Zhang and Z. F. Liu, Energy, 2014, 70, 612–617 CrossRef CAS.
- Z. H. Gao, H. Zhang, G. P. Cao, M. F. Han and Y. S. Yang, Electrochim. Acta, 2013, 87, 375–380 CrossRef CAS.
- F. Luan, G. Wang, Y. Ling, X. Lu, H. Wang, Y. Tong, X. X. Liu and Y. Li, Nanoscale, 2013, 5, 7984–7990 RSC.
- C. Z. Yuan, B. Gao and X. G. Zhang, J. Power Sources, 2007, 173, 606–612 CrossRef CAS.
- H. Wang, H. Yi, X. Chen and X. Wang, J. Mater. Chem. A, 2014, 2, 3223–3230 CAS.
- M. Yao, Z. Hu, Z. Xu, Y. Liu, P. Liu and Q. Zhang, J. Power Sources, 2015, 273, 914–922 CrossRef CAS.
- J. Huang, P. Xu, D. Cao, X. Zhou, S. Yang, Y. Li and G. Wang, J. Power Sources, 2014, 246, 371–376 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 120, 379–382 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: FESEM images of PG, schemes of the formation of NiO/NG and NiO/PG, CV and GCD measurements for NiO, NiO/NG and NiO/PG, and asymmetric device performance designed with NiO/PG//PG configuration, Fig. S1–S5. EIS fitting parameters of PG electrode, Table S1. See DOI: 10.1039/c6se00085a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Je Moon
Yun
a,
Je Moon
Yun
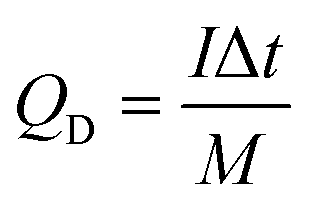
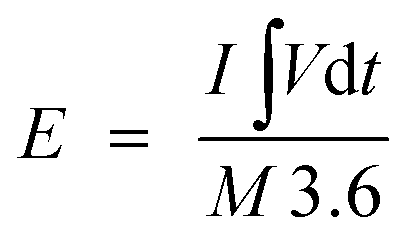
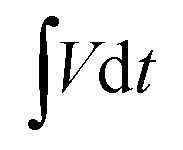 is the galvanostatic discharge current area, P (W kg−1) is the power density, and Δt (s) is the discharge time.
is the galvanostatic discharge current area, P (W kg−1) is the power density, and Δt (s) is the discharge time.