A reaction–diffusion kinetic model for the heterogeneous N-deacetylation step in chitin material conversion to chitosan in catalytic alkaline solutions
Received
14th August 2018
, Accepted 2nd October 2018
First published on 2nd October 2018
Abstract
This conversion study provides a new mechanistic insight into the modelling of the heterogeneous N-deacetylation step of α-chitin, obtained from waste crustacean shells, using a catalytic alkaline solution at different operating temperatures (25–80 °C) and concentrations (50–80 wt%). Transient particle-size distributions for two separate experiments with smaller powder or larger flake morphologies were obtained by applying an inline tracking system. The degree of deacetylation (DDA), polymer molecular mass and viscosity of the deacetylated raw resource were continuously monitored with time until the maximum DDA was reached. The mechanism of the conversion of an average biopolymer chain was described with a reaction–diffusion kinetic model, solved for all fraction intervals, and optimised. The effective diffusivity coefficient was estimated with regression analysis; the geometry of the particles was approximated as having spherical dimensions, and material isotropy was presumed. A second-order reaction process took place, since the content of the dissolved hydroxyl ions inside the pores was not considered constant. Additionally, several analytical tools such as scanning electron microscopy (SEM) was employed as well as specific porosity measurements to get a deeper phenomenological understanding of the material's morphological transformation. The selected mathematical relationship granted a relatively good agreement for the cumulative experimental data by regarding the kinetics in the initial consumption phase, as well as the subsequent transport resistance for OH−. The developed descriptive approach, together with online measuring methods, could enable a more comprehensive option for commercial productivity increase, as well as unit operations scale-up.
1. Introduction
Chitin is considered the second most abundant organic resource on Earth next to cellulose, with the estimated annual worldwide natural production rate of approximately 1011 tons. It is presented as ordered microfibrils, which is the major structural component in the exoskeletons of marine invertebrates, insects and the cell walls of fungi.1,2 The main commercial sources for its isolation are cuticles of various crustaceans, principally crabs and shrimps, which are available in abundance as waste from the food industry (6 million to 8 million tons are produced globally).3 Chitin is a nitrogenous polysaccharide, a copolymer of N-acetyl-D-glucosamine and D-glucosamine units linked by a β-(1–4) glycosidic bond, and it is structurally similar to cellulose, having acetamido groups at the C-2 positions instead of hydroxyl groups. The structure of chitin is hard and inelastic, and in nature exists in three different polymorphic forms α, β, and γ, which differ in the arrangement of the chains within the crystalline regions. α-Chitin is by far the most abundant form and is usually found where extreme hardness is required.4–6 This type of chitin is mainly used as a raw material to produce chitin-derived products such as: chitosan, chitin/chitosan derivatives, oligosaccharides and glucosamine. An increasing number of useful products derived from chitin continue to attract commercial development.7 Chitin has become of great interest not only as an under-utilised resource but also as a new functional biomaterial of high potential in various fields and as a huge potential nitrogen source.8 Chitosan represents the deacetylated form of chitin (that can have varying degrees of deacetylation), and in comparison to acetylated chitin it becomes soluble in acidic solutions (sometimes with difficulty). Chitosan has three types of reactive functional groups: an amino group and both primary and secondary hydroxyl groups at the C-2, C-3, and C-6 positions, respectively.9 Despite its biotechnologically relevant properties, chitosan can be further improved in terms of its bioactive features and physical–chemical behaviour. Improved properties from chemical modifications of chitosan have been reported in different fields of biomedical applications including wound dressings, tissue engineering, implant coatings and therapeutic agent delivery systems.4–10 Chitin N-deacetylation is possible either by enzymatic biotransformation or chemical hydrolysis.11–13 For industrial chitosan production, chemical deacetylation in heterogeneous media involving strong alkaline solutions remains the most studied one.14,15 Under such severe conditions, the chitin biopolymer undergoes hydrolysis, for which the reaction rate was strongly linked to the crystalline packing of the copolymer, and it is temperature dependent and oxygen-sensitive.16,17 So far, the complete deacetylation of chitin was not attained in a single-stage treatment with concentrated alkali. Several literature explanations were suggested for the decrease in the rate of chitin deacetylation in the alkaline solutions leading to the steady state regardless of the process time. The accessibility of N-acetylglucosamine (GlcNAc) sites was one of the key parameters considered in the deacetylation effectiveness, where the role of the alkali hydrates formed in solution, with respect to the alkali concentration and temperature, and their influence on activation energies, have been further considered.18 The kinetics of the alkaline N-deacetylation of chitin was mostly presented as a pseudo-first order reaction.19–21 However, some authors have reported that under different alkali concentrations, the heterogeneous deacetylation of chitin appeared to be more complicated and it might be controlled by a higher-ordered reaction and a diffusion-controlled process.22 In this study, the reaction–diffusion kinetic model of the heterogeneous N-deacetylation of α-chitin was developed and confirmed using the number and size of ground and powder chitin particles obtained using an in line particle tracking system in a series batch reactor. The governing equations for the reaction–diffusion model were solved for all measured particles. Quantitative analysis of two separated chitin morphologies was performed by applying different analytical tools: SEM, Brunauer–Emmett–Teller (BET) surface area analysis and Barrett–Joyner–Halenda (BJH) pore size and pore volume analysis and linked to the developed kinetic model. SEM analytical measurements were also employed to determine the thickness of the different chitosan samples. Several of the chitosan samples obtained at different temperatures were measured at multiple locations and the average flake thickness was obtained. The experimental observations for the heterogeneous N-deacetylation reaction and approximations with the spherical particle were used in the developed model, and effective diffusivity was used as a fitting parameter for the experimentally obtained data at different temperatures. Since the hydroxyl ions have to diffuse with the solvent inside chitin particle pores, the concentration was not considered as constant, and the reaction between the chitin's amide group and hydroxide ion was represented as a second order reversible reaction.
2. Results and discussion
2.1 Chitin particle morphology and particle size distribution
For the purpose of this work, chitin particle analysis, mainly focusing on the parameters, which could be useful for the mathematical model development, was performed. First, the selection of two distinct commercially available chitin samples was performed. In Fig. 1A and B, the typical shapes of the chitin in the form of fine powder and large chitin particles (flakes) are shown.
 |
| | Fig. 1 Representation of the particle shape and geometry of the commercial chitin samples in the form of A) powder and B) flakes. | |
Both types of chitin shapes were then analysed for their particle size distribution using a focused beam reflectance measurement (FBRM) probe. The FBRM probe was immersed in a flowing suspension of particles as described in section 3.5. Assessment of the particle size was performed by measuring the number of laser beam interruptions with constant circumferential speed, which gave the distribution of particle chord lengths. The FBRM probe was adjusted in the suspension of particles (in the range of 1–1000 μm). Intuitively, the counts of chord length in different channels of FBRM statistics can be associated with the number of particles with different sizes. However, as can be observed in Fig. 1A and B, the chitin samples have a roughly elliptical shape with an uneven surface. Even more important was the ratio between the length, width and depth of the particle, where the latter was much lower (≈40 μm), resulting in unreliable particle size estimation for the chitin particles. The particle size was measured in terms of the chord length, which is defined as the distance between the two edges of a particle.23 Therefore, the distribution of the chord lengths for both chitin samples corresponds well with the depth of the particles, which for the heterogeneous deacetylation of chitin corresponds to the shortest diffusion path of the NaOH molecules into the chitin polymer matrix. In Fig. 2, the comparison of the chord length distribution for chitin flakes and chitin powder is presented. Both samples have their highest chord length in the range of 10–50 μm, which lies in the range of the α-chitin thickness of 16 ± 3 μm (Table 1). Lower fractions correspond to the parts of the particles, which were detached from the flakes. The results in Fig. 2 also show that grinding of the shrimp shells leads to a more uniform distribution, with more than 50% of all measured lengths in the range of the typical chitin particle depth. Meanwhile, the non-mechanically treated chitin from the shrimp shells has a more broad distribution, especially in the lowest fraction 1–5 μm, which could correspond to the parts detached from the bulk particle. Additionally, some fine powder was observed on the particles in the form of flakes, which could also have been retained from the isolation step. For the systematic study of the deacetylation process used to obtain chitosan, the diffusion of concentrated alkaline inside the fiber is crucial and is inhibited by low porosity and high crystalline degree of the sample.24 Therefore, SEM analysis of the surface and cross-section of the chitin particle was performed. In Fig. 3A, the surface of the chitin is presented as a composition of microfibrils which are entangled together with large surface pores. The porous structure of chitin enables good reactivity and large surface availability for the reaction in the initial stage. After deacetylation, the structure of the chitosan surface is rough and scarred with irregular fibrils (Fig. 3B–D). The N-deacetylation process results in the redistribution of N-acetyl groups with amino groups, which form weaker connections and less porous structures, which is additionally examined by specific porosity and pore volume determination in section 2.2.25,26
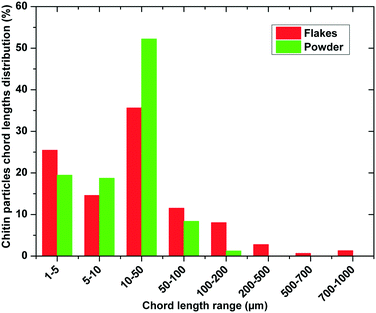 |
| | Fig. 2 Chitin particle chord length distribution in a suspension measured using an inline particle tracking (FBRM) probe. | |
Table 1 Experimental data of two types of commercial chitin samples after 12 h of deacetylation at different temperatures and NaOH concentrations
| Chitin |
Temperature (°C) |
Thickness after 12 h (μm) |
NaOH (wt%) |
DDA after 12 h (%) |
Chitosan molecular weight (kDa) |
Dynamic viscosity (PaS) |
| Flakes |
25 |
15 ± 1 |
50 |
41.2 ± 0.6 |
nd |
nd |
| Flakes |
25 |
15 ± 2 |
60 |
52 ± 1 |
nd |
nd |
| Flakes |
25 |
15 ± 1 |
70 |
77.6 ± 0.8 |
nd |
nd |
| Flakes |
25 |
16 ± 2 |
80 |
62.4 ± 0.2 |
nd |
nd |
| Flakes |
50 |
15 ± 2 |
70 |
72.3 ± 0.4 |
nd |
nd |
| Flakes |
60 |
13 ± 2 |
70 |
74.2 ± 0.8 |
430 |
767 |
| Flakes |
70 |
13 ± 2 |
70 |
76 ± 1 |
543 |
968 |
| Flakes |
80 |
12 ± 2 |
70 |
82.1 ± 0.7 |
619 |
1101 |
| Powder |
50 |
nd |
70 |
72.2 ± 0.4 |
nd |
nd |
| Powder |
60 |
nd |
70 |
79 ± 1 |
123 |
222 |
| Powder |
70 |
nd |
70 |
81.1 ± 0.9 |
129 |
230 |
| Powder |
80 |
nd |
70 |
89 ± 1 |
134 |
240 |
 |
| | Fig. 3 SEM images of the commercial chitin isolated from the shrimp shells: A) before deacetylation; B) after deacetylation (60 °C; DDA = 74.2%); C) after deacetylation (70 °C; DDA = 76.3%); D) after deacetylation (80 °C; DDA = 82.1%). | |
2.2 Alkaline N-deacetylation of α-chitin
For a better understanding of the morphology and changes in the chitin molecule itself during the deacetylation reaction, the molecular mass, viscosity and porosity of the samples were monitored, where it has been established that all the parameters affect the kinetics of alkaline N-deacetylation (Table 1). The N-acetyl groups in the chitin polymer are partly deacetylated to amino groups in the heterogeneous catalysis, where the degree of the deacetylation is mainly affected by the concentration of the alkali, temperature, reaction time, pre-treatment of chitin, particle size and chitin concentration.27 Similar to that case, literature data had shown that the increase of the NaOH concentration leads to an increase in the deacetylation degree to a certain limit value. Also, many authors studied the alkaline deacetylation of β-chitin from squid pens, which showed that at a temperature of 100 °C and at a concentration of 60% NaOH, the DDA reaches over 90% due to the weaker hydrogen bonds in this molecule. In the case of α-chitin, due to its stronger hydrogen bonds and tightly packed sugar rings, alkaline N-deacetylation is much slower and higher concentrations of NaOH are required for N-deacetylation.28,29 A higher concentration of NaOH leads to the degradation of the crystalline structure of the chitin molecule, since during the deacetylation process, the degradation of the main chain occurs, which leads to the considerable decrease in the biopolymer's molecular weight. This is due to the reduction of rigidity of the molecular chain and the increase in the electrostatic repulsion force of the ionic groups along the polyelectrolyte chain in the chitosan solution when the DDA of chitosan increases gradually.30 These data have been confirmed in our experiments and the highest DDA at 25 °C reached 77.6% at a concentration of 70 wt% alkali solution, while further increases resulted in a lower DDA (Table 1). The differences of both α-chitins were also in the molecular mass and viscosity of the chitosan solution, which in both cases (flakes and powder chitin) have increased with the increasing DDA. The powder samples had a lower molecular mass and smaller thickness compared with the flakes. Based on the increase in the DDA, the conclusion is that the NaOH path was much shorter and accessible in ground particles, where the N-deacetylation reaction being rapid and having a higher degree of deacetylation was achieved.28,29 For the determination of the Mw, eqn (4) was employed. Mark–Houwink parameters, which are necessary for calculating the molecular mass and are strongly dependent on the degree of deacetylation, temperature, and solvent type, were obtained from the literature.30 For example, constant a has been reported to decrease from 1.12 to 0.81 while Km increased from 0.1 to 16 × 10−5 [dL g−1] as the degree of deacetylation increased from 69 to 100%. Based on eqn (4), Mw strongly depends on the sample's polydispersity, especially the Km constant. For a given set of values for constants a and Km, it was shown that Km is underestimated when the polydispersity of the sample increases, thus leading to overestimated molecular weights.30 In the study performed by Bajaj et al. (2011),31 the viscosities and molecular mass of chitin and chitosan extracted from the shrimp shells were highly dependent on the method of preparation, even when the degrees of deproteination and decalcification were similar to those of the end product. In addition, several factors in the production of chitosan, such as high temperature, concentration of alkali, reaction time, previous treatment of the chitin, particle size, chitin concentration, dissolved oxygen concentration and shear stress may also influence the molecular weight of chitosan.31 The huge surface availability and large specific surface area in the initial chitin sample and the further decrease in the porosity, which has an impact on the kinetics of deacetylation, has been explained by using BET surface analysis.
From the results, (Table 1) it can be concluded that the temperature has a positive effect on the increase in the DDA; there is a decrease in the specific surface area of the chitin/chitosan biopolymer matrix, including all pores on the surface. The average pore volume per gram of sample indicates a decrease in the porosity of the surface. The reduction of porosity on the surface of the treated chitin samples is due to the redistribution and weakening of hydrogen bonds. Additionally, this observation was confirmed by the cross-section analysis of the flakes with SEM. As can be seen in Table 2 and Fig. 3, the thickness of the flakes is reduced from 16 ± 2 μm in the chitin to 12 ± 2 μm with the higher DDA obtained. The treatment of chitin molecules at higher temperatures also reduced the pore diameter at insignificant values, observing the average diameter for all samples obtained at one temperature by increasing the relative absorption pressure (p/p0). Investigations on the water vapour sorption by the chitosan samples revealed that the sorption ability decreases with the increasing deacetylation degree of the samples. This behaviour was explained by the influence of the crystallinity of the samples on the sorption ability.32
Table 2 BET surface area, average pore volume and average thickness of the chitosan obtained from the flakes by deacetylation with 70 wt% NaOH solution
|
T (°C) |
DDA after 12 h (%) |
BET specific surface area (m2 g−1) |
Total pore volume (cm3 g−1) |
Thickness of the particles (μm) |
| 60 |
74 ± 1 |
2.19 ± 0.06 |
0.008 ± 0.002 |
13 ± 3 |
| 70 |
76 ± 1 |
1.92 ± 0.05 |
0.008 ± 0.002 |
13 ± 2 |
| 80 |
82.0 ± 0.7 |
1.32 ± 0.04 |
0.004 ± 0.004 |
12 ± 2 |
By the N-deacetylation reaction, the crystal structure in the chitin molecule is disturbed. There is a destruction of hydrogen bonds that maintain the properly packaged and regulated N-acetyl glucosamine and D-glucosamine molecules in the chitin polymer. The elevated temperature and the concentrated NaOH solution led to an increase in the amorphous region and an irregular orientation of the N-acetylated chitin chains.33
2.3 Kinetics of α-chitin N-deacetylation and validation of the reaction–diffusion model
The highest number of the particle chord lengths measured was, in both samples (Fig. 1), in the range of 10–50 μm. Therefore the experimental data for both chitin samples after 3 hours of N-deacetylation are very similar, which is explained by the wetting of the surface and diffusion into the smaller particles, followed by N-deacetylation. A slower conversion of chitin to chitosan is due to the reaction of larger unreacted particles or particles with closed pores, which require more time for the deacetylation of all groups, which are also more tightly connected due to higher Mw via hydrogen bonding. This is also confirmed by the higher DDA obtained for the powder sample (Table 1). Amide hydrolysis was thoroughly studied because of its importance in biochemistry. Numerous studies focused on the mechanism of reaction.34–36 The reversibility of the base amide hydrolysis was proposed for the reaction in poly (N-vinylformamide).37 Other authors38–40 usually assumed that the reaction is of pseudo-first order based on that the concentration of NaOH is significantly higher than that of chitin and stays practically constant during the course of the reaction. Because we used a reaction–diffusion approach and the concentration of OH− inside the chitin particle is not constant, a second order reaction was applied. The mathematical model and reaction are discussed in section 4.1. The parameters used in the model are presented in Table 3. In Fig. 4, the decrease in the unreacted chitin fraction with time at three different temperatures is shown. The concentration profiles obtained from the model are presented using lines, and the experimentally determined fraction of unreacted chitin is presented using experimental points. The values for effective diffusivity and both reaction rate constants were fitted to the experimental data, shown in Fig. 4, at 60 °C for the powder and flake samples. The modelled chitin conversion with the value obtained for the effective diffusion coefficient at 60 °C also gave a good agreement with the experiments at 70 and 80 °C, which is why no further temperature dependence of the parameter was investigated in this narrow temperature range. It should also be noted that the change in the effective diffusion coefficient with the change in the material density and porosity during the reaction was not taken into account since it would be difficult to obtain a correlation with acceptable precision. The activation energy for the forward reaction was obtained from the literature,40 while the activation energy for the backward reaction was also fitted to the experimental data. Reaction rate constants were the same in both cases but the effective diffusivity was different. For the other two temperatures, at 70 and 80 °C, the reaction rate constants were obtained using the modified Arrhenius equation (eqn (11) and (12)). The values of the kinetic constants in Table 3 show that the reaction rate of the N-deacetylation increases with increasing temperature, which is in accordance with the experimental data. A good agreement between the model and experimental data for both flakes and powder can be seen in Fig. 4, especially for the first three hours where the reaction is fast, due to the minimal open amorphous regions of the α-chitin particles on the surface. Thus, the selected kinetic model can be used to properly describe the N-deacetylation reaction of α-chitin. The effective diffusivity is higher for the bigger particles (chitin in flakes). The effective diffusivity coefficient is proportional to the porosity, therefore the flakes could have higher porosity, but since the parameter was fitted and an approximation of the spherical particles was used, this may be a consequence of approximation. In reality, the path of diffusion is probably shorter than the radius of spherical particles, because, by approximating large particles with spheres, the realistic geometry with large “pores and holes” in them is not considered, due to which the sodium hydroxide solution can quickly penetrate inside, and the diffusion of OH− ions can proceed from the wetted surface area within the particle. This way the path for diffusion is shorter than it is in the model, leading to the higher effective diffusivity coefficient obtained by fitting, which is more pronounced in the case of larger particles. The diffusion of OH− ions proceeds from the wetted surface area within the particle. This way the path for diffusion is shorter than it is assumed in the model, leading to a higher effective diffusivity coefficient obtained by fitting. The chitin powder particles are better approximated with a sphere, and the effective diffusivity coefficient should be closer to the real value. Furthermore, the Mw of the chitin in the form of chitin powder was also lower compared to the one in flakes, which could also play a vital role in the particle diffusion transformation during N-deacetylation; here, the mass of the particles is reduced, due to the formation of the sodium acetate as the side product.
Table 3 Parameters used in the reaction–diffusion model
| Chitin |
D
eff (m2 s−1) |
k
f (m3 mol−1 s−1) |
k
r (m3 mol−1 s−1) |
E
af (kJ mol−1) |
E
ar (kJ mol−1) |
| Powder |
3.77 × 10−11 |
2.3 × 10−4 |
1.5 × 10−4 |
50 |
10 |
| Flakes |
7.93 × 10−10 |
2.3 × 10−4 |
1.5 × 10−4 |
50 |
10 |
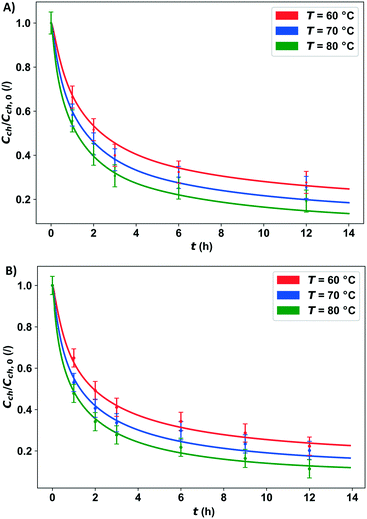 |
| | Fig. 4 Experimental data corresponding to the developed reaction–diffusion model of the α-chitin concentration profiles for N-deacetylation with an alkaline solution using: A) small chitin particles (powder) and B) large chitin particles (flakes) at 60, 70 and 80 °C. | |
From the results of the modelling presented herein, it can be concluded that the morphology and porosity of the chitin particles play an important role in the deacetylation process. Additionally, it was confirmed that the reaction inside the chitin particles can be considered as a reversible one; nonetheless, the thermodynamic (and kinetic) process analyses have indicated that, while it is reversible indeed, the backward mechanistic step is most probably desorption-determined due to transition state energetics.
The experimental data in Fig. 4 show the influence of the temperature on the N-deacetylation reaction. As previously mentioned, temperature is a significant factor for this reaction. With the temperature increase, the N-deacetylation reaction is faster; also, the DDA is higher at higher temperatures for the same reaction time. At the temperature of 80 °C, the highest DDA of 82.1% was achieved after 12 h, in the case of the flakes, while a DDA of 89.4% was obtained for the same time period for the powder. By studying the influence of the α-chitin particle size on the reaction rate (Fig. 4), it can be concluded that the small particle fractions in both commercial chitin samples react the fastest. More than 50% of chitin is reacted in 1.6 h at 60 °C in both cases. With higher temperature, a higher degree of deacetylation can be achieved in both cases, as can be concluded from Fig. 4, where the fraction of unreacted chitin N-acetylated groups decreases with temperature. Because the reaction is reversible, the rate of reaction decreases with time and the concentration of amide groups in chitin approaches equilibrium. According to the model, the final unreacted chitin percentage at 80 °C would be 19% and 11% for the flakes and powder, respectively.
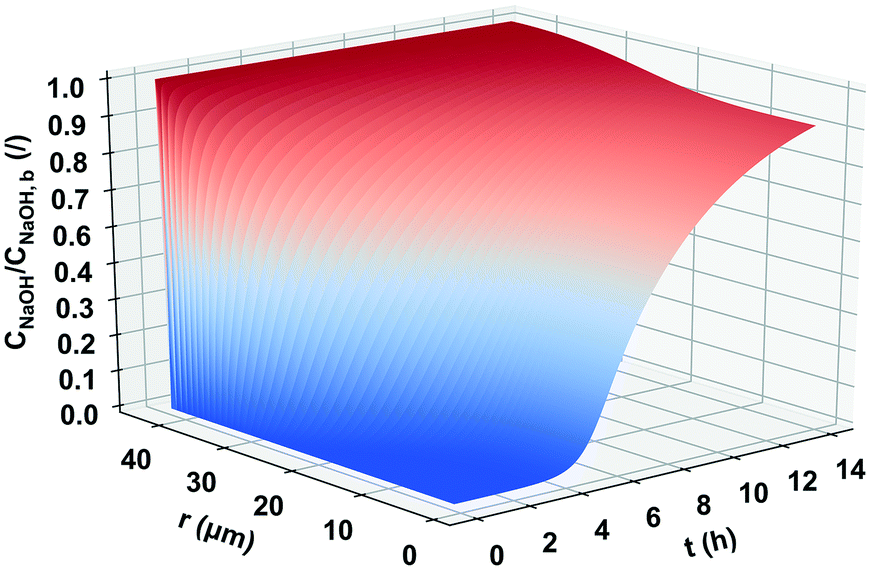
One of the factors that were also examined in order to optimize the reaction of N-deacetylation was the mixing rate. The applied stirrer speed ranged from 200–800 rpm, and as it is shown in Table 4, the mixing speed has no significant effect on the reaction, since the DDA did not change. Since the mixing rate did not have any effect on the overall reaction rate, any attrition occurring due to stirring could be considered negligible. Based on the explained and the measured morphology transformation of the chitin during the deacetylation, the heterogeneous N-deacetylation took place preferentially in the amorphous region of the particle, afterwards proceeding from the edge to the inside to the crystalline region and reaching equilibrium due to the transport limitation. The amount of N-acetyl-D-glucosamine residues continued decreasing during deacetylation, whereas D-glucosamine residues continued to increase in the α-chitin. The degradation of the polymer chain occurs simultaneously with the deacetylation, and may interfere and compete with the deacetylation, resulting in the dependence of the N-acetylation on the concentration and temperature and the levelling off of the DDA. Therefore, we can confirm that the mechanism of the heterogeneous N-deacetylation must be controlled by both the reaction and diffusion.41,42 In Fig. 5, the typical concentration profile of the OH− ions inside a chitin particle is shown. The particular profile was calculated for the particle with a diameter of 68 μm in the small particle size distribution (powder). It can be seen that for this relatively large particle size, 14 hours is enough for the hydroxide ions to reach the centre of the particle and react with chitin, but not to a reach steady state. From the beginning of the reaction to the first 4 hours, there are practically no hydroxide ions in the centre of the particle and therefore the reaction cannot occur. Similarly, a steady state is not achieved for the largest particles in this particle size distribution. For smaller particle size classes in the particle size distribution from the powder sample, a steady state is reached within 14 hours (not shown). The powder with a narrower size distribution should therefore be used for deacetylation so that the reactant could diffuse throughout the particles in approximately the same time for all particles. This way uniform conversion of chitin to chitosan could be achieved in all of the particles. If a large particle size distribution is used, a small number of large particles represent the majority of the chitin mass and at the end of reaction time; a large portion of unreacted chitin can be retained inside those particles. The overall conversion of chitin can thus be quite small even though chitin in the smallest particle is completely N-deacetylated to chitosan.
Table 4 Influence of the mixing speed on the DDA of the chitin/chitosan samples. The concentration of NaOH was 70% and T = 80 °C
| RPM |
DDA 1 h (%) |
DDA 3 h (%) |
DDA 6 h (%) |
DDA (12 h) (%) |
| 200 |
34 ± 1 |
52 ± 1 |
62.8 ± 0.9 |
89.4 ± 0.4 |
| 400 |
33.8 ± 0.6 |
51.9 ± 0.8 |
62.8 ± 0.1 |
88.5 ± 0.2 |
| 500 |
31.5 ± 0.9 |
52.0 ± 0.4 |
62.8 ± 0.2 |
88.9 ± 0.1 |
| 700 |
33.4 ± 0.1 |
51.9 ± 0.2 |
62.8 ± 0.4 |
88.4 ± 0.6 |
| 800 |
33.1 ± 0.8 |
51.9 ± 0.6 |
62.8 ± 0.2 |
89.1 ± 0.2 |
 |
| | Fig. 5 Concentration profile of NaOH diffusing into and reacting inside the α-chitin particle (spherical shape) with a radius of 34 μm. | |
3. Experimental
3.1 Materials and methods
Chitin from shrimps (practical grade, powder), chitosan (high molecular weight), and chitin from shrimp shells (practical grade, coarse flakes) were purchased from Sigma Aldrich (Steinheim, Germany). Sodium hydroxide pellets, hydrochloric acid (32%), and acetic acid (glacial 100%, anhydrous), all of analytical grade, were purchased from Merck KGaA (Darmstadt, Germany).
3.2 Heterogeneous N-deacetylation of the shrimp shell chitin biopolymer in an alkaline solution
First the different concentrations of NaOH solution (50–80 wt%) were prepared and 1 g of chitin was added to a flask. The mixture was kept at room temperature (24 °C) for 24 hours. Chitin samples were taken every 3 hours. Each sample was washed and dried at the temperature of 40 °C. After complete drying, the samples were analyzed, determining the degree of deacetylation as described in section 3.3.
The ratio of chitin and NaOH solution (70 wt%) was 1 g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 mL (w/v) and was used for all the kinetic experiments. The N-deacetylation reaction was performed for 12 h in 100 mL parallel batch reactors (Radleys, Manchester, UK) at different mixing speeds (200–800 rpm) and different temperatures (50–80 °C). Samples (cca. 200 mg) of the solids were taken every 3 h, washed with MQ water and dried in an oven at the temperature of 40 °C. The samples were analyzed using the methods described in sections 3.3 to 3.8.
:14 mL (w/v) and was used for all the kinetic experiments. The N-deacetylation reaction was performed for 12 h in 100 mL parallel batch reactors (Radleys, Manchester, UK) at different mixing speeds (200–800 rpm) and different temperatures (50–80 °C). Samples (cca. 200 mg) of the solids were taken every 3 h, washed with MQ water and dried in an oven at the temperature of 40 °C. The samples were analyzed using the methods described in sections 3.3 to 3.8.
3.3 Determination of the degree of deacetylation (DDA)
0.125 g of soluble chitin (chitosan) was dissolved in 25 mL of 0.1 mol dm−3 hydrochloric acid and stirred using a magnetic stirrer for 30 min until all particles were totally dissolved. The solution was titrated with 0.1 mol dm−3 standard solution NaOH using two drops of phenolphthalein as an acid–base titration indicator. The degree of deacetylation was calculated using (eqn (1))| |  | (1) |
where: c1 is the HCl concentration in mol dm−3, c2 is the NaOH concentration in mol dm−3, v1 is the volume of the HCl solution in cm3, v2 is the volume of the NaOH solution in cm3, 0.016 is the molecular weight of NH2 in 1 cm3 0.1 mol dm−3 HCl in g, G is the sample weight in g, and W is the amount of water present in the sample in %.
DD (%) – degree of acetylation:
| |  | (2) |
DDA (%) – degree of deacetylation:
where 9.94% is the theoretical NH
2 percentage.
If the obtained chitin was not soluble in the 0.1 mol dm−3 hydrochloric acid, the DDA was determined by using a calibration curve, which is prepared from standard samples of chitin and chitosan with the degree of deacetylation in the range of 6–100%.
3.4 Scanning electron microscopy (SEM)
Chitin and chitosan samples were structurally characterized by using scanning electron microscopy (SEM) (SUPRA 35 VP, Carl Zeiss, Jena, Germany) operating at 1 kV. Electron microscopy was performed at high magnifications; it generated high-resolution images and was used to precisely measure very small changes of sample features.
3.5 Particle chord length distribution
The distribution of powder and ground chitin particles was determined based on in-line particle measurements using a METTLER TOLEDO FBRM® G400 (focused beam reflectance measurement) probe (Mettler Toledo, Columbus, Ohio, USA), which was inserted directly into the suspension of chitin particles in the form of fine powder and flakes (1 g per 50 mL of solvent). The particle size distribution was measured immediately after the addition of the particles using iC FBRM™ software, and the number of particles in a selected size range was acquired. At least three experiments were performed and the average values were calculated.
3.6 Determination of surface area, pore volume and pore size of chitin and chitosan
N2 adsorption–desorption experiments were carried out using an ASAP 2020 instrument (Micromeritics) in order to determine the specific surface area and porosity of the samples. The powder samples (cca. 0.1 g) was outgassed for about 24 h at 50 °C in a vacuum (residual pressure 1.5 × 104 Pa) to ensure complete removal of atmospheric contaminants from the surface before analysis.
3.7 Viscosity of the chitosan solutions
For the determination of the intrinsic viscosity of the chitin samples, 0.5 g chitin was dissolved in 50 mL of 0.1 mol dm−3 acetic acid solution and stirred for 1 h until it is dissolved entirely. The viscosity of the chitin–acetic acid solution was measured using a rotational Fungilab S.A. viscometer. A standard TL7 spindle with a rotation speed of 50 rpm for 10 s at room temperature was used.
3.8 Molecular weight determination of the chitosan solutions
The average molecular weights (Mw) of chitosan were calculated from the measured intrinsic viscosities using the Mark–Houwink relationship (eqn (4))where η is the intrinsic viscosity, Mw is the viscosity average molecular weight and Km and a are constants for the given solute–solvent system and temperature. The viscosity of the chitosan samples was measured as described in section 3.7. The values of the constants a = 0.89 and Km = 2 × 10−5 (dL g−1) were used based on the available light scattering data and literature data of chitosans with initial molecular weights and degrees of acetylation close to those of our sample.43
4.1 Reaction–diffusion model development
A kinetic modelling approach for the heterogeneous reaction of solid chitin with hydroxide ions was based on the reaction–diffusion equation (eqn (5)). For the reaction to occur on the N-acetyl group of the chitin biopolymer, the NaOH solution (OH− and Na+ ions) must first diffuse into the particle. Even though particles of chitin, in reality, have non-uniform geometrical shapes, approximation with a spherical particle was used for the purpose of modelling. The transient reaction–diffusion equation in spherical symmetry, for hydroxyl ions, can be written as follows:| |  | (5) |
where COH− is the molar concentration of the hydroxide ions inside the spherical chitin particles, r is the radial distance, Deff is the effective diffusivity of the hydroxide ions in solid chitin and Rc is the reaction term. The initial condition is:| |  | (6) |
where R is the radius of the chitin particle. Because NaOH in the bulk solution was in excess in the performed experiments, the concentration of OH− in the bulk solution is assumed to be constant and is denoted by COH−,b. Thus, the boundary condition at r = R is:| | | COH−(t,R) = COH−,b, t > 0 | (7) |
At r = 0, the concentration profile for the OH− ions has a minimum because of the symmetry and position in centre of the particle, therefore:
| |  | (8) |
According to experimental observations, the reaction between the chitin's amide group and hydroxide ion was represented as a second order reversible reaction:
| R1 − NHCOCH3 + OH− ⇌ R1 − NH2 + CH3COO− |
and the reaction term can be expressed as
eqn (9):
| | | Rc = kfCchCOH− − kr(Cch,0 − Cch)2 | (9) |
where
Cch,0 denotes the chitin concentration in the solid particles at time
t = 0 and
Cch is the chitin concentration which depends on time and radial distance. The two kinetic rate constants are dependent on the temperature according to the Arrhenius law. In general:
| |  | (10) |
where
Rg is the gas constant. The kinetic rate constant was obtained by fitting the experimental data at 60 °C (333.15 K). The reaction rate constants at other temperatures can be calculated from the modified Arrhenius equations:
| |  | (11) |
| |  | (12) |
where
kf is the reaction rate constant for the forward reaction (
eqn (11)) and
kr is the reaction rate constant for the reverse reaction (
eqn (12)) and
T60 = 333.15 K.
Because only OH− diffuses into the particles, chitin consumption by the reaction can be written as (eqn (13)):
| |  | (13) |
with the initial conditions:
t = 0, 0 ≤
r ≤
R,
Cch =
Cch,0.
4.2 Numerical solution
The governing equations were solved for all particles in a batch reactor. Particle size distributions in two separate experiments with flakes and with powder were obtained using an in-line particle tracking probe. Both particle size distributions were used and compared with the corresponding experimental data. The partial differential equation (eqn (5)) was spatially discretized by using the Chebyshev spectral collocation method,24 and the implicit Euler method was used for temporal discretization. 50 points (n = 50) were used for the former and 1000 points for the latter discretization. The computational domain for the particle with radius Rj was (t,r) ∈ [0,tmax] × [0,Rj] where tmax was 14 h. The resulting algebraic equations were solved for all particle size classes in the particle size distribution. The Python 3.5 programing language with the NumPy package was used to solve the equations. The solution for the chitin concentration inside the particles for every particle class was calculated in each time step by the explicit forward Euler method, since the reaction term is nonlinear in Cch. For a certain particle size one can get the concentration profile Cch(t,r). There are m different particle sizes with an average radius of Rj where 1 ≤ j ≤ m. The number of particles of that size is denoted by Nj. The average concentration of chitin in one particle with radius Rj can be calculated according to equation (eqn (14)):| |  | (14) |
where  .
.
This must be done for all size classes of particles. The average concentration in all of the particles can be calculated according to following equation:
| |  | (15) |
where
Vch is the total volume of chitin in the reactor,
Nj is the number of particles and
Cch,avg(
t) is a function of time only.
5. Conclusions
Chitosan transformation by N-deacetylation from chitin in a heterogeneous reaction system, which is the most frequently used commercial method, has been described by a unique novel model based approach, which integrates both kinetics and transport phenomena. Additionally, the data for particle morphology and their size distribution in the batch reactor system were incorporated in the model. The selected mathematical relationship was in accordance with the mechanisms, granting a relatively good agreement for the cumulative experimental data by regarding the kinetics in the initial consumption phase and the subsequent transport resistance for OH−. The developed descriptive approach, together with inline measuring methods, could enable a more comprehensive option for commercial productivity increase, as well as unit operations scale-up.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was facilitated by the Mar3Bio project, financed through the first call of the Marine Biotechnology ERA-NET (funded under the European Commission's Seventh Framework Programme) and the BioApp project (Interreg V-A Italy-Slovenia 2014–2020), its operation is co-funded by the European Regional Development Fund; the authors also acknowledge the financial support from the Slovenian Research Agency (research core funding No. P2-0152) as well as Ana Bjelić's SEM analyses.
References
-
D. L. Kaplan, Biopolymers from Renewable Resources, Springer-Verlag Berlin Heidelberg, New York, 1st edn, 2008 Search PubMed.
-
S. K. Kim, Chitin, Chitosan, Oligosaccharides and Their Derivatives:Biological Activities and Applications, 1st edition, CRC Press, Taylor and Francis, Boka Raton, FL, USA, 2010 Search PubMed.
-
FAO, The State of World Fisheries and Aquaculture 2014, Rome, 2014, p. 223 Search PubMed.
- D. Elieh-Ali-Komi and M. R. Hamblin, Chitin and Chitosan: Production and Application of Versatile Biomedical Nanomaterials, Int. J. Adv. Res., 2016, 4, 411–427 CAS.
- C. J. Brine and P. R. Austin, Chitin variability with species and method of preparation, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1981, 69, 283–286 CrossRef.
- E. F. Franca, R. D. Lins, L. C. G. Freitas and T. P. Straatsma, Characterization of Chitin and Chitosan Molecular Structure in Aqueous Solution, J. Chem. Theory Comput., 2008, 4, 2141–2149 CrossRef CAS PubMed.
- F. Khoushab and M. Yamabhai, Chitin Research Revisited, Mar. Drugs, 2010, 8, 1988–2012 CrossRef CAS PubMed.
-
G. Lodhi, Y. S. Kim, J. W. Hwang, S. K. Kim, Y. J. Jeon, J. Y. Je, C. B. Ahn, S. H. Moon, B. T. Jeon and P. J. Park, Chitooligosaccharide and Its Derivatives: Preparation and Biological Applications, BioMed Research International, 2014, vol. 13, pp. 380–701 Search PubMed.
- Z. Mohammedi, Chitosan and Chitosan Oligosaharides: Aplications in Medicine, Agriculture and Biotechnology, Int. J. Mol. Sci., 2017, 2, 102–106 Search PubMed.
- B. Bellich, I. D'Agostino, S. Semeraro, A. Gamini and A. Cesaro, “The Good, the Bad and the Ugly” of Chitosans, Mar. Drugs., 2016, 14, 99 CrossRef PubMed.
- S. Aiba, Preparation of N-acetylchitooligosaccharides by hydrolysis of chitosan with chitinase followed by N-acetylation, Carbohydr. Res., 1994, 265, 323–328 CrossRef CAS PubMed.
- I. Younes and M. Rinaudo, Chitin and chitosan preparation from marine sources. Structure, properties and applications, Mar. Drugs., 2015, 13, 1133–1174 CrossRef CAS PubMed.
- B. K. Park and M. M. Kim, Applications of chitin and its derivatives in biological medicine, Int. J. Mol. Sci., 2010, 11, 5152–5164 CrossRef CAS PubMed.
- V. Eijsink, I. Hoell and G. V. Kolstada, Structure and function of enzymes acting on chitin and chitosan, Biotechnol. Genet. Eng. Rev., 2010, 27, 331–366 CrossRef PubMed.
- G. Lamarque, G. Chaussard and A. Domard, Thermodynamic Aspects of the Heterogeneous Deacetylation of α-Chitin: Reaction Mechanisms, Biomacromolecules, 2007, 8, 1942–1950 CrossRef CAS PubMed.
- G. Lamarque, C. Viton and A. Domard, Comparative study of the second and third heterogeneous deacetylations of alpha- and beta-chitins in a multistep process, Biomacromolecules, 2004, 5, 1899–1907 CrossRef CAS PubMed.
- G. Lamarque, M. Cretenet, C. Viton and A. Domard, New route of deacetylation of alpha- and beta-chitins by means of freeze-pump out-thaw cycles, Biomacromolecules, 2005, 6, 1380–1388 CrossRef CAS PubMed.
- D. Zahn, On the Role of Water in Amide Hydrolysis, Eur. J. Org. Chem., 2004, 19, 4020–4023 CrossRef.
- P. Methacanon, M. Prasitsilp, T. Pothsree and J. Pattaraarchachai, Heterogeneous N-deacetylation of squid chitin in alkaline solution, Carbohydr. Polym., 2003, 52, 119–123 CrossRef CAS.
- H. Ahlafi, H. Moussout, F. Boukhlifi and M. Echetna, Kinetics of N-Deacetylation of Chitin Extracted from Shrimp Shells Collected from Coastal Area of Morocco, Mediterr. J. Chem., 2013, 2, 503–513 CrossRef CAS.
- T. G. Liu, B. Li, W. Huang, B. Lv, J. Chen, J. X. Zhang and L. P. Zhu, Effects and kinetics of a novel temperature cycling treatment on the N-deacetylation of chitin in alkaline solution, Carbohydr. Polym., 2009, 77, 110–117 CrossRef CAS.
- K. L. B. Chang, G. Tsai, J. Lee and W. Fu, Heterogeneous N-deacetylation of chitin in alkaline solution, Carbohydr. Polym., 1997, 303, 327–332 CrossRef CAS.
- M. Rinaudo, Chitin and chitosan, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- G. Cárdenas, G. Cabrera, E. Taboada and S. P. Miranda, Chitin characterisation by SEM, TIR, XRD and C cross polarisation/Mass Ange Spinning NMR, Appl. Polym. Sci., 2004, 93, 1876–1885 CrossRef.
- N. Raab and I. Bachelet, Resolving biofilm topography by native scanning electron microscopy, J. Biol. Methods., 2017, 4, e70, DOI:10.14440/jbm.2017.173.
- D. Raabe, C. Sachs and P. Romano, The crustacean exoskeleton as an example of a structurally and mechanically graded biological nanocomposite material, Acta Mater., 2005, 53, 4281–4292 CrossRef CAS.
- C. Liu, G. Wang, W. Sui, L. An and Ch. Si, Preparation and Characterization of Chitosan by a Novel Deacetylation Approach Using Glycerol as Green Reaction Solvent, ACS Sustainable Chem. Eng., 2017, 5, 4690–4698 CrossRef CAS.
- P. Mathacanon, M. Prasitsilp, T. Pothsree and J. Pattaraarahacai, Heterogeneous N-deacetylation of squid chitin in alkaline solution, Carbohydr. Polym., 2003, 52, 119–123 CrossRef.
- X. F. Guo, F. K. Kikuchi, Y. Matahir, K. Sakai and K. K. Ogawa, Water soluble chitin of low degree of deacetylation, J. Carbohydr. Chem., 2002, 21, 149–161 CrossRef CAS.
- S. Baxter, S. Zivanovic and J. Weiss, Molecular weight and degree of acetylation of high-intensity ultrasonicated chitosan, Food Hydrocolloids, 2005, 19, 821–830 CrossRef CAS.
- M. Bajaj, J. Winter and C. Gallert, Effect of deproteination and deacetylation conditions on the viscosity of chitin and chitosan from Crangon crangon shrimp shells, Biochem. Eng. J., 2011, 56, 51–62 CrossRef CAS.
- L. J. H. Foster, J. Hook, M. Basuki and H. Marcal, Chitosan as a Biomaterial: Influence of Degree of Deacetylation on Its Physiochemical, Material and Biological Properties, PLoS One, 2015, 10, 140–220 Search PubMed.
- Y. Zhang, C. Xue, Y. Xue, R. Gao and X. Zhong, Determination of the degree of deacetylation of chitin and chitosan by X ray powder diffraction, Carbohydr. Res., 2005, 340, 1914–1917 CrossRef CAS PubMed.
- D. Bakowies and P. A. Kollman, Theoretical Study of Base-Catalyzed Amide Hydrolysis : Gas- and Aqueous-Phase Hydrolysis of Formamide, J. Am. Chem. Soc., 1999, 121, 5712–5726 CrossRef CAS.
- R. S. Brown, A. J. Bamnet and H. S. Tilk, Recent perspectives concerning the mechanism of H3O+and hydroxide-promoted amide hydrolysis, J. Phys. Chem., 1992, 25, 338–341 Search PubMed.
- X. Lopez, J. In, G. M. Blackburn, M. Karplus, T. Uni, S. Sheffield and L. D. C. Biophysique, Alkaline Hydrolysis of Amide Bonds : Effect of Bond Twist and Nitrogen Pyramidalization, J. Phys. Chem. A, 2003, 107, 2304–2315 CrossRef CAS.
- Y. Xiong and C. Zhan, Theoretical Studies of the Transition-State Structures and Free Energy Barriers for Base-Catalyzed Hydrolysis of Amides, J. Phys. Chem. A, 2006, 110, 12644–12652 CrossRef CAS PubMed.
- L. Gu, S. Zhu and A. N. Hrymak, Acidic and Basic Hydrolysis of Poly (N-vinylformamide), J. Appl. Polym. Sci., 2002, 86, 3412–3419 CrossRef CAS.
- K. K. Liang, B. Chang, G. Tsai, J. Lee and W. Fu, Heterogeneous N-deacetylation of chitin in alkaline solution, Carbohydr. Res., 1997, 303, 327–332 CrossRef.
- H. Wang, G. G. Robin and D. Rogers, Dissolution of Biomass Using Ionic Liquids, Green Chem., 2013, 11, 11128–11138 Search PubMed.
- Daniel Elieh-Ali-Komi and M. R. Hamblin, Chitin and chitosan: Production and Application of Versatile Biomedical Nanomaterials, Int. J. Adv. Res., 2016, 4, 411–427 CAS.
- G. G. Miralles, I. Panos, A. Santiago and A. Heras, N-Deacetylation and depolymerization reactions of chitin/chitosan: Influence of the source of chitin, Carbohydr. Polym., 2005, 62, 316–320 CrossRef.
- M. E. M. Sameeh and M. E. Gamel, Numerical solution of singular two-point boundary value problems by the collocation method with the Chebyshev bases, SeMA Journal, 2016, 74, 627–641 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Blaž
Likozar
* and
Blaž
Likozar













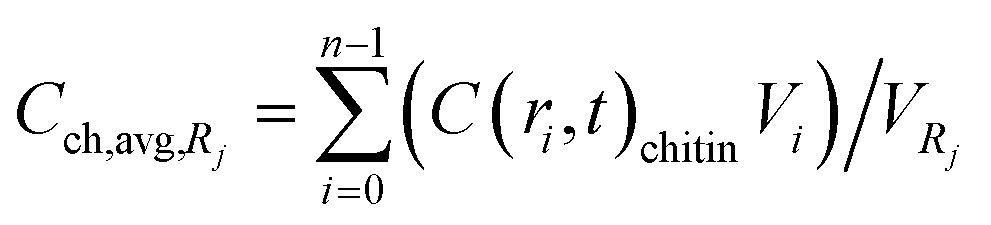
 .
.
